
Overview of Bat and Wildlife Coronavirus Surveillance in Africa: A Framework for Global Investigations

 , , , ,
, , , ,
Abstract
:1. Introduction
2. The Importance of Viral Taxonomy
3. Biosurveillance Studies Based on Nucleic Acid Detection in Africa
3.1. Surveillance in African Bats
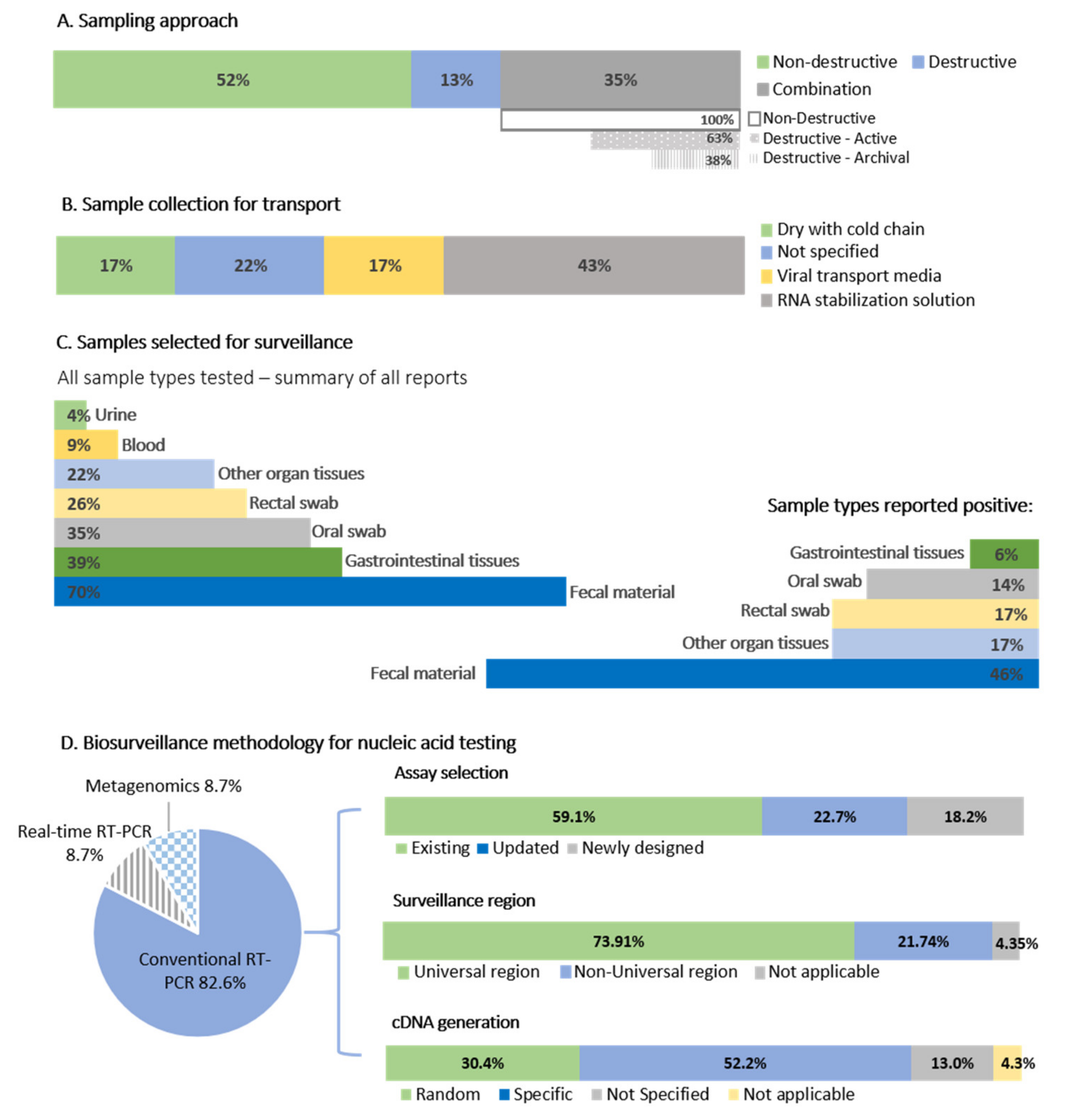
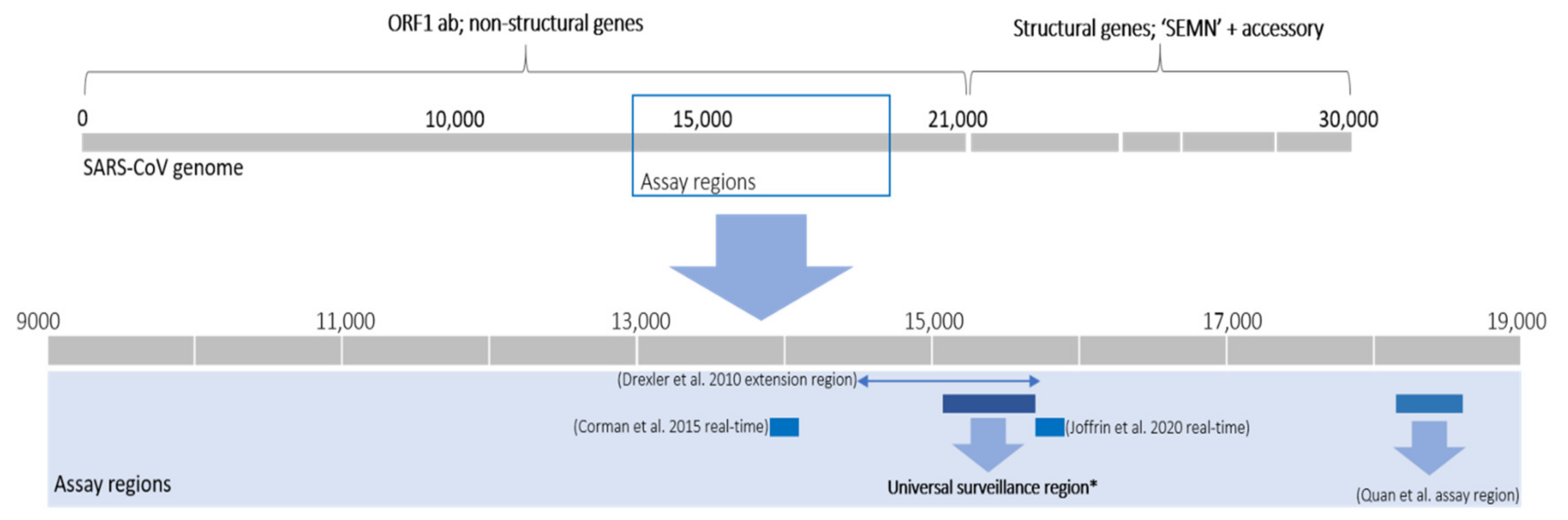
3.1.1. Sampling Approaches and Methodologies of Bat Coronavirus Surveillance
3.1.2. Summary of Sample Sizes and Bat Species Tested
3.1.3. Importance of Accurate Bat Species Identification
3.1.4. Characterization of Bat Coronavirus Genomes and Virus Isolation Attempts
3.1.5. Coronavirus RNA Identified in African Bats
3.1.6. Investigating Factors Affecting the Maintenance of Bat Coronaviruses
3.2. Surveillance in Other Wildlife and Domestic Animals (Livestock)
4. Coronavirus Serosurveillance
5. Factors Associated with the Potential Emergence of Coronaviruses
6. The Future of Coronavirus Surveillance
7. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Appendix A
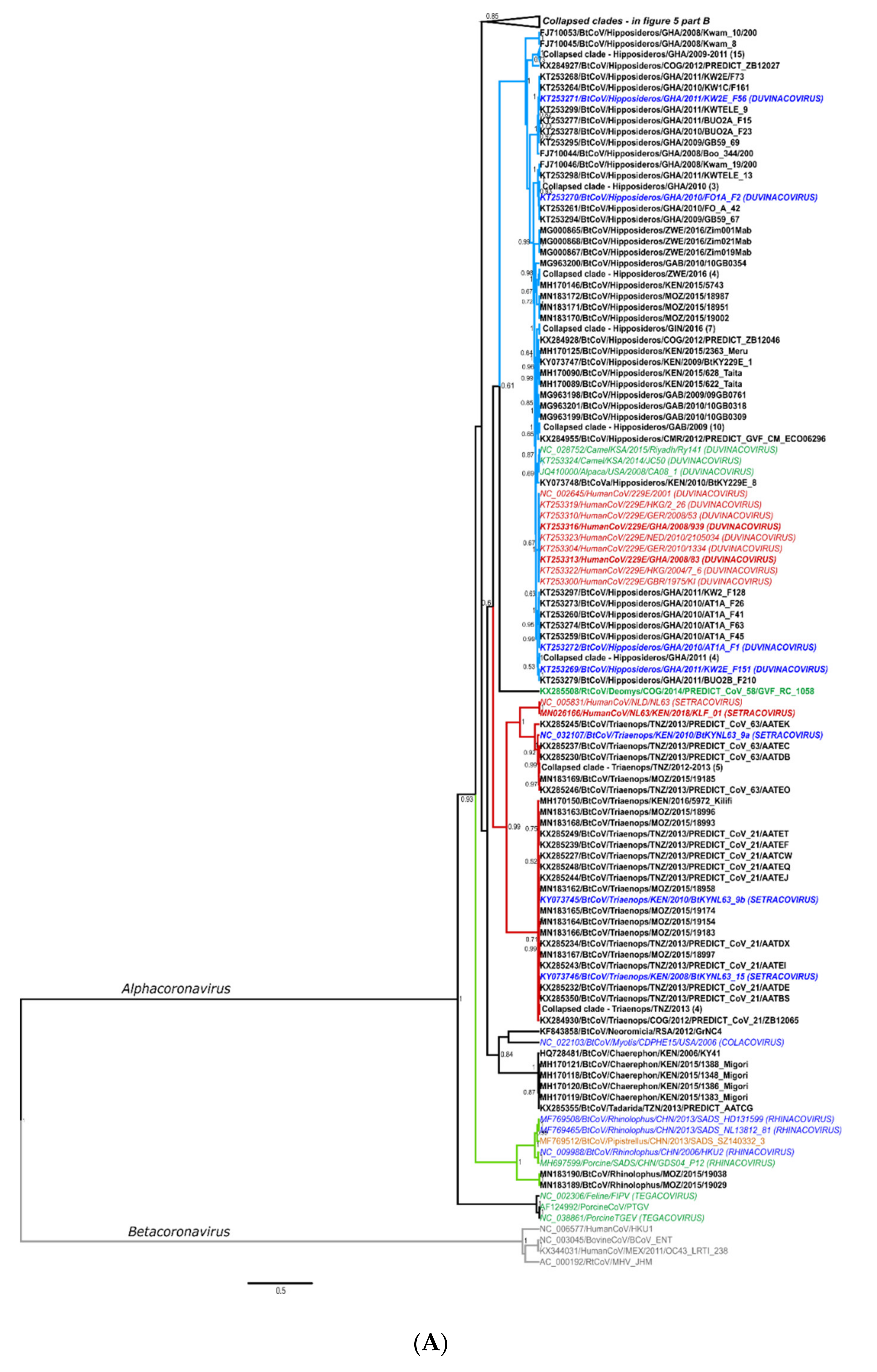
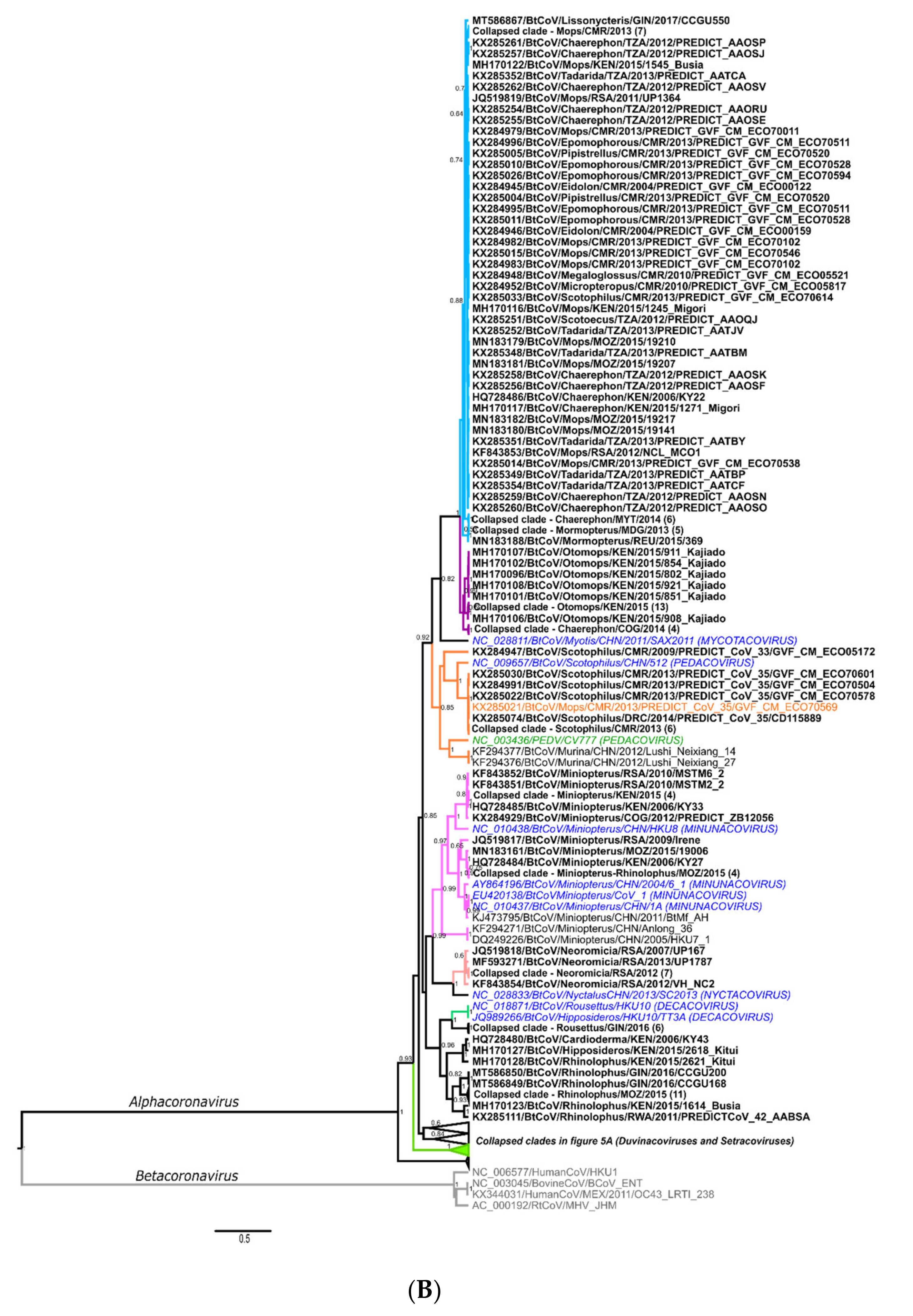
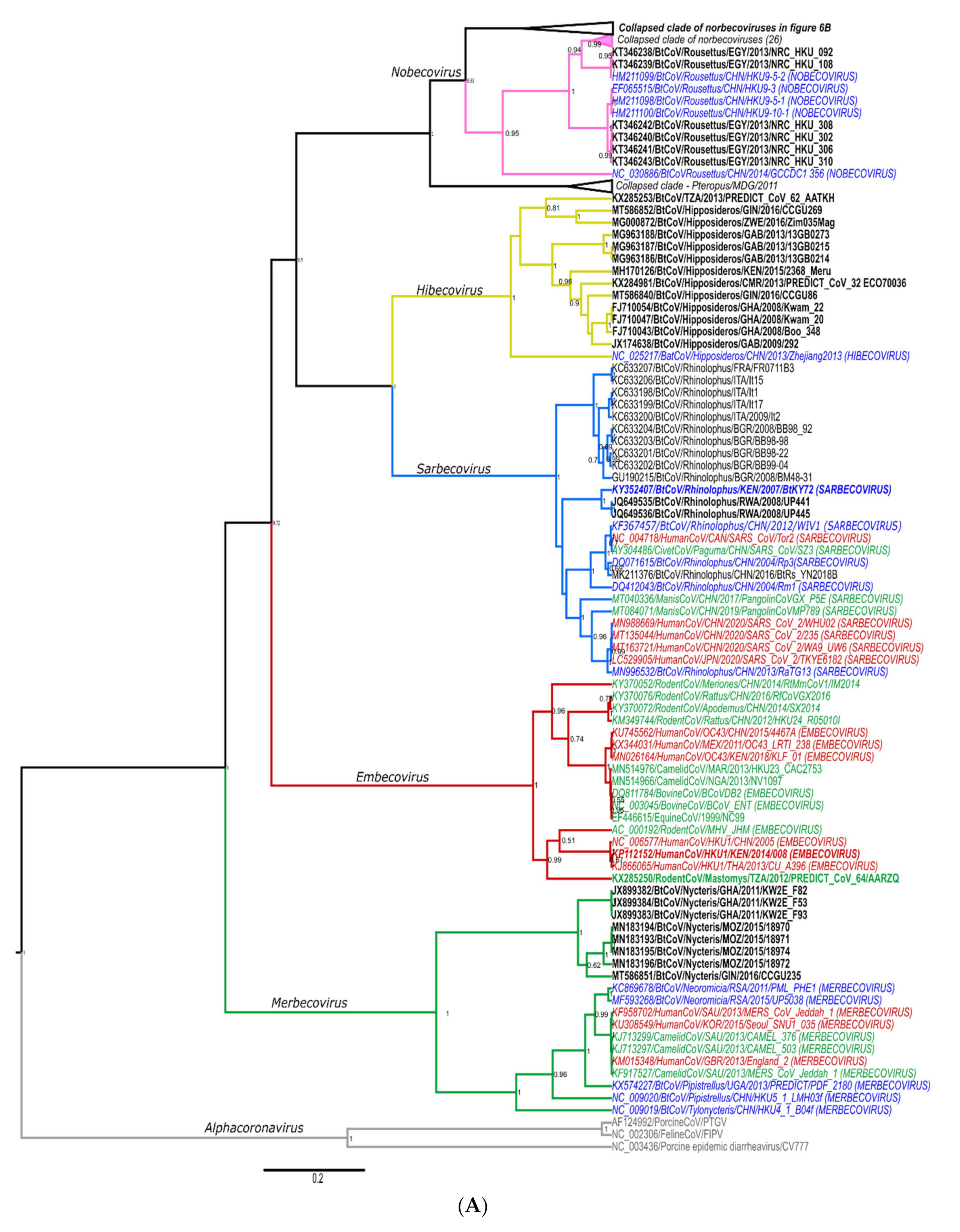
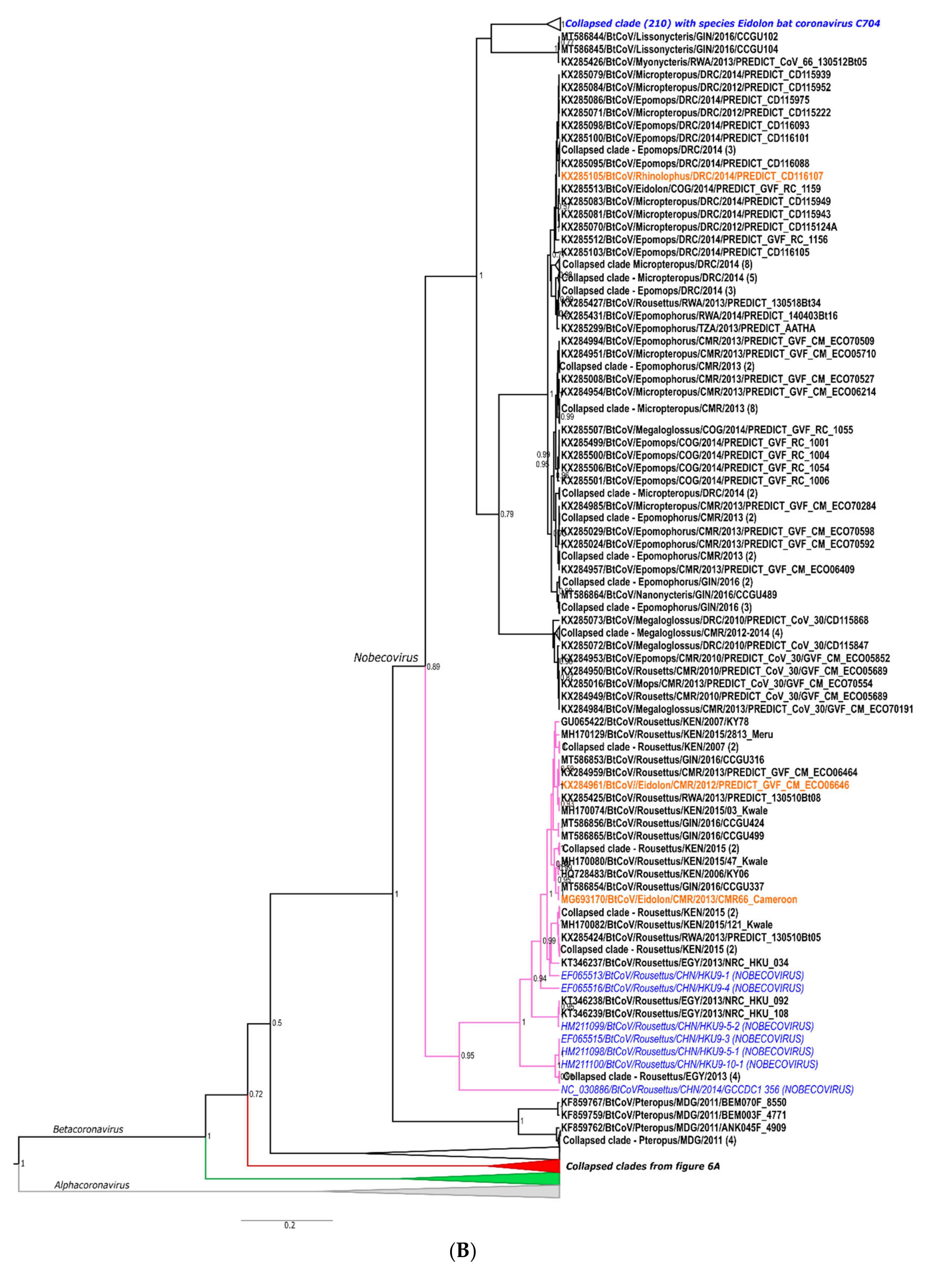
Details of Phylogenetics
References
- Ksiazek, T.G.; Erdman, D.; Goldsmith, C.S.; Zaki, S.R.; Peret, T.; Emery, S.; Tong, S.; Urbani, C.; Comer, J.A.; Lim, W.; et al. A novel coronavirus associated with severe acute respiratory syndrome. N. Engl. J. Med. 2003, 348, 1953–1966. [Google Scholar] [CrossRef] [PubMed]
- Berry, M.; Gamieldien, J.; Fielding, B.C. Identification of new respiratory viruses in the new millennium. Viruses 2015, 7, 996–1019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, P.; Fan, H.; Lan, T.; Yang, X.L.; Shi, W.F.; Zhang, W.; Zhu, Y.; Zhang, Y.W.; Xie, Q.M.; Mani, S.; et al. Fatal swine acute diarrhoea syndrome caused by an HKU2-related coronavirus of bat origin. Nature 2018, 556, 255–259. