
Patterns in Genotype Composition of Indian Isolates of the Bombyx mori Nucleopolyhedrovirus and Bombyx mori Bidensovirus

 ,
,  , and
, and
Abstract
:1. Introduction
2. Materials and Methods
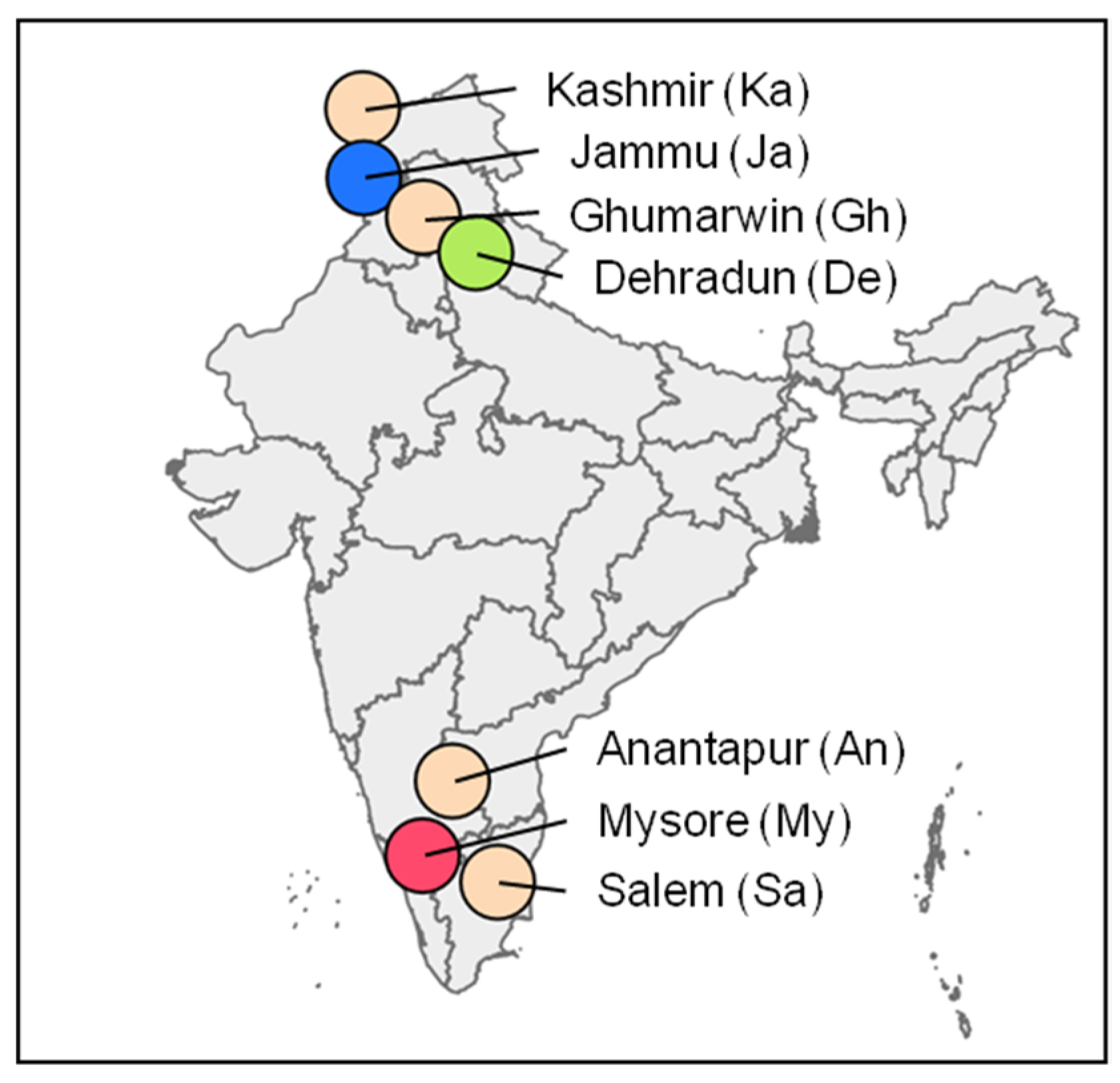
2.1. Insects and Viruses
2.2. Extraction of Viral DNA
2.3. Partial Gene Analysis
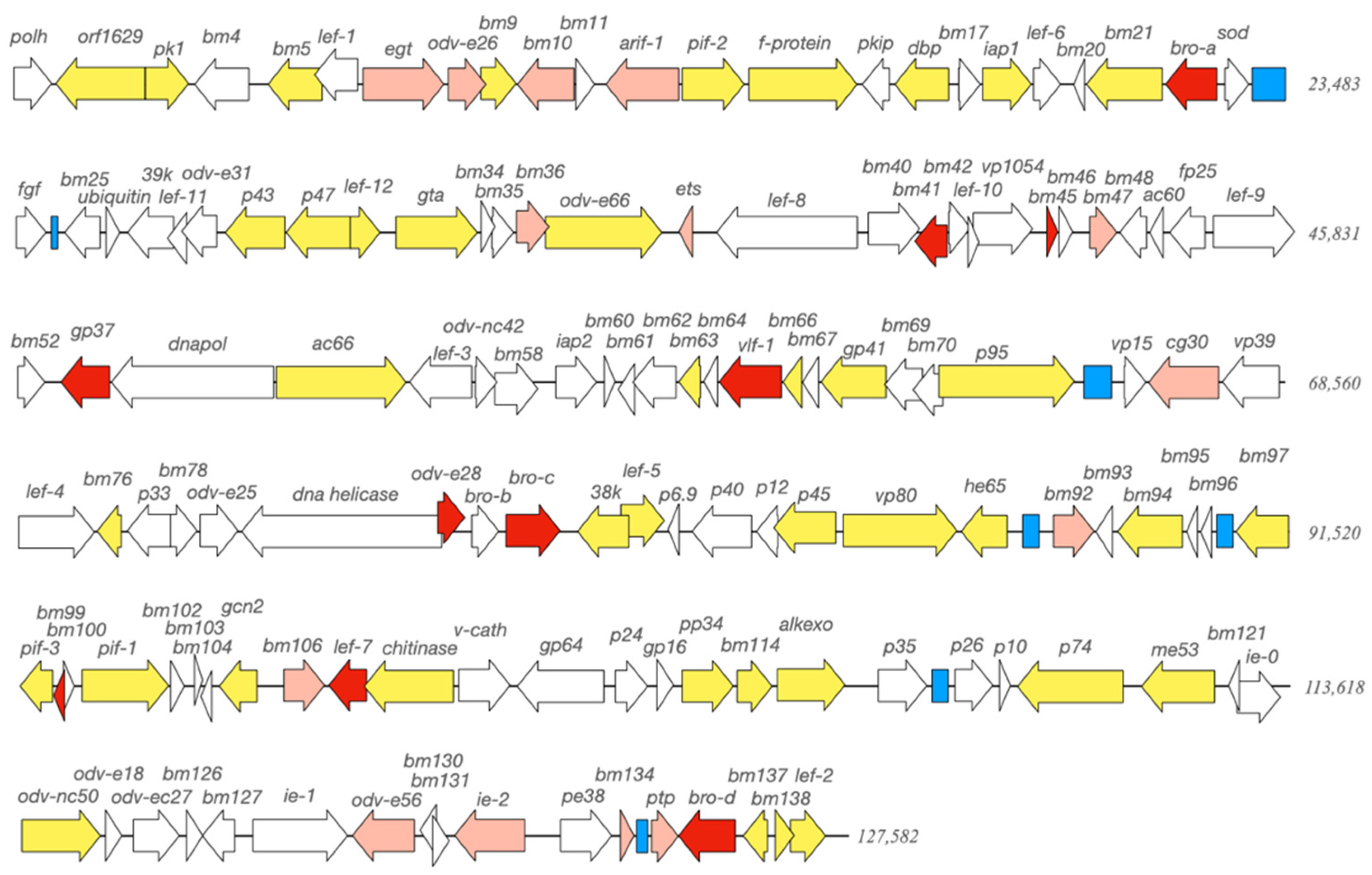
2.4. Whole Genome Sequencing
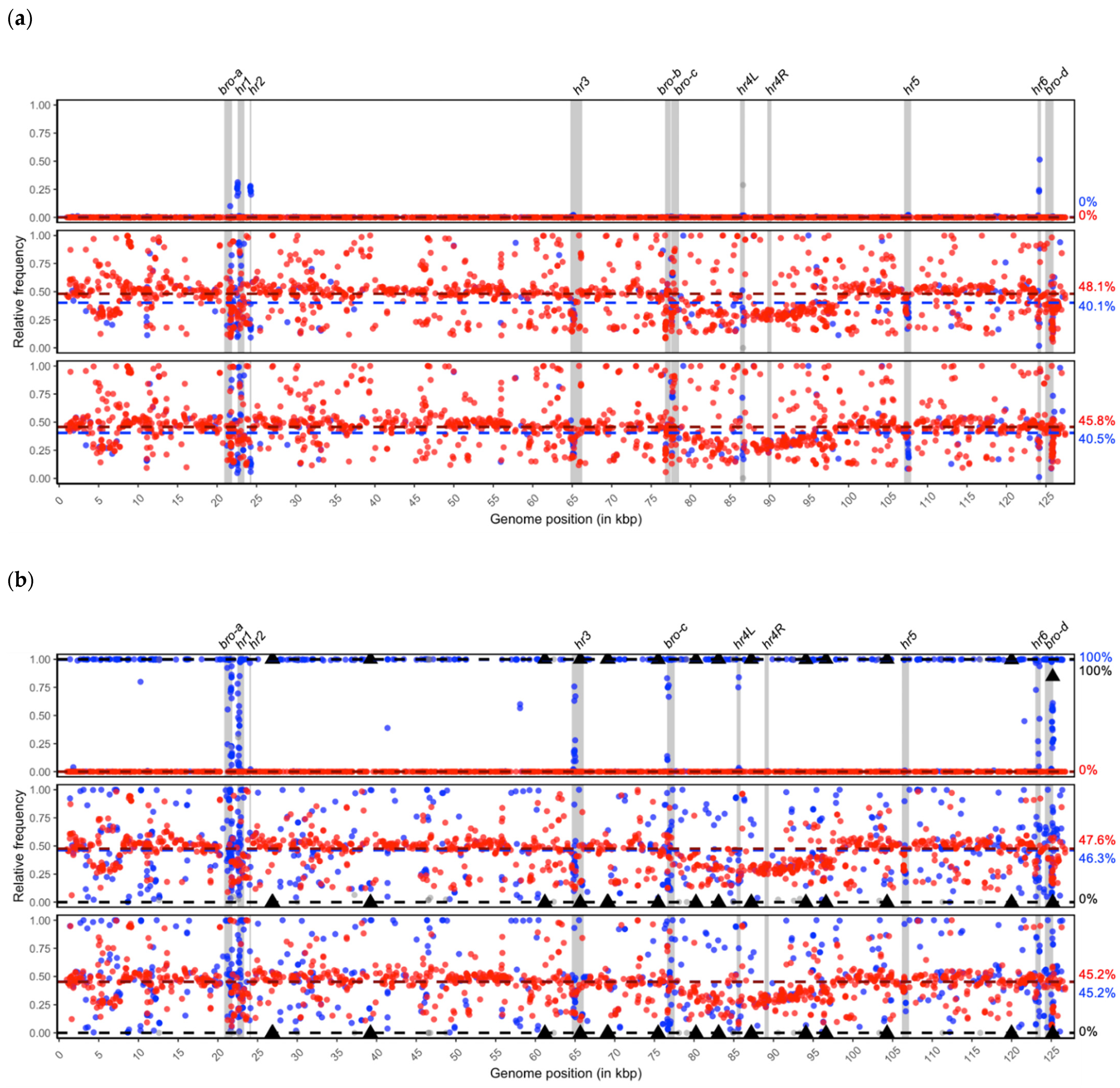
2.5. SNP Genotyping of BmNPV and BmBDV Isolates
