
ITN—VIROINF: Understanding (Harmful) Virus-Host Interactions by Linking Virology and Bioinformatics

 , , , , , , , , , , , , , , , , and
, , , , , , , , , , , , , , , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
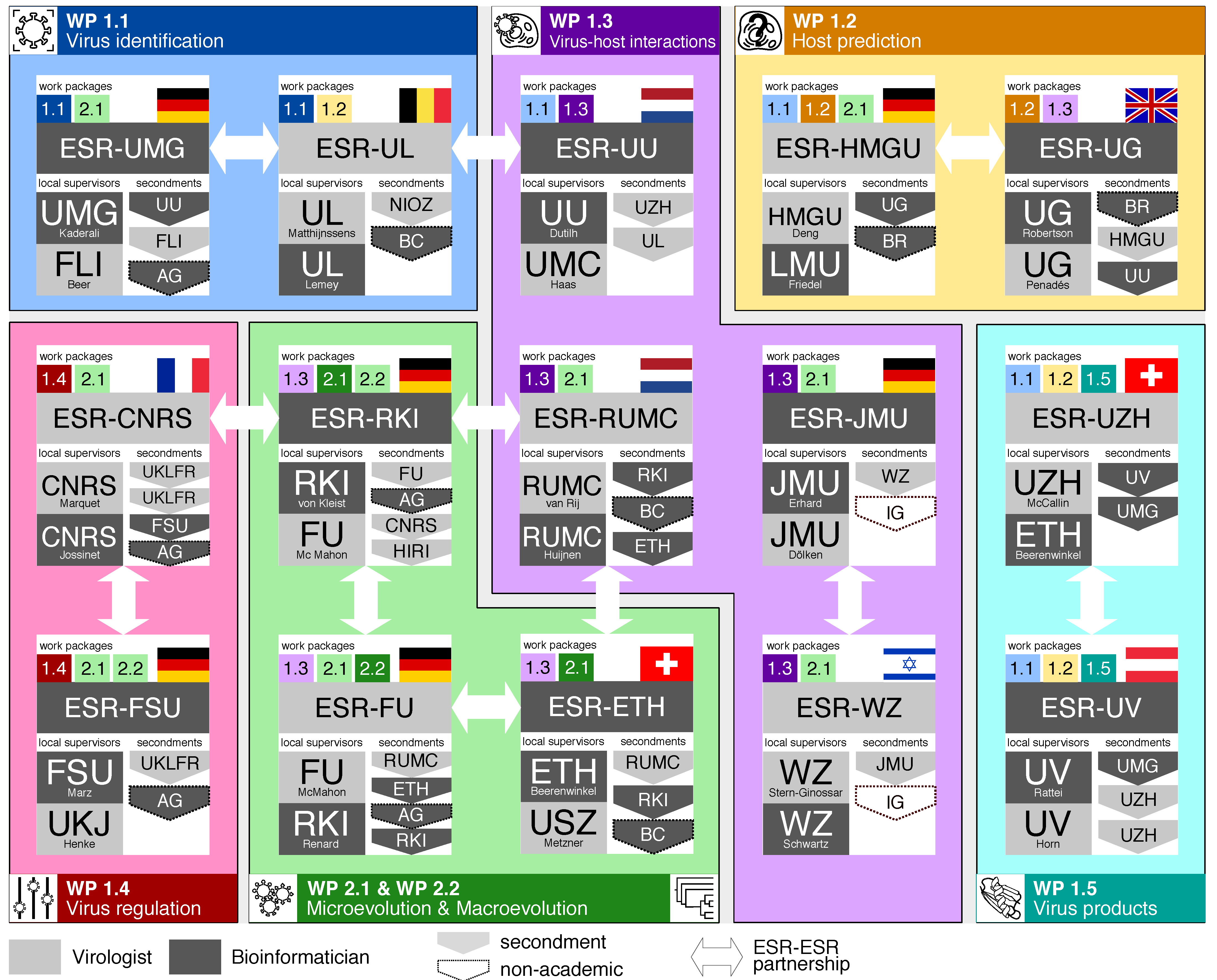
2. The Research Programme
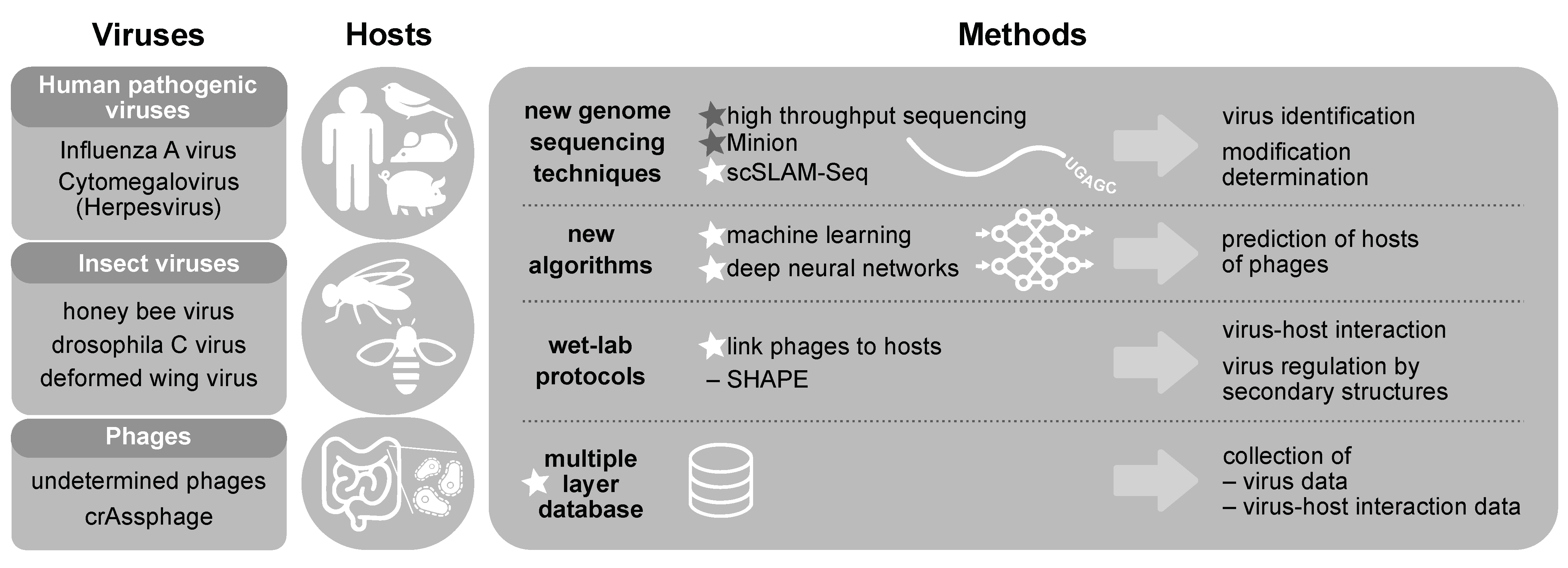
3. Modelling Virus-Host Interactions
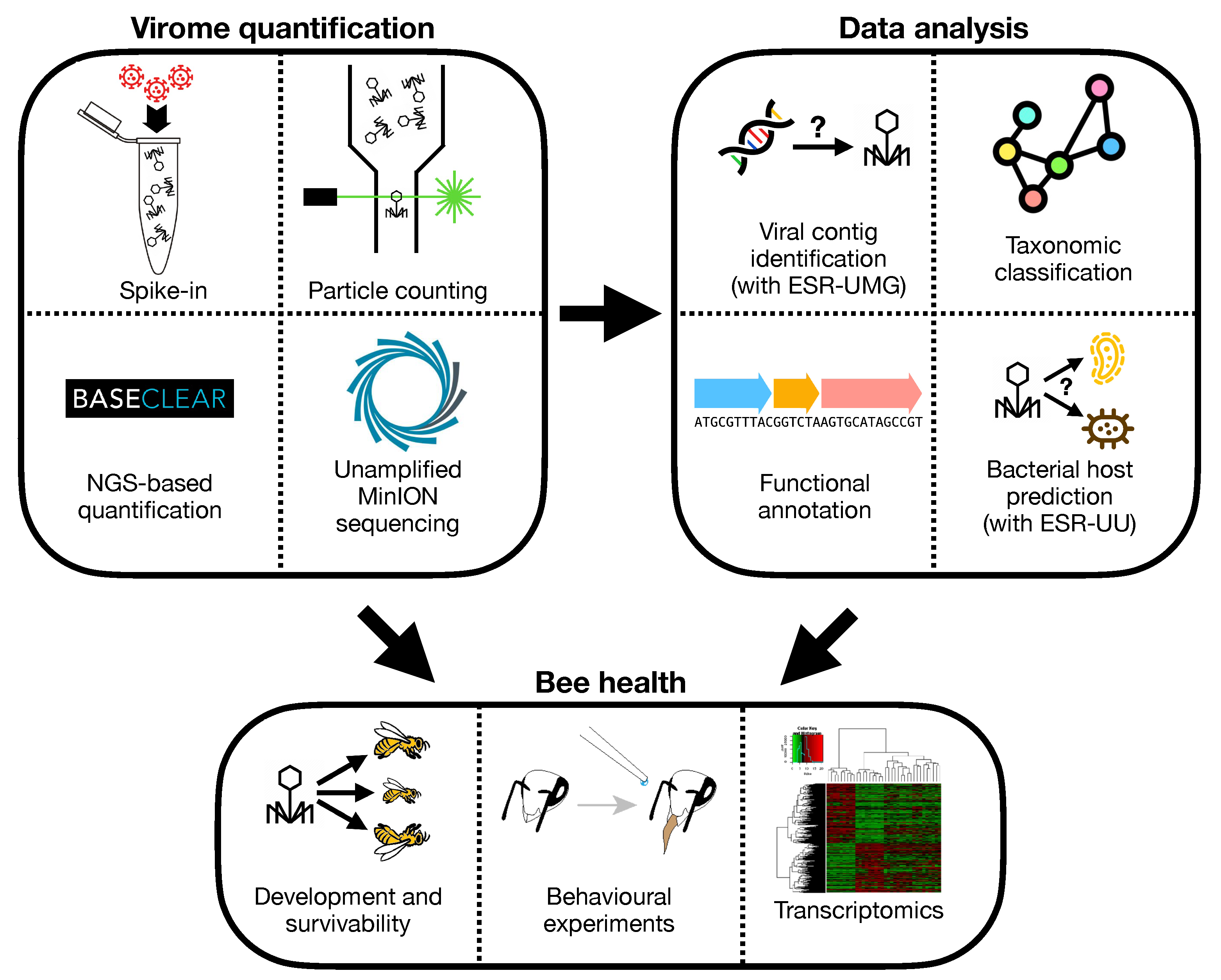
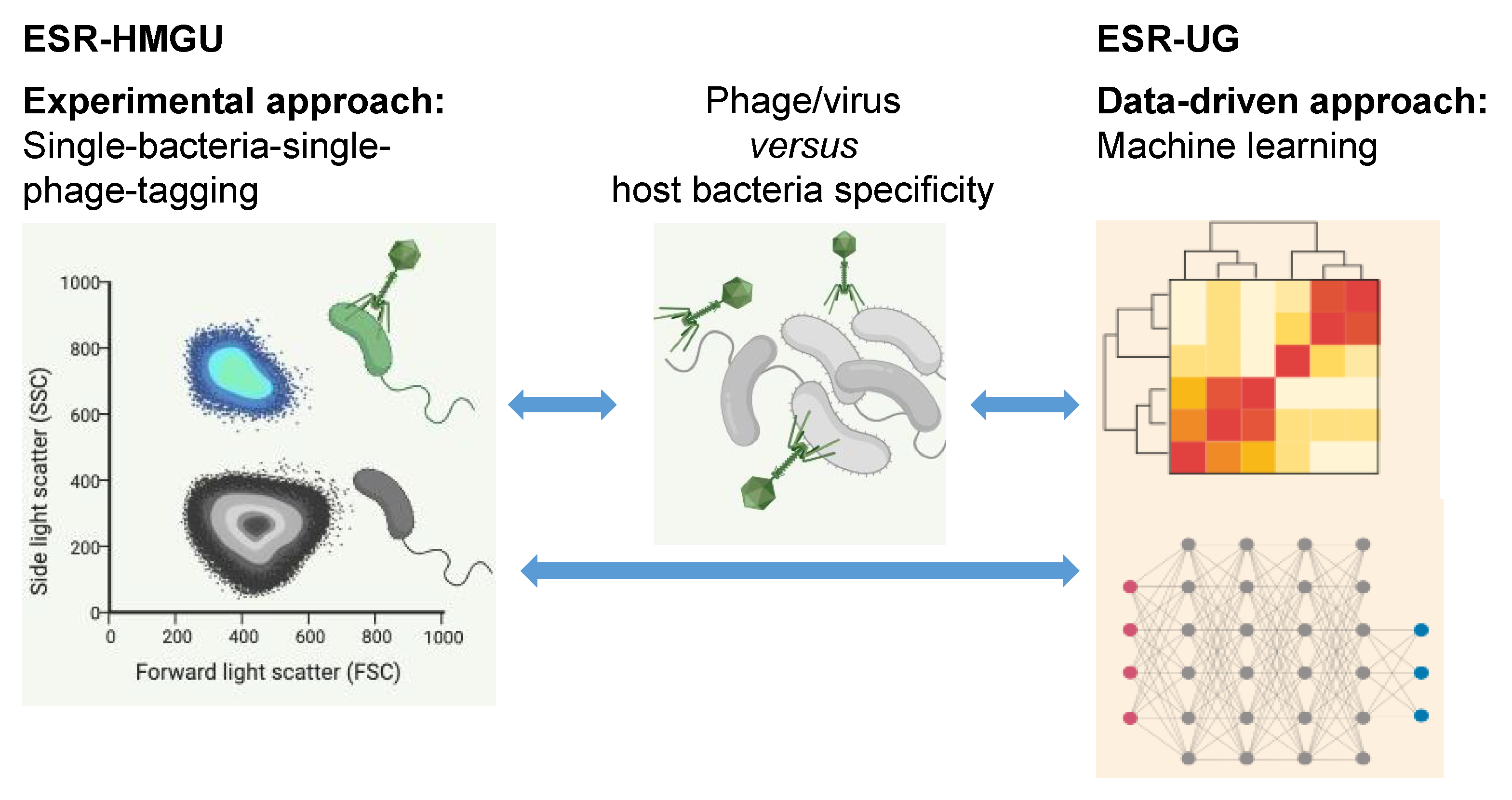
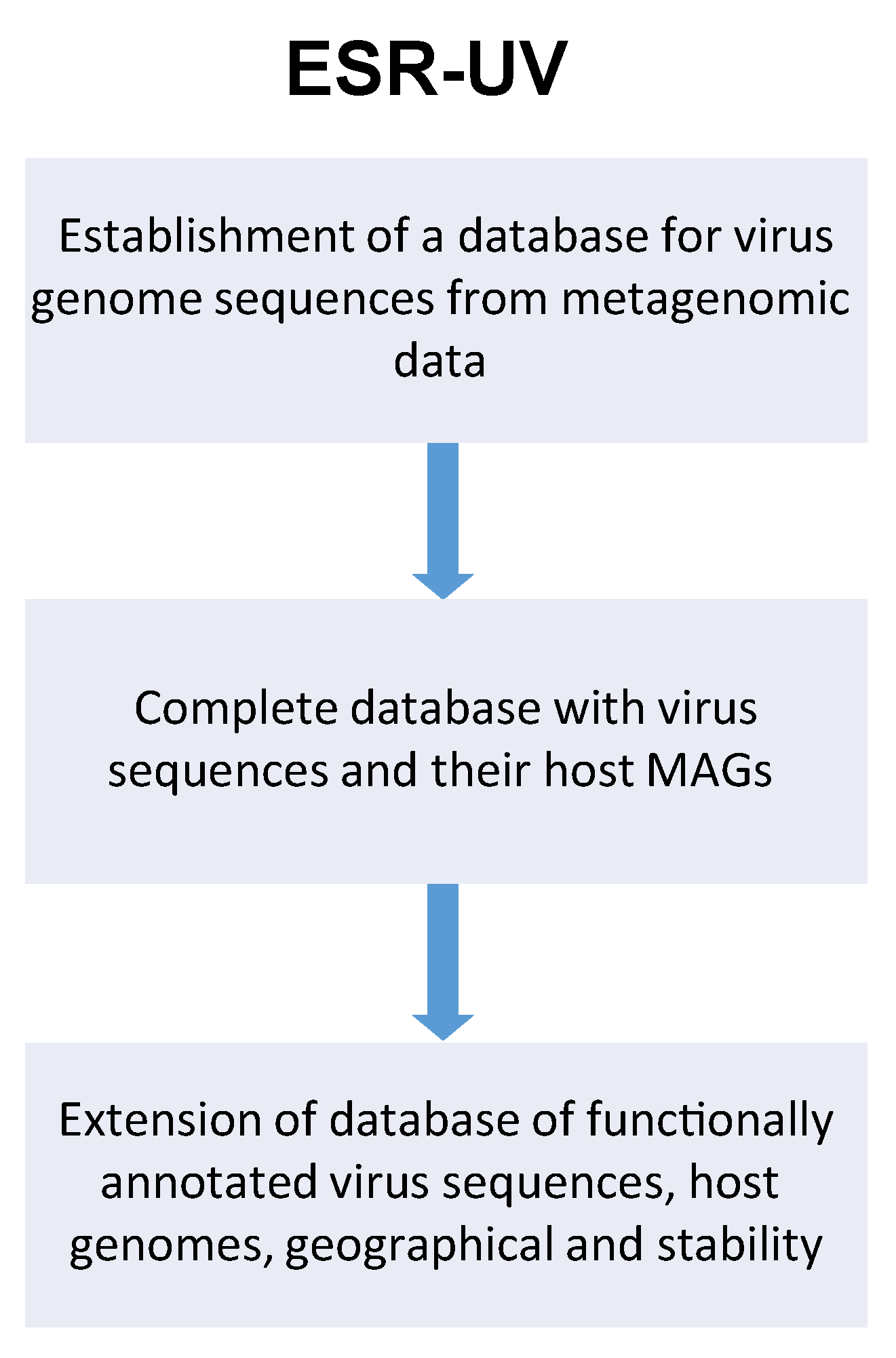
3.1. Virus Identification
3.2. Host Prediction
3.3. Virus-Host Interactions
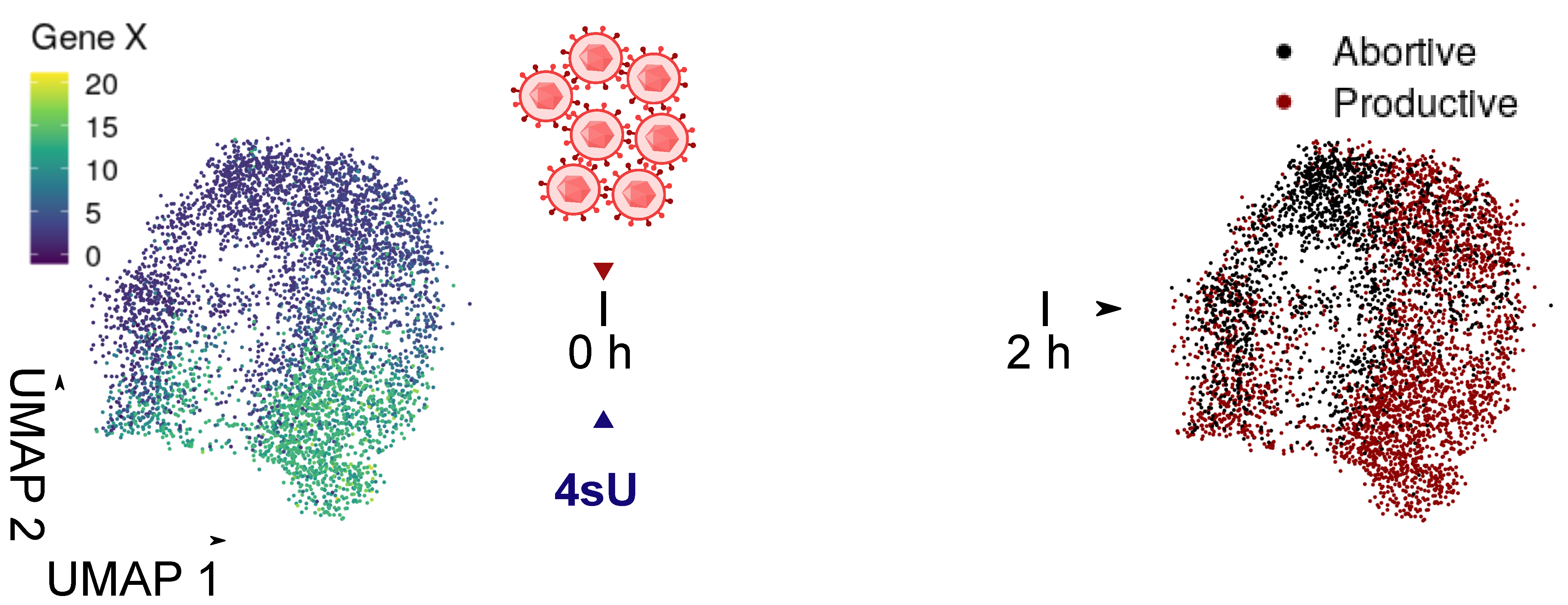
3.4. Virus Regulation
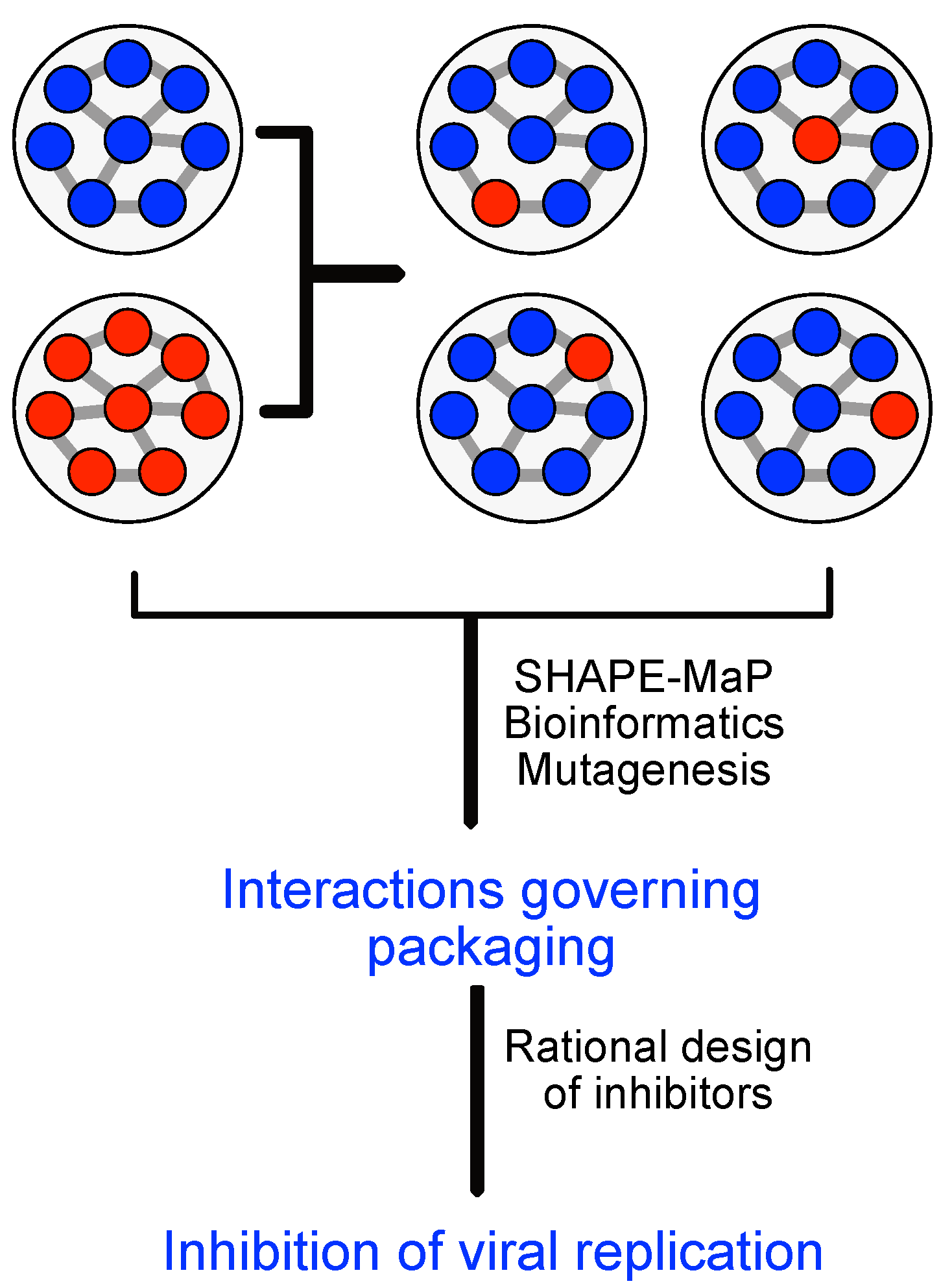
3.5. Virus Products
4. Modelling Virus Evolution in Hosts