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Li, Q.N.; Su, J.N.; Chen, G.H.; Wu, Z.X.; Luo, Y.; Wu, R.T.; Sun, Y.; Lan, T.; Ma, J.Y. The re-emerging of SADS-CoV infection in pig herds in Southern China. Transbound. Emerg. Dis. 2019, 66, 2180–2183. [Google Scholar] [CrossRef] [Green Version]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorbalenya, A.E.; Baker, S.C.; Baric, R.S.; de Groot, R.J.; Drosten, C.; Gulyaeva, A.A.; Haagmans, B.L.; Lauber, C.; Leontovich, A.M.; Neuman, B.W.; et al. The species Severe acute respiratory syndrome-related coronavirus: Classifying 2019-nCoV and naming it SARS-CoV-2. Nat. Microbiol. 2020, 5, 536–544. [Google Scholar] [CrossRef] [Green Version]
- Zaki, A.M.; Van Boheemen, S.; Bestebroer, T.M.; Osterhaus, A.D.M.E.; Fouchier, R.A.M. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N. Engl. J. Med. 2012, 367, 1814–1820. [Google Scholar] [CrossRef]
- WHO. WHO MERS-CoV Situation Update, January 2020; World Health Organization: Geneva, Switzerland, 2020; Licence CC BY-NC-SA 3.0 IGO. [Google Scholar]
- Cui, J.; Li, F.; Shi, Z.L. Origin and evolution of pathogenic coronaviruses. Nat. Rev. Microbiol. 2019, 17, 181–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lam, T.T.-Y.; Jia, N.; Zhang, Y.-W.; Shum, M.H.-H.; Jiang, J.-F.; Zhu, H.-C.; Tong, Y.-G.; Shi, Y.-X.; Ni, X.-B.; Liao, Y.-S.; et al. Identifying SARS-CoV-2-related coronaviruses in Malayan pangolins. Nature 2020, 583, 282–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hemida, M.G.; Elmoslemany, A.; Al-Hizab, F.; Alnaeem, A.; Almathen, F.; Faye, B.; Chu, D.K.W.; Perera, R.A.P.M.; Peiris, M. Dromedary camels and the transmission of Middle East respiratory syndrome coronavirus (MERS-CoV). Transbound. Emerg. Dis. 2015, 64, 344–353. [Google Scholar] [CrossRef] [Green Version]