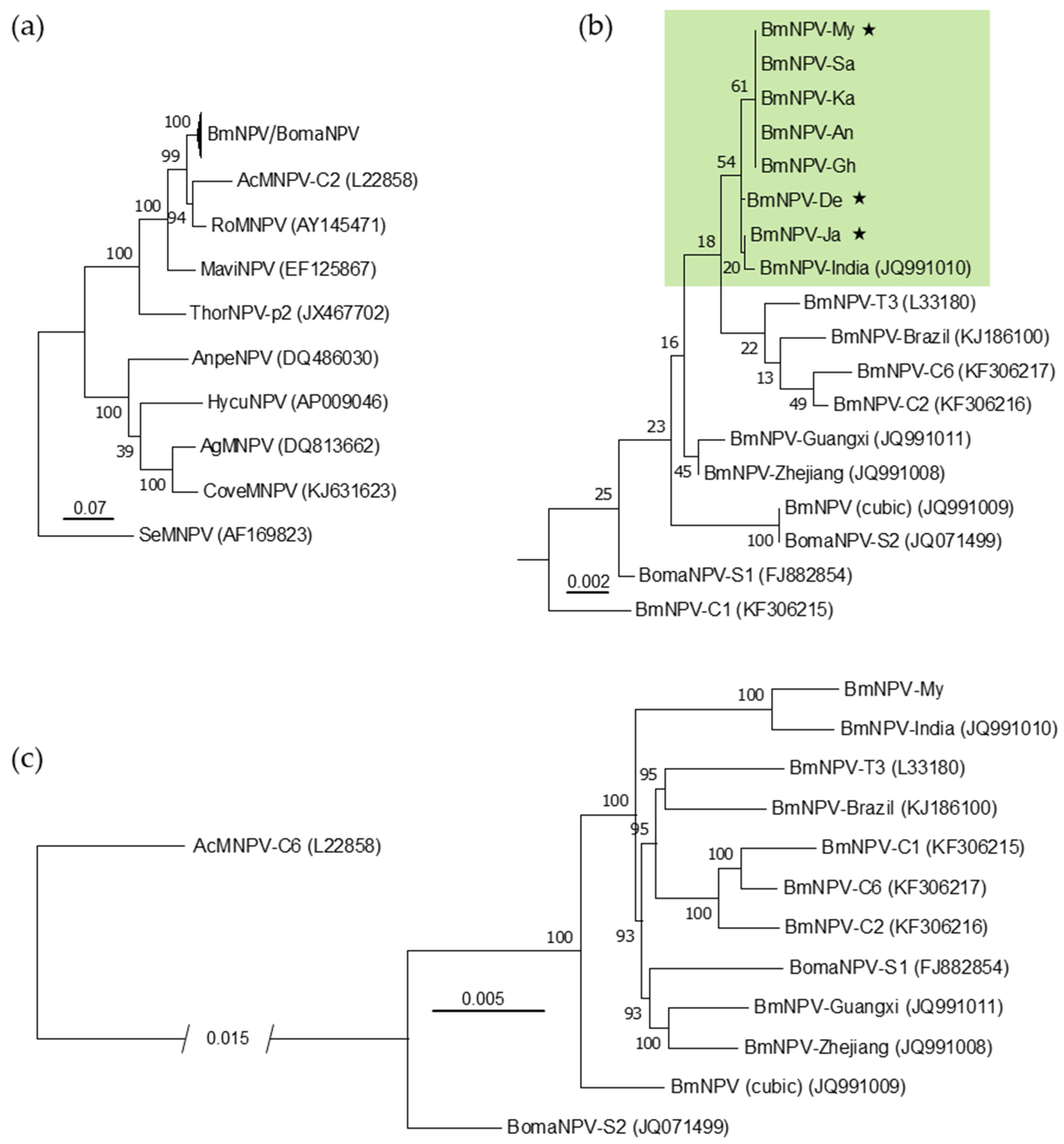
2.6. Genome-Based Phylogeny
3. Results
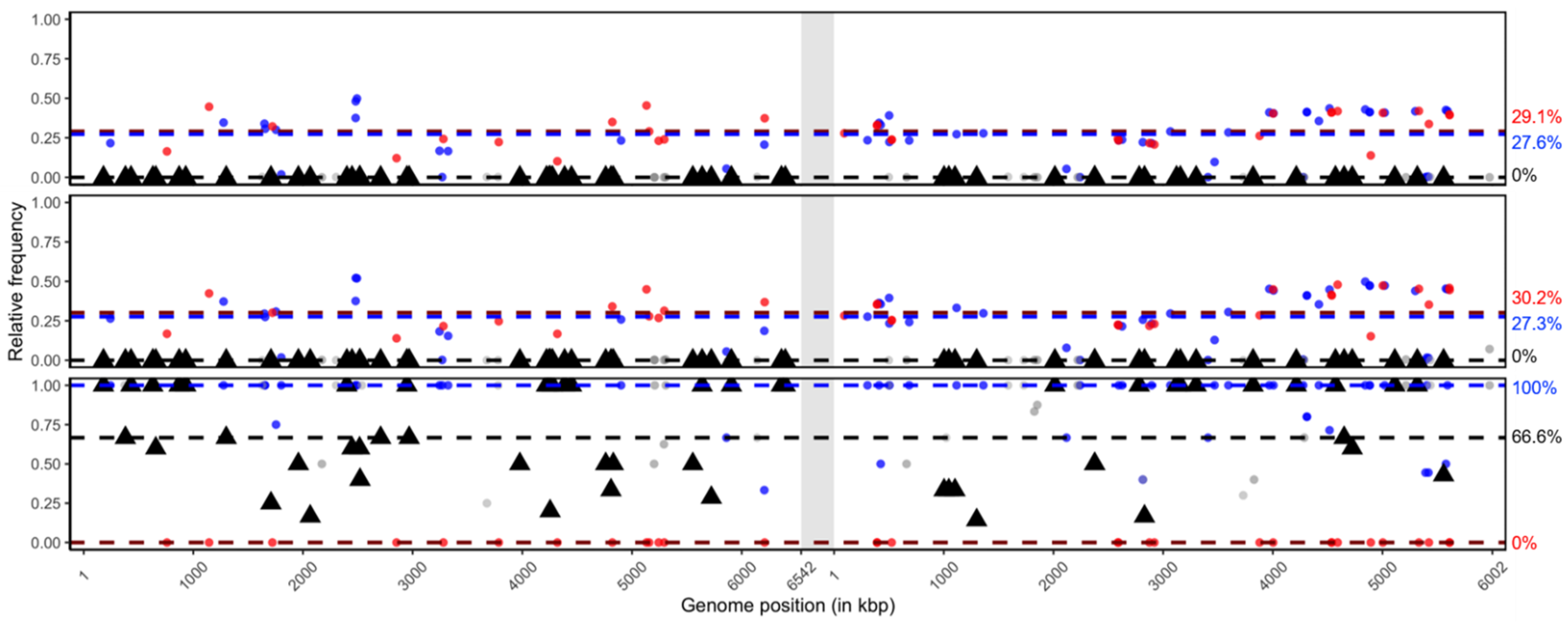
3.1. Genotype Composition of BmNPV Isolates
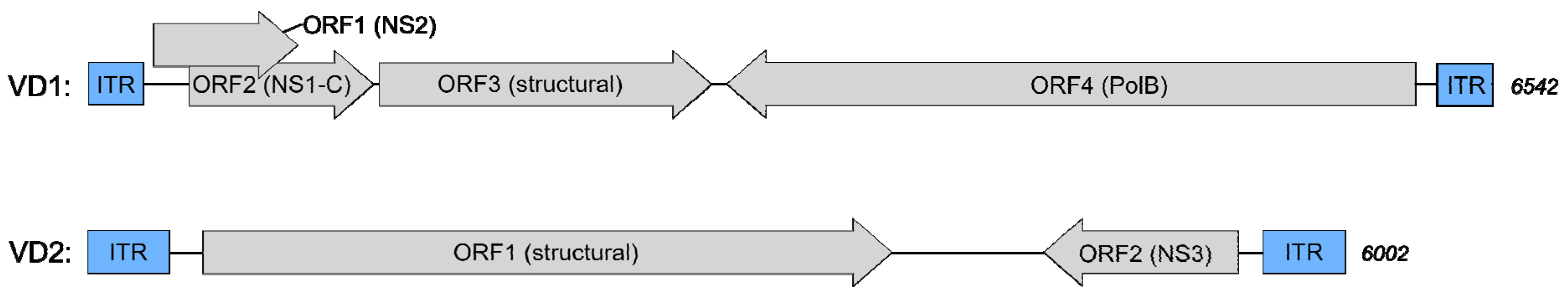
3.2. Isolate Composition of BmBDV-Ja
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sundari, K.T.; Ramalakshmi, P. Silk production: The global scenario. Asian Rev. Soc. Sci. 2018, 7, 22–24. [Google Scholar]