4.1. Microevolution: Virus Quasispecies
4.2. Natural Selection of Viruses
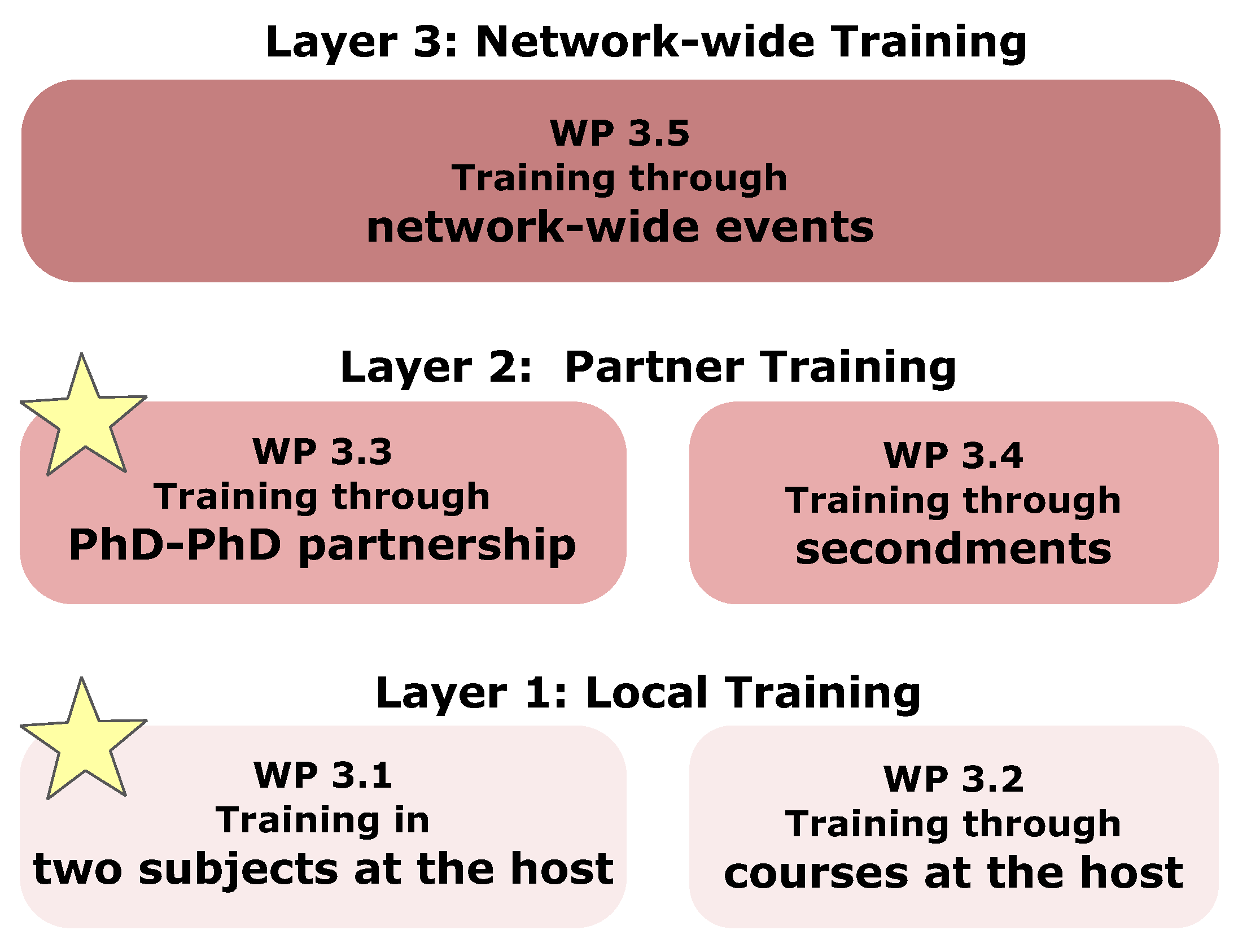
5. Training
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Hufsky, F.; Ibrahim, B.; Modha, S.; Clokie, M.R.J.; Deinhardt-Emmer, S.; Dutilh, B.E.; Lycett, S.; Simmonds, P.; Thiel, V.; Abroi, A.; et al. The Third Annual Meeting of the European Virus Bioinformatics Center. Viruses 2019, 11, 420. [Google Scholar] [CrossRef] [Green Version]
- Ibrahim, B.; Arkhipova, K.; Andeweg, A.C.; Posada-Cespedes, S.; Enault, F.; Gruber, A.; Koonin, E.V.; Kupczok, A.; Lemey, P.; McHardy, A.C.; et al. Bioinformatics Meets Virology: The European Virus Bioinformatics Center’s Second Annual Meeting. Viruses 2018, 10, 256. [Google Scholar] [CrossRef] [Green Version]