- Haagmans, B.L.; Al Dhahiry, S.H.S.; Reusken, C.B.E.M.; Raj, V.S.; Galiano, M.; Myers, R.; Godeke, G.; Jonges, M.; Farag, E.; Diab, A.; et al. Middle East respiratory syndrome coronavirus in dromedary camels: An outbreak investigation. Lancet Infect. Dis. 2014, 14, 140–145. [Google Scholar] [CrossRef] [Green Version]
- Andersen, K.G.; Rambaut, A.; Lipkin, W.I.; Holmes, E.C.; Garry, R.F. The proximal origin of SARS-CoV-2. Nat. Med. 2020, 26, 450–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poon, L.L.M.; Chu, D.K.W.; Chan, K.H.; Wong, O.K.; Ellis, T.M.; Leung, Y.H.C.; Lau, S.K.P.; Woo, P.C.Y.; Suen, K.Y.; Yuen, K.Y.; et al. Identification of a novel coronavirus in bats. J. Virol. 2005, 79, 2001–2009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Shi, Z.; Yu, M.; Ren, W.; Smith, C.; Hu, Z.; Zhang, H.; Zhang, J.; Eaton, B.T.; Zhang, S.; et al. Bats are natural reservoirs of SARS-Like coronaviruses. Science 2005, 310, 676–679. [Google Scholar] [CrossRef]
- Lau, S.K.P.; Woo, P.C.Y.; Li, K.S.M.; Huang, Y.; Tsoi, H.-W.; Wong, B.H.L.; Wong, S.S.Y.; Leung, S.-Y.; Chan, K.-H.; Yuen, K.-Y. Severe acute respiratory syndrome coronavirus-like virus in Chinese horseshoe bats. Proc. Natl. Acad. Sci. USA 2005, 102, 14040–14045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woo, P.C.Y.; Lau, S.K.P.; Huang, Y.; Yuen, K.-Y. Coronavirus diversity, phylogeny and interspecies jumping. Exp. Biol. Med. 2009, 234, 1117–1127. [Google Scholar] [CrossRef] [Green Version]
- Lau, S.K.P.; Woo, P.C.Y.; Li, K.S.M.; Tsang, A.K.L.; Fan, R.Y.Y.; Luk, H.K.H.; Cai, J.-P.; Chan, K.-H.; Zheng, B.-J.; Wang, M.; et al. Discovery of a novel coronavirus, China Rattus coronavirus HKU24, from Norway rats supports murine origin of Betacoronavirus 1 with implications on the ancestor of Betacoronavirus lineage A. J. Virol. 2015, 89, 3076–3092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao, Y.; Shi, M.; Chommanard, C.; Queen, K.; Zhang, J.; Markotter, W.; Kuzmin, I.V.; Holmes, E.C.; Tong, S. Surveillance of bat coronaviruses in Kenya identifies relatives of human coronaviruses NL63 and 229E and their recombination history. J. Virol. 2017, 91, e01953-16. [Google Scholar] [CrossRef] [Green Version]