- Hamamura, Y. Food selection by silkworm larvæ. Nature 1959, 183, 1746–1747. [Google Scholar] [CrossRef]
- Zhang, Z.-J.; Zhang, S.-S.; Niu, B.-L.; Ji, D.-F.; Liu, X.-J.; Li, M.-W.; Bai, H.; Palli, S.R.; Wang, C.-Z.; Tan, A.-J. A determining factor for insect feeding preference in the silkworm, Bombyx mori. PLoS Biol. 2019, 17, e3000162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khurad, A.; Mahulikar, A.; Rathod, M.; Rai, M.; Kanginakudru, S.; Nagaraju, J. Vertical transmission of nucleopolyhedrovirus in the silkworm, Bombyx mori L. J. Invertebr. Pathol. 2004, 87, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Ravikumar, G.; Urs, S.R.; Prakash, N.V.; Rao, C.; Vardhana, K. Development of a multiplex polymerase chain reaction for the simultaneous detection of microsporidians, nucleopolyhedrovirus, and densovirus infecting silkworms. J. Invertebr. Pathol. 2011, 107, 193–197. [Google Scholar] [CrossRef] [PubMed]
- Gani, M.; Josepha, A.V.M.; Satish, L.; Chouhan, S.; Gadwala, M.; Ahmad, M.N.; Sivaprasad, V. Prevalence and molecular characterization of viruses causing diseases in Bombyx mori L. (Lepidoptera: Bombycidae) from different climatic regions of India. J. Biol. Control. 2018, 32, 252–256. [Google Scholar] [CrossRef]
- Kumar, P.; Baig, M.; Sengupta, K. Handbook on Pest and Disease Control of Mulberry and Silkworm. Available online: https://digitallibrary.un.org/record/167892 (accessed on 26 April 2021).