- Marz, M.; Beerenwinkel, N.; Drosten, C.; Fricke, M.; Frishman, D.; Hofacker, I.L.; Hoffmann, D.; Middendorf, M.; Rattei, T.; Stadler, P.F.; et al. Challenges in RNA virus bioinformatics. Bioinformatics 2014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conceicao-Neto, N.; Yinda, K.C.; Van Ranst, M.; Matthijnssens, J. NetoVIR: Modular Approach to Customize Sample Preparation Procedures for Viral Metagenomics. Methods Mol. Biol. 2018, 1838, 85–95. [Google Scholar] [CrossRef]
- Lauber, C.; Seitz, S.; Mattei, S.; Suh, A.; Beck, J.; Herstein, J.; Börold, J.; Salzburger, W.; Kaderali, L.; Briggs, J.A.G.; et al. Deciphering the Origin and Evolution of Hepatitis B Viruses by Means of a Family of Non-enveloped Fish Viruses. Cell Host Microbe 2017, 22, 387–399.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krupovic, M.; Prangishvili, D.; Hendrix, R.W.; Bamford, D.H. Genomics of bacterial and archaeal viruses: Dynamics within the prokaryotic virosphere. Microbiol. Mol. Biol. Rev. MMBR 2011, 75, 610–635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, L.; Ignacio-Espinoza, J.C.; Gregory, A.C.; Poulos, B.T.; Weitz, J.S.; Hugenholtz, P.; Sullivan, M.B. Viral tagging reveals discrete populations in Synechococcus viral genome sequence space. Nature 2014, 513, 242–245. [Google Scholar] [CrossRef] [PubMed]