- Anthony, S.J.; Epstein, J.H.; Murray, K.A.; Navarrete-Macias, I.; Zambrana-Torrelio, C.M.; Solovyov, A.; Ojeda-Flores, R.; Arrigo, N.C.; Islam, A.; Khan, A.; et al. A strategy to estimate unknown viral diversity in mammals. MBio 2013, 4, e00598-13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salkeld, D.J.; Stapp, P.; Tripp, D.W.; Gage, K.L.; Lowell, J.; Webb, C.T.; Brinkerhoff, R.J.; Antolin, M.F. Ecological traits driving the outbreaks and emergence of zoonotic pathogens. Bioscience 2016, 66, 118–129. [Google Scholar] [CrossRef] [PubMed]
- Allen, T.; Murray, K.A.; Zambrana-Torrelio, C.; Morse, S.S.; Rondinini, C.; Di Marco, M.; Breit, N.; Olival, K.J.; Daszak, P. Global hotspots and correlates of emerging zoonotic diseases. Nat. Commun. 2017, 8, 1124. [Google Scholar] [CrossRef]
- Markotter, W.; Coertse, J.; De Vries, L.; Geldenhuys, M.; Mortlock, M. Bat-borne viruses in Africa: A critical review. J. Zool. 2020, 311, 77–98. [Google Scholar] [CrossRef]
- Gorbalenya, A.E.; Krupovic, M.; Mushegian, A.R.; Kropinski, A.M.; Siddell, S.G.; Varsani, A.; Adams, M.J.; Davison, A.J.; Dutilh, B.E.; Harrach, B.; et al. The new scope of virus taxonomy: Partitioning the virosphere into 15 hierarchical ranks. Nat. Microbiol. 2020, 5, 668–674. [Google Scholar] [CrossRef]
- Ziebuhr, J.; Baker, S.; Baric, R.S.; De Groot, R.J.; Drosten, C.; Gulyaeva, A.A.; Haagmans, B.L.; Neuman, B.W.; Perlman, S.; Poon, L.L.M.; et al. Create Eight New Species in the Subfamily Orthocoronavirinae of the Family Coronaviridae and Four New Species and a New Genus in the Subfamily Serpentovirinae of the Family Tobaniviridae. 2020. International Committee on Taxonomy of Viruses Website. Available online: https://talk.ictvonline.org/files/ictv_official_taxonomy_updates_since_the_8th_report/m/animal-ssrna-viruses/9495 (accessed on 6 May 2020).