- Reddy, B.K.; Rao, J.V. Seasonal occurrence and control of silkworm diseases, grasserie, flacherie and muscardine and insect pest, uzi fly in Andhra Pradesh, India. Int. J. Ind. Entomol. 2009, 18, 57–61. [Google Scholar]
- Acharya, A.; Sriram, S.; Sehrawat, S.; Rahman, M.; Sehgal, D.; Gopinathan, K. Bombyx mori nucleopolyhedrovirus: Molecular biology and biotechnological applications for large-scale synthesis of recombinant proteins. Curr. Sci. 2002, 83, 455–465. [Google Scholar]
- Ardisson-Araújo, D.M.P.; Melo, F.L.; Andrade, M.D.S.; Brancalhão, R.M.C.; Báo, S.N.; Ribeiro, B.M. Complete genome sequence of the first non-Asian isolate of Bombyx mori nucleopolyhedrovirus. Virus Genes 2014, 49, 477–484. [Google Scholar] [CrossRef] [PubMed]
- Gani, M.; Chouhan, S.; Babulal; Gupta, R.K.; Khan, G.; Bharath Kumar, N.; Saini, P.; Ghosh, M.K. Bombyx mori nucleopolyhedrovirus (BmNPV): Its impact on silkworm rearing and management strategies. J. Biol. Control 2018, 31, 189. [Google Scholar] [CrossRef]
- McCook, A.R.T.; Fernández, B.C.; Ruiz, A. Description of the main aspects influencing Bombyx mori L. (Lepidoptera: Bombycidae) rearing. Cuba. J. Agric. Sci. 2021, 5, 1–13. [Google Scholar]
- Harrison, R.L.; Herniou, E.A.; Jehle, J.A.; Theilmann, D.A.; Burand, J.P.; Becnel, J.J.; Krell, P.J.; Van Oers, M.M.; Mowery, J.D.; Bauchan, G.R.; et al. ICTV Virus Taxonomy Profile: Baculoviridae. J. Gen. Virol. 2018, 99, 1185–1186. [Google Scholar] [CrossRef] [PubMed]
- Jehle, J.A.; Lange, M.; Wang, H.; Hu, Z.; Wang, Y.; Hauschild, R. Molecular identification and phylogenetic analysis of baculoviruses from Lepidoptera. Virology 2006, 346, 180–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wennmann, J.T.; Keilwagen, J.; Jehle, J.A. Baculovirus Kimura two-parameter species demarcation criterion is confirmed by the distances of 38 core gene nucleotide sequences. J. Gen. Virol. 2018, 99, 1307–1320. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.-P.; Gu, L.-Z.; Lou, Y.-H.; Cheng, R.-L.; Xu, H.-J.; Wang, W.-B.; Zhang, C.-X. A baculovirus isolated from wild silkworm encompasses the host ranges of Bombyx mori nucleopolyhedrosis virus and Autographa californica multiple nucleopolyhedrovirus in cultured cells. J. Gen. Virol. 2012, 93, 2480–2489. [Google Scholar] [CrossRef] [PubMed]
- Cheng, R.-L.; Xu, Y.-P.; Zhang, C.-X. Genome Sequence of a Bombyx mori Nucleopolyhedrovirus Strain with Cubic Occlusion Bodies. J. Virol. 2012, 86, 10245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, H.-W.; Zhang, X.-C.; Xu, Y.-P.; Cheng, X.-W.; Zhang, C.-X. Genome of a Bombyx mori Nucleopolyhedrovirus Strain Isolated from India. J. Virol. 2012, 86, 11941. [Google Scholar] [CrossRef] [Green Version]
- Gomi, S.; Majima, K.; Maeda, S. Sequence analysis of the genome of Bombyx mori nucleopolyhedrovirus. J. Gen. Virol. 1999, 80, 1323–1337. [Google Scholar] [CrossRef]