- Penadés, J.R.; Chen, J.; Quiles-Puchalt, N.; Carpena, N.; Novick, R.P. Bacteriophage-mediated spread of bacterial virulence genes. Curr. Opin. Microbiol. 2015, 23, 171–178. [Google Scholar] [CrossRef]
- Khan Mirzaei, M.; Xue, J.; Costa, R.; Ru, J.; Schulz, S.; Taranu, Z.E.; Deng, L. Challenges of Studying the Human Virome—Relevant Emerging Technologies. Trends Microbiol. 2020. [Google Scholar] [CrossRef]
- Deng, L.; Gregory, A.; Yilmaz, S.; Poulos, B.T.; Hugenholtz, P.; Sullivan, M.B. Contrasting Life Strategies of Viruses that Infect Photo- and Heterotrophic Bacteria, as Revealed by Viral Tagging. mBio 2012, 3. [Google Scholar] [CrossRef] [Green Version]
- Dzunkova, M.; Low, S.J.; Daly, J.N.; Deng, L.; Rinke, C.; Hugenholtz, P. Defining the human gut host-phage network through single-cell viral tagging. Nat. Microbiol. 2019, 4, 2192–2203. [Google Scholar] [CrossRef]
- Dutilh, B.E.; Cassman, N.; McNair, K.; Sanchez, S.E.; Silva, G.G.Z.; Boling, L.; Barr, J.J.; Speth, D.R.; Seguritan, V.; Aziz, R.K.; et al. A highly abundant bacteriophage discovered in the unknown sequences of human faecal metagenomes. Nat. Commun. 2014, 5, 4498. [Google Scholar] [CrossRef] [Green Version]
- Edwards, R.A.; Vega, A.A.; Norman, H.M.; Ohaeri, M.; Levi, K.; Dinsdale, E.A.; Cinek, O.; Aziz, R.K.; McNair, K.; Barr, J.J.; et al. Global phylogeography and ancient evolution of the widespread human gut virus crAssphage. Nat. Microbiol. 2019, 4, 1727–1736. [Google Scholar] [CrossRef] [Green Version]
- Shkoporov, A.N.; Khokhlova, E.V.; Fitzgerald, C.B.; Stockdale, S.R.; Draper, L.A.; Ross, R.P.; Hill, C. ΦCrAss001 represents the most abundant bacteriophage family in the human gut and infects Bacteroides intestinalis. Nat. Commun. 2018, 9, 4781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shkoporov, A.N.; Stockdale, S.R.; Adriaenssens, E.M.; Yutin, N.; Koonin, E.; Dutilh, B.E.; Krupovic, M.; Edwards, R.A.; Tolstoy, I.; Hill, C. Create One New Order (Crassvirales) Including Six New Families, Ten New Subfamilies, 78 New Genera and 279 New Species. ICTV Taxonomy Proposal 2020.039B. 2020. Available online: https://talk.ictvonline.org/files/proposals/taxonomy_proposals_prokaryote1 (accessed on 15 April 2021).