- Tong, S.; Conrardy, C.; Ruone, S.; Kuzmin, I.V.; Guo, X.; Tao, Y.; Niezgoda, M.; Haynes, L.; Agwanda, B.; Breiman, R.F.; et al. Detection of novel SARS-like and other coronaviruses in bats from Kenya. Emerg. Infect. Dis. 2009, 15, 482–485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shehata, M.M.; Chu, D.K.W.W.; Gomaa, M.R.; AbiSaid, M.; Shesheny, R.E.; Kandeil, A.; Bagato, O.; Chan, S.M.S.S.; Barbour, E.K.; Shaib, H.S.; et al. Surveillance for coronaviruses in bats, Lebanon and Egypt, 2013–2015. Emerg. Infect. Dis. 2016, 22, 148–150. [Google Scholar] [CrossRef] [Green Version]
- Leopardi, S.; Oluwayelu, D.; Meseko, C.; Marciano, S.; Tassoni, L.; Bakarey, S.; Monne, I.; Cattoli, G.; De Benedictis, P. The close genetic relationship of lineage D Betacoronavirus from Nigerian and Kenyan straw-colored fruit bats (Eidolon helvum) is consistent with the existence of a single epidemiological unit across sub-Saharan Africa. Virus Genes 2016, 52, 573–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waruhiu, C.; Ommeh, S.; Obanda, V.; Agwanda, B.; Gakuya, F.; Ge, X.; Yang, X.; Wu, L.; Zohaib, A.; Hu, B.; et al. Molecular detection of viruses in Kenyan bats and discovery of novel astroviruses, caliciviruses and rotaviruses. Virol. Sin. 2017, 32, 101–114. [Google Scholar] [CrossRef] [Green Version]
- Anthony, S.J.; Johnson, C.K.; Greig, D.J.; Kramer, S.; Che, X.; Wells, H.; Hicks, A.L.; Joly, D.O.; Wolfe, N.D.; Daszak, P.; et al. Global patterns in coronavirus diversity. Virus Evol. 2017, 3, vex012. [Google Scholar] [CrossRef] [PubMed]
- Bourgarel, M.; Pfukenyi, D.M.; Boué, V.; Talignani, L.; Chiweshe, N.; Diop, F.; Caron, A.; Matope, G.; Missé, D.; Liégeois, F. Circulation of Alphacoronavirus, Betacoronavirus and Paramyxovirus in Hipposideros bat species in Zimbabwe. Infect. Genet. Evol. 2018, 58, 253–257. [Google Scholar] [CrossRef]
- Geldenhuys, M.; Mortlock, M.; Weyer, J.; Bezuidt, O.; Seamark, E.; Kearney, T.; Gleasner, C.; Erkkila, T.; Cui, H.; Markotter, W. A metagenomic viral discovery approach identifies potential zoonotic and novel mammalian viruses in Neoromicia bats within South Africa. PLoS ONE 2018, 13, e0194527. [Google Scholar] [CrossRef]
- Ar Gouilh, M.; Puechmaille, S.J.; Diancourt, L.; Vandenbogaert, M.; Serra-Cobo, J.; Lopez Roïg, M.; Brown, P.; Moutou, F.; Caro, V.; Vabret, A.; et al. SARS-CoV related Betacoronavirus and diverse Alphacoronavirus members found in western old-world. Virology 2018, 517, 88–97. [Google Scholar] [CrossRef] [PubMed]
- Yinda, C.K.; Ghogomu, S.M.; Conceição-Neto, N.; Beller, L.; Deboutte, W.; Vanhulle, E.; Maes, P.; Van Ranst, M.; Matthijnssens, J. Cameroonian fruit bats harbor divergent viruses, including rotavirus H, bastroviruses, and picobirnaviruses using an alternative genetic code. Virus Evol. 2018, 4, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Markotter, W.; Geldenhuys, M.; Jansen van Vuren, P.; Kemp, A.; Mortlock, M.; Mudakikwa, A.; Nel, L.; Nziza, J.; Paweska, J.; Weyer, J. Paramyxo- and Coronaviruses in Rwandan Bats. Trop. Med. Infect. Dis. 2019, 4, 99. [Google Scholar] [CrossRef] [Green Version]
- Nziza, J.; Goldstein, T.; Cranfield, M.; Webala, P.; Nsengimana, O.; Nyatanyi, T.; Mudakikwa, A.; Tremeau-Bravard, A.; Byarugaba, D.; Tumushime, J.C.; et al. Coronaviruses detected in bats in close contact with humans in Rwanda. Ecohealth 2019, 17, 152–159. [Google Scholar] [CrossRef] [PubMed]
- Pfefferle, S.; Oppong, S.; Drexler, J.F.; Gloza-Rausch, F.; Ipsen, A.; Seebens, A.; Müller, M.A.; Annan, A.; Vallo, P.; Adu-Sarkodie, Y.; et al. Distant relatives of severe acute respiratory syndrome coronavirus and close relatives of human coronavirus 229E in bats, Ghana. Emerg. Infect. Dis. 2009, 15, 1377–1384. [Google Scholar] [CrossRef] [PubMed]
- Joffrin, L.; Goodman, S.M.; Wilkinson, D.A.; Ramasindrazana, B.; Lagadec, E.; Gomard, Y.; Le Minter, G.; Dos Santos, A.; Corrie Schoeman, M.; Sookhareea, R.; et al. Bat coronavirus phylogeography in the Western Indian Ocean. Sci. Rep. 2020, 10, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Lacroix, A.; Vidal, N.; Keita, A.K.; Thaurignac, G.; Esteban, A.; De Nys, H.; Diallo, R.; Toure, A.; Goumou, S.; Soumah, A.K.; et al. Wide diversity of Coronaviruses in frugivorous and insectivorous bat species: A pilot study in Guinea, West Africa. Viruses 2020, 12, 855. [Google Scholar] [CrossRef]
- Maganga, G.D.; Pinto, A.; Mombo, I.M.; Madjitobaye, M.; Mbeang Beyeme, A.M.; Boundenga, L.; Ar Gouilh, M.; N’Dilimabaka, N.; Drexler, J.F.; Drosten, C.; et al. Genetic diversity and ecology of coronaviruses hosted by cave-dwelling bats in Gabon. Sci. Rep. 2020, 10, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Quan, P.-L.; Firth, C.; Street, C.; Henriquez, J.A.; Petrosov, A.; Tashmukhamedova, A.; Hutchison, S.K.; Egholm, M.; Osinubi, M.O.V.; Niezgoda, M.; et al. Identification of a severe acute respiratory syndrome coronavirus-like virus in a leaf-nosed bat in Nigeria. MBio 2010, 1, 00208-10. [Google Scholar] [CrossRef] [Green Version]
- Geldenhuys, M.; Weyer, J.; Nel, L.H.; Markotter, W. Coronaviruses in South African bats. Vector Borne Zoonotic Dis. 2013, 13, 516–519. [Google Scholar] [CrossRef] [PubMed]
- Ithete, N.L.; Stoffberg, S.; Corman, V.M.; Cottontail, V.M.; Richards, L.R.; Schoeman, M.C.; Drosten, C.; Drexler, J.F.; Preiser, W. Close relative of human Middle East respiratory syndrome coronavirus in bat, South Africa. Emerg. Infect. Dis. 2013, 19, 1697–1699. [Google Scholar] [CrossRef] [PubMed]
- Annan, A.; Baldwin, H.J.; Corman, V.M.; Klose, S.M.; Owusu, M.; Nkrumah, E.E.; Badu, E.K.; Anti, P.; Agbenyega, O.; Meyer, B.; et al. Human Betacoronavirus 2c EMC/2012-related viruses in bats, Ghana and Europe. Emerg. Infect. Dis. 2013, 19, 456–459. [Google Scholar] [CrossRef]