- Hu, Z.; Li, G.; Li, G.; Yao, Q.; Chen, K. Bombyx mori bidensovirus: The type species of the new genus Bidensovirus in the new family Bidnaviridae. Chin. Sci. Bull. 2013, 58, 4528–4532. [Google Scholar] [CrossRef]
- Gupta, T.; Ito, K.; Kadono-Okuda, K.; Murthy, G.N.; Gowri, E.V.; Ponnuvel, K.M. Characterization and genome comparison of an Indian isolate of bidensovirus infecting the silkworm Bombyx mori. Arch. Virol. 2017, 163, 125–134. [Google Scholar] [CrossRef]
- Hayakawa, T.; Kojima, K.; Nonaka, K.; Nakagaki, M.; Sahara, K.; Asano, S.-I.; Iizuka, T.; Bando, H. Analysis of proteins encoded in the bipartite genome of a new type of parvo-like virus isolated from silkworm—structural protein with DNA polymerase motif. Virus Res. 2000, 66, 101–108. [Google Scholar] [CrossRef]
- Wang, Y.J.; Yao, Q.; Chen, K.P.; Lu, J.; Han, X.; Wang, Y. Characterization of the genome structure of Bombyx mori densovirus (China isolate). Virus Genes 2006, 35, 103–108. [Google Scholar] [CrossRef]
- Chateigner, A.; Bézier, A.; Labrousse, C.; Jiolle, D.; Barbe, V.; Herniou, E.A. Ultra deep sequencing of a baculovirus population reveals widespread genomic variations. Viruses 2015, 7, 3625–3646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, J.; A Jehle, J.; Wennmann, J.T. Population structure of Cydia pomonella granulovirus isolates revealed by quantitative analysis of genetic variation. Virus Evol. 2021, 7, veaa073. [Google Scholar] [CrossRef] [PubMed]
- Masson, T.; Fabre, M.L.; Pidre, M.L.; Niz, J.M.; Berretta, M.F.; Romanowski, V.; Ferrelli, M.L. Genomic diversity in a population of Spodoptera frugiperda nucleopolyhedrovirus. Infect. Genet. Evol. 2021, 90, 104749. [Google Scholar] [CrossRef]
- Liang, X.; Lu, Z.-L.; Wei, B.-X.; Feng, J.-L.; Qu, D.; Luo, T.R. Phylogenetic analysis of Bombyx mori nucleopolyhedrovirus polyhedrin and p10 genes in wild isolates from Guangxi Zhuang Autonomous Region, China. Virus Genes 2012, 46, 140–151. [Google Scholar] [CrossRef] [PubMed]
- Alletti, G.G.; Sauer, A.J.; Weihrauch, B.; Fritsch, E.; Undorf-Spahn, K.; Wennmann, J.T.; Jehle, J.A. Using next generation sequencing to identify and quantify the genetic composition of resistance-breaking commercial isolates of Cydia pomonella granulovirus. Viruses 2017, 9, 250. [Google Scholar] [CrossRef] [Green Version]
- Sambrook, J.; Russel, D.W. Molecular Cloning: A Laboratory Manual, 3rd ed.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2001. [Google Scholar]
- Lange, M.; Wang, H.; Zhihong, H.; Jehle, J.A. Towards a molecular identification and classification system of lepidopteran-specific baculoviruses. Virology 2004, 325, 36–47. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [Green Version]
- Krueger, F. Trim Galore: A Wrapper Tool around Cutadapt and FastQC to Consistently Apply Quality and Adapter Trimming to FastQ Files. Available online: https://github.com/FelixKrueger/TrimGalore (accessed on 24 April 2021).