- Yutin, N.; Benler, S.; Shmakov, S.A.; Wolf, Y.I.; Tolstoy, I.; Rayko, M.; Antipov, D.; Pevzner, P.A.; Koonin, E.V. Unique genomic features of crAss-like phages, the dominant component of the human gut virome. bioRxiv 2020. [Google Scholar] [CrossRef]
- Erhard, F.; Baptista, M.A.P.; Krammer, T.; Hennig, T.; Lange, M.; Arampatzi, P.; Jurges, C.S.; Theis, F.J.; Saliba, A.E.; Dolken, L. scSLAM-seq reveals core features of transcription dynamics in single cells. Nature 2019, 571, 419–423. [Google Scholar] [CrossRef]
- Jürges, C.; Dölken, L.; Erhard, F. Dissecting newly transcribed and old RNA using GRAND-SLAM. Bioinformatics 2018, 34, i218–i226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bronkhorst, A.W.; van Rij, R.P. The long and short of antiviral defense: Small RNA-based immunity in insects. Curr. Opin. Virol. 2014, 7, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Palmer, W.H.; Hadfield, J.D.; Obbard, D.J. RNA-Interference Pathways Display High Rates of Adaptive Protein Evolution in Multiple Invertebrates. Genetics 2018, 208, 1585–1599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Obbard, D.J.; Linton, Y.M.; Jiggins, F.M.; Yan, G.; Little, T.J. Population genetics of Plasmodium resistance genes in Anopheles gambiae: No evidence for strong selection. Mol. Ecol. 2007, 16, 3497–3510. [Google Scholar] [CrossRef]
- van Rij, R.P.; Saleh, M.C.; Berry, B.; Foo, C.; Houk, A.; Antoniewski, C.; Andino, R. The RNA silencing endonuclease Argonaute 2 mediates specific antiviral immunity in Drosophila melanogaster. Genes Dev. 2006, 20, 2985–2995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Mierlo, J.T.; Bronkhorst, A.W.; Overheul, G.J.; Sadanandan, S.A.; Ekstrom, J.O.; Heestermans, M.; Hultmark, D.; Antoniewski, C.; van Rij, R.P. Convergent evolution of argonaute-2 slicer antagonism in two distinct insect RNA viruses. PLoS Pathog. 2012, 8, e1002872. [Google Scholar] [CrossRef] [PubMed]
- Desirò, D.; Hölzer, M.; Ibrahim, B.; Marz, M. SilentMutations (SIM): A tool for analyzing long-range RNA-RNA interactions in viral genomes and structured RNAs. Virus Res. 2019, 260, 135–141. [Google Scholar] [CrossRef]
- Li, A.X.; Marz, M.; Qin, J.; Reidys, C.M. RNA-RNA interaction prediction based on multiple sequence alignments. Bioinformatics 2011, 27, 456–463. [Google Scholar] [CrossRef]
- Fancello, L.; Raoult, D.; Desnues, C. Computational tools for viral metagenomics and their application in clinical research. Virology 2012, 434, 162–174. [Google Scholar] [CrossRef] [Green Version]
- Huson, D.H.; Auch, A.F.; Qi, J.; Schuster, S.C. MEGAN analysis of metagenomic data. Genome Res. 2007, 17, 377–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, T.S.; Mohammed, M.H.; Komanduri, D.; Mande, S.S. ProViDE: A software tool for accurate estimation of viral diversity in metagenomic samples. Bioinformation 2011, 6, 91–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edwards, R.A.; Rohwer, F. Viral metagenomics. Nat. Rev. Microbiol. 2005, 3, 504–510. [Google Scholar] [CrossRef]
- Barzon, L.; Lavezzo, E.; Militello, V.; Toppo, S.; Palu, G. Applications of next-generation sequencing technologies to diagnostic virology. Int. J. Mol. Sci. 2011, 12, 7861–7884. [Google Scholar] [CrossRef] [Green Version]
- Smyth, R.P.; Despons, L.; Huili, G.; Bernacchi, S.; Hijnen, M.; Mak, J.; Jossinet, F.; Weixi, L.; Paillart, J.C.; von Kleist, M.; et al. Mutational interference mapping experiment (MIME) for studying RNA structure and function. Nat. Methods 2015, 12, 866–872. [Google Scholar] [CrossRef]
- Smyth, R.P.; Smith, M.R.; Jousset, A.C.; Despons, L.; Laumond, G.; Decoville, T.; Cattenoz, P.; Moog, C.; Jossinet, F.; Mougel, M.; et al. In cell mutational interference mapping experiment (in cell MIME) identifies the 5’ polyadenylation signal as a dual regulator of HIV-1 genomic RNA production and packaging. Nucleic Acids Res. 2018, 46, e57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, M.R.; Smyth, R.P.; Marquet, R.; von Kleist, M. MIMEAnTo: Profiling functional RNA in mutational interference mapping experiments. Bioinformatics 2016, 32, 3369–3370. [Google Scholar] [CrossRef]
- Ronquist, F.; Huelsenbeck, J.P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 2003, 19, 1572–1574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- McMahon, D.P.; Wilfert, L.; Paxton, R.J.; Brown, M.J.F. Emerging Viruses in Bees: From Molecules to Ecology. Adv. Virus Res. 2018, 101, 251–291. [Google Scholar] [CrossRef]













Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Goettsch, W.; Beerenwinkel, N.; Deng, L.; Dölken, L.; Dutilh, B.E.; Erhard, F.; Kaderali, L.; von Kleist, M.; Marquet, R.; Matthijnssens, J.; et al. ITN—VIROINF: Understanding (Harmful) Virus-Host Interactions by Linking Virology and Bioinformatics. Viruses 2021, 13, 766. https://0-doi-org.brum.beds.ac.uk/10.3390/v13050766
Goettsch W, Beerenwinkel N, Deng L, Dölken L, Dutilh BE, Erhard F, Kaderali L, von Kleist M, Marquet R, Matthijnssens J, et al. ITN—VIROINF: Understanding (Harmful) Virus-Host Interactions by Linking Virology and Bioinformatics. Viruses. 2021; 13(5):766. https://0-doi-org.brum.beds.ac.uk/10.3390/v13050766
Chicago/Turabian StyleGoettsch, Winfried, Niko Beerenwinkel, Li Deng, Lars Dölken, Bas E. Dutilh, Florian Erhard, Lars Kaderali, Max von Kleist, Roland Marquet, Jelle Matthijnssens, and et al. 2021. "ITN—VIROINF: Understanding (Harmful) Virus-Host Interactions by Linking Virology and Bioinformatics" Viruses 13, no. 5: 766. https://0-doi-org.brum.beds.ac.uk/10.3390/v13050766