- Maganga, G.D.; Bourgarel, M.; Vallo, P.; Dallo, T.D.; Ngoagouni, C.; Drexler, J.F.; Drosten, C.; Nakouné, E.R.; Leroy, E.M.; Morand, S. Bat distribution size or shape as determinant of viral richness in African bats. PLoS ONE 2014, 9, e100172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corman, V.M.; Baldwin, H.J.; Tateno, A.F.; Zerbinati, R.M.; Annan, A.; Owusu, M.; Nkrumah, E.E.; Maganga, G.D.; Oppong, S.; Adu-Sarkodie, Y.; et al. Evidence for an ancestral association of human coronavirus 229E with bats. J. Virol. 2015, 89, 11858–11870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Razanajatovo, N.H.; Nomenjanahary, L.A.; Wilkinson, D.A.; Razafimanahaka, J.H.; Goodman, S.M.; Jenkins, R.K.; Jones, J.P.G.; Heraud, J.-M. Detection of new genetic variants of Betacoronaviruses in endemic frugivorous bats of Madagascar. Virol. J. 2015, 12, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patrono, L.V.; Samuni, L.; Corman, V.M.; Nourifar, L.; Röthemeier, C.; Wittig, R.M.; Drosten, C.; Calvignac-Spencer, S.; Leendertz, F.H. Human coronavirus OC43 outbreak in wild chimpanzees, Côte d’Ivoire, 2016 correspondence. Emerg. Microbes Infect. 2018, 7, 1–4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Duah, P.; Meyer, B.; Sylverken, A.; Owusu, M.; Gottula, L.T.; Yeboah, R.; Lamptey, J.; Frimpong, Y.O.; Burimuah, V.; Folitse, R.; et al. Development of a whole-virus ELISA for serological evaluation of domestic livestock as possible hosts of human coronavirus nl63. Viruses 2019, 11, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Duah, P.; Sylverken, A.; Owusu, M.; Yeboah, R.; Lamptey, J.; Frimpong, Y.O.; Burimuah, V.; Antwi, C.; Folitse, R.; Agbenyega, O.; et al. Potential intermediate hosts for coronavirus transmission: No evidence of Clade 2c coronaviruses in domestic livestock from Ghana. Trop. Med. Infect. Dis. 2019, 4, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burimuah, V.; Sylverken, A.; Owusu, M.; El-Duah, P.; Yeboah, R.; Lamptey, J.; Frimpong, Y.O.; Agbenyega, O.; Folitse, R.; Tasiame, W.; et al. Sero-prevalence, cross-species infection and serological determinants of prevalence of Bovine coronavirus in cattle, sheep and goats in Ghana. Vet. Microbiol. 2020, 241, 108544. [Google Scholar] [CrossRef] [PubMed]
- Reusken, C.B.E.M.; Haagmans, B.L.; Müller, M.A.; Gutierrez, C.; Godeke, G.-J.; Meyer, B.; Muth, D.; Raj, V.S.; Vries, L.S.-D.; Corman, V.M.; et al. Middle East respiratory syndrome coronavirus in dromedary camels: An outbreak investigation. Lancet 2013, 3099, 1–6. [Google Scholar]
- Hammer, A.S.; Quaade, M.L.; Rasmussen, T.B.; Fonager, J.; Rasmussen, M.; Mundbjerg, K.; Lohse, L.; Strandbygaard, B.; Jørgensen, C.S.; Alfaro-Núñez, A.; et al. SARS-CoV-2 transmission between mink (Neovison vison) and humans, Denmark. Emerg. Infect. Dis. 2021, 27, 547–551. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Jiang, J.Z.; Wan, X.F.; Hua, Y.; Li, L.; Zhou, J.; Wang, X.; Hou, F.; Chen, J.; Zou, J.; et al. Are pangolins the intermediate host of the 2019 novel coronavirus (SARS-CoV-2)? PLoS Pathog. 2020, 16, e1008421. [Google Scholar] [CrossRef]
- Xiao, K.; Zhai, J.; Feng, Y.; Zhou, N.; Zhang, X.; Zou, J.J.; Li, N.; Guo, Y.; Li, X.; Shen, X.; et al. Isolation of SARS-CoV-2-related coronavirus from Malayan pangolins. Nature 2020, 583, 286–289. [Google Scholar] [CrossRef] [PubMed]
- HealthMap. Predict USAID Data. Available online: https://www.healthmap.org/predict/ (accessed on 25 August 2020).
- Tao, Y.; Tang, K.; Shi, M.; Conrardy, C.; Li, K.S.M.; Lau, S.K.P.; Anderson, L.J.; Tong, S. Genomic characterization of seven distinct bat coronaviruses in Kenya. Virus Res. 2012, 167, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Corman, V.M.; Ithete, N.L.; Richards, R.; Schoeman, M.C.; Preiser, W.; Drosten, C.; Drexler, J.F.; Raposo, R.A.S.; Abdel-Mohsen, M.; Deng, X.; et al. Rooting the phylogenetic tree of Middle East respiratory syndrome coronavirus by characterization of a conspecific virus from an African bat. J. Virol. 2014, 88, 11297–11303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anthony, S.J.; Gilardi, K.; Menachery, V.D.; Goldstein, T.; Ssebide, B.; Mbabazi, R.; Navarrete-Macias, I.; Liang, E.; Wells, H.; Hicks, A.; et al. Further evidence for bats as the evolutionary source of Middle East respiratory syndrome coronavirus. MBio 2017, 8, e00373-17. [Google Scholar] [CrossRef] [Green Version]
- Montecino-Latorre, D.; Goldstein, T.; Gilardi, K.; Wolking, D.; Van Wormer, E.; Kazwala, R.; Ssebide, B.; Nziza, J.; Sijali, Z.; Cranfield, M.; et al. Reproduction of East-African bats may guide risk mitigation for coronavirus spillover. One Health Outlook 2020, 2, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drexler, J.F.; Corman, V.M.; Drosten, C. Ecology, evolution and classification of bat coronaviruses in the aftermath of SARS. Antivir. Res. 2014, 101, 45–56. [Google Scholar] [CrossRef] [PubMed]
- De Souza Luna, L.K.; Heiser, V.; Regamey, N.; Panning, M.; Drexler, J.F.; Mulangu, S.; Poon, L.; Baumgarte, S.; Haijema, B.J.; Kaiser, L.; et al. Generic detection of coronaviruses and differentiation at the prototype strain level by reverse transcription-PCR and nonfluorescent low-density microarray. J. Clin. Microbiol. 2007, 45, 1049–1052. [Google Scholar] [CrossRef] [Green Version]
- Woo, P.C.Y.; Lau, S.K.P.; Chu, C.; Chan, K.; Tsoi, H.; Huang, Y.; Wong, B.H.L.; Poon, R.W.S.; Cai, J.J.; Luk, W.; et al. Characterization and complete genome sequence of a novel Coronavirus HKU1, from patients with pneumonia. J. Virol. 2005, 79, 884–895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, S.; Masangkay, J.S.; Nagata, N.; Morikawa, S.; Mizutani, T.; Fukushi, S.; Alviola, P.; Omatsu, T.; Ueda, N.; Iha, K.; et al. Bat coronaviruses and experimental infection of bats, the Philippines. Emerg. Infect. Dis. 2010, 16, 1217–1223. [Google Scholar] [CrossRef]
- Chu, D.K.W.; Leung, C.Y.H.; Gilbert, M.; Joyner, P.H.; Ng, E.M.; Tse, T.M.; Guan, Y.; Peiris, J.S.M.; Poon, L.L.M. Avian coronavirus in wild aquatic birds. J. Virol. 2011, 85, 12815–12820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drexler, J.F.; Gloza-Rausch, F.; Glende, J.; Corman, V.M.; Muth, D.; Goettsche, M.; Seebens, A.; Niedrig, M.; Pfefferle, S.; Yordanov, S.; et al. Genomic characterization of severe acute respiratory syndrome-related coronavirus in European bats and classification of coronaviruses based on partial RNA-dependent RNA polymerase gene sequences. J. Virol. 2010, 84, 11336–11349. [Google Scholar] [CrossRef] [Green Version]