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 2011, 27, 2987–2993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 Genome project data processing subgroup the sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; O Pollard, M.; Whitwham, A.; Keane, T.; A McCarthy, S.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. GigaScience 2021, 10, 008. [Google Scholar] [CrossRef]
- Wennmann, J.T.; Fan, J.; Jehle, J.A. Bacsnp: Using single nucleotide polymorphism (SNP) specificities and frequencies to identify genotype composition in baculoviruses. Viruses 2020, 12, 625. [Google Scholar] [CrossRef]
- Krupovic, M.; Koonin, E.V. Evolution of eukaryotic single-stranded DNA viruses of the Bidnaviridae family from genes of four other groups of widely different viruses. Sci. Rep. 2014, 4, 5347. [Google Scholar] [CrossRef] [Green Version]
- Lange, M.; Jehle, J.A. The genome of the Cryptophlebia leucotreta granulovirus. Virology 2003, 317, 220–236. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.-P.; Cheng, R.-L.; Xi, Y.; Zhang, C.-X. Genomic diversity of Bombyx mori nucleopolyhedrovirus strains. Genomics 2013, 102, 63–71. [Google Scholar] [CrossRef] [Green Version]
- Hill, T.; Unckless, R.L. Recurrent evolution of high virulence in isolated populations of a DNA virus. eLife 2020, 9. [Google Scholar] [CrossRef]
- Fan, J.; Wennmann, J.T.; Wang, D.; Jehle, J.A. Novel diversity and virulence patterns found in new isolates of Cydia pomonella granulovirus from China. Appl. Environ. Microbiol. 2019, 86, 86. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Wennmann, J.T.; Wang, D.; Jehle, J.A. Single nucleotide polymorphism (SNP) frequencies and distribution reveal complex genetic composition of seven novel natural isolates of Cydia pomonella granulovirus. Virology 2020, 541, 32–40. [Google Scholar] [CrossRef]
- Li, G.; Hu, Z.; Guo, X.; Li, G.; Tang, Q.; Wang, P.; Chen, K.; Yao, Q. Identification of Bombyx mori bidensovirus VD1-ORF4 reveals a novel protein associated with viral structural component. Curr. Microbiol. 2013, 66, 527–534. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Isolate | City, Region or State | GenBank Accession No. | ||
|---|---|---|---|---|
| Polh | lef-8 | lef-9 | ||
| BmNPV-An | Anantapur, Andhra Pradesh | MK415967 | MK415970 | MK415956 |
| BmNPV-De | Dehradun, Uttarakhand | MK415968 | MK415975 | MK415957 |
| BmNPV-Gh | Ghumarwin, Himachal Pradesh | MK415966 | MK415974 | MK415958 |
| BmNPV-Ja | Jammu, Jammu and Kashmir | MK415964 | MK415976 | MK415959 |
| BmNPV-Ka | Kashmir, Jammu and Kashmir | MK415969 | MK415973 | MK415960 |
| BmNPV-My | Mysore, Karnataka | MK415963 | MK415971 | MK415961 |
| BmNPV-Sa | Salem, Tamil Nadu | MK415965 | MK415972 | MK415962 |
| OB Stock | Total Reads Sequenced | Phred ≥ 30 | No. Reads Mapping to BmNPV * (%) | BmNPV Mean Read Depth ± SD * | No. Reads Mapping to BmBDV-Ja ** VD1 (%)/VD2 (%) | BmBDV-Ja Mean Read Depth ± SD ** | RPKM of VD1/VD2 | Ratio of VD1/VD2 (~Fragment Ratio) |
|---|---|---|---|---|---|---|---|---|
| My | 528,646 | 491,996 | 468,827 (95%) | 507 ± 70 | 275 (<0.1%)/ 283 (<0.1%) | 5 ± 2 6 ± 4 | 75,334/ 84,500 | 0.89 (~9:10) |
| De | 946,752 | 855,476 | 659,811 (77%) | 661 ± 126 | 63,593 (7.4%)/ 97,297(11.4%) | 453 ± 89 696 ± 127 | 63,593/ 97,297 | 0.65 (~2:3) |
| Ja | 797,784 | 704,544 | 248,770 (35%) *** | 270 ± 39 | 62,277 (8.8%)/ 98,732 (14.0%) | 689 ± 116 1109 ± 192 | 62,277/ 98,732 | 0.63 (~3:5) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gani, M.; Senger, S.; Lokanath, S.; Saini, P.; Bali, K.; Gupta, R.; Sivaprasad, V.; Jehle, J.A.; Wennmann, J.T. Patterns in Genotype Composition of Indian Isolates of the Bombyx mori Nucleopolyhedrovirus and Bombyx mori Bidensovirus. Viruses 2021, 13, 901. https://0-doi-org.brum.beds.ac.uk/10.3390/v13050901
Gani M, Senger S, Lokanath S, Saini P, Bali K, Gupta R, Sivaprasad V, Jehle JA, Wennmann JT. Patterns in Genotype Composition of Indian Isolates of the Bombyx mori Nucleopolyhedrovirus and Bombyx mori Bidensovirus. Viruses. 2021; 13(5):901. https://0-doi-org.brum.beds.ac.uk/10.3390/v13050901
Chicago/Turabian StyleGani, Mudasir, Sergei Senger, Satish Lokanath, Pawan Saini, Kamlesh Bali, Rakesh Gupta, Vankadara Sivaprasad, Johannes A. Jehle, and Jörg T. Wennmann. 2021. "Patterns in Genotype Composition of Indian Isolates of the Bombyx mori Nucleopolyhedrovirus and Bombyx mori Bidensovirus" Viruses 13, no. 5: 901. https://0-doi-org.brum.beds.ac.uk/10.3390/v13050901