- AfricanBast NPC. ACR African Chiroptera Report 2019; AfricanBats NPC: Pretoria, South Africa, 2019; ISBN 1990-6471. [Google Scholar]
- Monadjem, A.; Demos, T.C.; Dalton, D.L.; Webala, P.W.; Musila, S.; Kerbis Peterhans, J.C.; Patterson, B.D. A revision of pipistrelle-like bats (Mammalia: Chiroptera: Vespertilionidae) in East Africa with the description of new genera and species. Zool. J. Linn. Soc. 2020, 191, 1114–1146. [Google Scholar] [CrossRef]
- Foley, N.M.; Goodman, S.M.; Whelan, C.V.; Puechmaille, S.J.; Teeling, E. Towards navigating the Minotaur’s Labyrinth: Cryptic diversity and taxonomic revision within the speciose genus Hipposideros (Hipposideridae). Acta Chiropterol. 2017, 19, 1–18. [Google Scholar] [CrossRef]
- Monadjem, A.; Shapiro, J.T.; Richards, L.R.; Karabulut, H.; Crawley, W.; Nielsen, I.B.; Hansen, A.; Bohmann, K.; Mourier, T. Systematics of West African miniopterus with the description of a new species. Acta Chiropterol. 2020, 21, 237–256. [Google Scholar] [CrossRef]
- Taylor, P.J.; MacDonald, A.; Goodman, S.M.; Kearney, T.; Cotterill, F.P.D.; Stoffberg, S.; Monadjem, A.; Schoeman, M.C.; Guyton, J.; Naskrecki, P.; et al. Integrative taxonomy resolves three new cryptic species of small southern African horseshoe bats (Rhinolophus). Zool. J. Linn. Soc. 2019, 187, 535–537. [Google Scholar] [CrossRef] [Green Version]
- Simmons, N.B.; Cirranello, A.L. Bat Species of the World: A Taxonomic and Geographic Database. Available online: https://www.batnames.org/ (accessed on 18 November 2020).
- Folmer, O.; Black, M.; Hoeh, W.; Lutz, R.; Vrijenhoek, R. DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol. Mar. Biol. Biotechnol. 1994, 3, 294–299. [Google Scholar]
- Bickham, J.W.; Patton, J.C.; Schlitter, D.A.; Rautenbach, I.L.; Honeycutt, R.L. Molecular phylogenetics, karyotypic diversity, and partition of the genus Myotis (Chiroptera: Vespertilionidae). Mol. Phylogenet. Evol. 2004, 33, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Geldenhuys, M.; Coertse, J.; Mortlock, M.; Markotter, W. In vitro isolation of bat viruses using commercial and bat-derived cell lines. In Bats and Viruses: Current Research and Future Trends; Corrales-Aguilar, E., Schwemmle, M., Eds.; Caister Academic Press: Norfolk, UK, 2020; pp. 149–180. [Google Scholar]
- Ge, X.; Li, J.; Yang, X.; Chmura, A.A.; Zhu, G.; Epstein, J.H.; Mazet, J.K.; Hu, B.; Zhang, W.; Peng, C.; et al. Isolation and characterization of a bat SARS-like coronavirus that uses the ACE2 receptor. Nature 2013, 503, 535–538. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.-L.; Hu, B.; Wang, B.; Wang, M.-N.; Zhang, Q.; Zhang, W.; Wu, L.-J.; Ge, X.-Y.; Zhang, Y.-Z.; Daszak, P.; et al. Isolation and characterization of a novel bat coronavirus closely related to the direct progenitor of Severe Acute Respiratory Syndrome coronavirus. J. Virol. 2016, 90, 3253–3256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leopardi, S.; Holmes, E.C.; Gastaldelli, M.; Tassoni, L.; Priori, P.; Scaravelli, D.; Zamperin, G.; De Benedictis, P. Interplay between co-divergence and cross-species transmission in the evolutionary history of bat coronaviruses. Infect. Genet. Evol. 2018, 58, 279–289. [Google Scholar] [CrossRef] [PubMed]
- Wong, A.C.P.; Li, X.; Lau, S.K.P.; Woo, P.C.Y. Global epidemiology of bat coronaviruses. Viruses 2019, 11, 174. [Google Scholar] [CrossRef] [Green Version]
- Lau, S.K.P.; Li, K.S.M.; Huang, Y.; Shek, C.-T.; Tse, H.; Wang, M.; Choi, G.K.Y.; Xu, H.; Lam, C.S.F.; Guo, R.; et al. Ecoepidemiology and complete genome comparison of different strains of severe acute respiratory syndrome-related Rhinolophus bat coronavirus in China reveal bats as a reservoir for acute, self-limiting infection that allows recombination events. J. Virol. 2010, 84, 2808–2819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, R.J.; Wang, J.; Peacey, M.; Moore, N.E.; McInnes, K.; Tompkins, D.M. New Alphacoronavirus in Mystacina tuberculata bats, New Zealand. Emerg. Infect. Dis. 2014, 20, 697–700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Latinne, A.; Hu, B.; Olival, K.J.; Zhu, G.; Zhang, L.; Li, H.; Chmura, A.A.; Field, H.E.; Zambrana-Torrelio, C.; Epstein, J.H.; et al. Origin and cross-species transmission of bat coronaviruses in China. Nat. Commun. 2020, 11, 1–15. [Google Scholar] [CrossRef]
- Woo, P.C.Y.; Wang, M.; Lau, S.K.P.; Xu, H.; Poon, R.W.S.; Guo, R.; Wong, B.H.L.; Gao, K.; Tsoi, H.-W.; Huang, Y.; et al. Comparative analysis of twelve genomes of three novel group 2c and group 2d coronaviruses reveals unique group and subgroup features. J. Virol. 2007, 81, 1574–1585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salemi, M.; Fitch, W.M.; Ciccozzi, M.; Ruiz-Alvarez, M.J.; Rezza, G.; Lewis, M.J. Severe acute respiratory syndrome coronavirus sequence characteristics and evolutionary rate estimate from maximum likelihood analysis. J. Virol. 2004, 78, 1602–1603. [Google Scholar] [CrossRef] [Green Version]
- Vijaykrishna, D.; Smith, G.J.D.; Zhang, J.X.; Peiris, J.S.M.; Chen, H.; Guan, Y. Evolutionary insights into the ecology of coronaviruses. J. Virol. 2007, 81, 4012–4020. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.; Liu, W.J.; Xu, W.; Jin, T.; Zhao, Y.; Song, J.; Shi, Y.; Ji, W.; Jia, H.; Zhou, Y.; et al. A bat-derived putative cross-family recombinant coronavirus with a reovirus gene. PLoS Pathog. 2016, 12, e1005883. [Google Scholar] [CrossRef]
- De Groot, R.J. Structure, function and evolution of the hemagglutinin-esterase proteins of corona- and toroviruses. Glycoconj. J. 2006, 23, 59–72. [Google Scholar] [CrossRef]
- Hu, B.; Zeng, L.P.; Yang, X.L.; Ge, X.Y.; Zhang, W.; Li, B.; Xie, J.Z.; Shen, X.R.; Zhang, Y.Z.; Wang, N.; et al. Discovery of a rich gene pool of bat SARS-related coronaviruses provides new insights into the origin of SARS coronavirus. PLoS Pathog. 2017, 13, 1–28. [Google Scholar] [CrossRef] [PubMed]
- Graham, R.L.; Baric, R.S. Recombination, reservoirs, and the modular spike: Mechanisms of coronavirus cross-species transmission. J. Virol. 2010, 84, 3134–3146. [Google Scholar] [CrossRef] [Green Version]
- Benda, P.; Vallo, P. Taxonomic revision of the genus Triaenops (Chiroptera: Hipposideridae) with description of a new species from southern arabia and definitions of a new genus and tribe. Folia Zool. 2009, 58, 1–45. [Google Scholar]
- Monadjem, A.; Shapiro, J. Triaenops afer, African Trident Bat. Iucn Red List Threat. Species 2017, e.T81081036A95642225. [Google Scholar] [CrossRef]
- De Groot, R.J.; Baker, S.C.; Baric, R.S.; Brown, C.S.; Drosten, C.; Enjuanes, L.; Fouchier, R.A.M.; Galiano, M.; Gorbalenya, A.E.; Memish, Z.A.; et al. Middle East Respiratory Syndrome Coronavirus (MERS-CoV): Announcement of the Coronavirus Study Group. J. Virol. 2013, 87, 7790–7792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dighe, A.; Jombart, T.; Van Kerkhove, M.D.; Ferguson, N. A systematic review of MERS-CoV seroprevalence and RNA prevalence in dromedary camels: Implications for animal vaccination. Epidemics 2019, 29, 100350. [Google Scholar] [CrossRef] [PubMed]
- Anthony, S.J.; Ojeda-Flores, R.; Rico-Chavez, O.; Navarrete-Macias, I.; Zambrana-Torrelio, C.M.; Rostal, M.K.; Epstein, J.H.; Tipps, T.; Liang, E.; Sanchez-Leon, M.; et al. Coronaviruses in bats from Mexico. J. Gen. Virol. 2013, 94, 1028–1038. [Google Scholar] [CrossRef]
- Memish, Z.A.; Mishra, N.; Olival, K.J.; Fagbo, S.F.; Kapoor, V.; Epstein, J.H.; Alhakeem, R.; Asmari, M.A.; Islam, A.; Kapoor, A.; et al. Middle East Respiratory Syndrome coronavirus in bats, Saudi Arabia. Emerg. Infect. Dis. 2013, 19, 1819–1823. [Google Scholar] [CrossRef] [Green Version]
- Monadjem, A.; Taylor, P.J.; Jacobs, D.; Cotterill, F. Neoromicia capensis, Cape Bat. Iucn Red List Threat. Species 2017, e.T44918A22048372. [Google Scholar] [CrossRef]
- Kearney, T.C.; Keith, M.; Markotter, W.; Pretorius, M.; Seamark, E.C.J. Bat species (Mammalia: Chiroptera) occurring at Telperion Nature Reserve. Ann. Ditsong Natl. Mus. Nat. Hist. 2019, 8, 30–42. [Google Scholar]
- Piraccini, R. Pipistrellus hesperidus. Iucn Red List Threat. Species 2016, e.T136741A22035802. [Google Scholar] [CrossRef]
- Zhou, H.; Chen, X.; Hu, T.; Li, J.; Song, H.; Liu, Y.; Wang, P.; Liu, D.; Yang, J.; Holmes, E.C.; et al. A novel bat coronavirus closely related to SARS-CoV-2 contains natural insertions at the S1/S2 cleavage site of the spike protein. Curr. Biol. 2020, 30, 2196–2203.e3. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.; Tong, S. Complete genome sequence of a severe acute respiratory syndrome-related coronavirus from Kenyan bats. Microbiol. Resour. Announc. 2019, 8, e00548-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monadjem, A.; Taylor, P.J.; Jacobs, D.; Kock, D.; Amr, Z.S.S.; Cotterill, F. Rhinolophus clivosus, Geoffroy’s Horseshoe Bat. IUCN Red List Threat Species 2017, e.T19531A21980500. [Google Scholar] [CrossRef]
- Jacobs, D.; Cohen, L.; Richards, L.R.; Monadjem, A.; Schoeman, C.; MacEwan, K.; Sethusa, T.; Taylor, P.J. A conservation assessment of Rhinolophus clivosus. In The Red List of Mammals of South Africa, Swaziland and Lesotho South; Child, M.F., Roxburgh, L., Do Linh San, E., Raimondo, D., Davies-Mostert, H.T., Eds.; South African National Biodiversity Institute and Endangered Wildlife Trust: Gauteng, South Africa, 2016; pp. 1–5. [Google Scholar]
- Mendenhall, I.H.; Borthwick, S.; Neves, E.S.; Low, D.; Linster, M.; Liang, B.; Skiles, M.; Jayakumar, J.; Han, H.; Gunalan, V.; et al. Identification of a lineage D Betacoronavirus in Cave Nectar Bats (Eonycteris spelaea) in Singapore and an overview of Lineage D reservoir ecology in SE Asian Bats. Transbound. Emerg. Dis. 2017, 64, 1790–1800. [Google Scholar] [CrossRef] [PubMed]
- Kamins, A.O.O.; Restif, O.; Ntiamoa-Baidu, Y.; Suu-Ire, R.; Hayman, D.T.S.T.S.; Cunningham, A.A.A.; Wood, J.L.N.L.N.; Rowcliffe, J.M.M. Uncovering the fruit bat bushmeat commodity chain and the true extent of fruit bat hunting in Ghana, West Africa. Biol. Conserv. 2011, 144, 3000–3008. [Google Scholar] [CrossRef] [Green Version]
- Markotter, W.; MacEwan, K.; White, W.; Cohen, L.; Jacobs, D.; Monadjem, A.; Richards, L.R.; Schoeman, C.; Sethusa, T.; Taylor, P.J. A conservation assessment of Rousettus aegyptiacus. In The Red List of Mammals of South Africa, Swaziland and Lesotho; Child, M.F., Roxburgh, L., Do Linh San, E., Raimondo, D., Davies-Mostert, H.T., Eds.; South African National Biodiversity Institute and Endangered Wildlife Trust: Gauteng, South Africa, 2016; pp. 1–5. [Google Scholar]
- Mortlock, M.; Dietrich, M.; Weyer, J.; Paweska, J.T.; Markotter, W. Co-circulation and excretion dynamics of diverse rubula-and related viruses in Egyptian rousette bats from South Africa. Viruses 2019, 11, 37. [Google Scholar] [CrossRef] [Green Version]
- Amman, B.R.; Carroll, S.A.; Reed, Z.D.; Sealy, T.K.; Balinandi, S.; Swanepoel, R.; Kemp, A.; Erickson, B.R.; Comer, J.A.; Campbell, S.; et al. Seasonal pulses of Marburg virus circulation in juvenile Rousettus aegyptiacus bats coincide with periods of increased risk of human infection. PLoS Pathog. 2012, 8, e1002877. [Google Scholar] [CrossRef]
- Drexler, J.F.; Corman, V.M.; Wegner, T.; Tateno, A.F.; Zerbinati, R.M.; Gloza-Rausch, F.; Seebens, A.; Müller, M.A.; Drosten, C. Amplification of emerging viruses in a bat colony. Emerg. Infect. Dis. 2011, 17, 449–456. [Google Scholar] [CrossRef]
- Wacharapluesadee, S.; Duengkae, P.; Chaiyes, A.; Kaewpom, T.; Rodpan, A.; Yingsakmongkon, S.; Petcharat, S.; Phengsakul, P.; Maneeorn, P.; Hemachudha, T. Longitudinal study of age-specific pattern of coronavirus infection in Lyle’s flying fox (Pteropus lylei) in Thailand. Virol. J. 2018, 15, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lučan, R.K.; Bartonička, T.; Benda, P.; Bilgin, R.; Jedlička, P.; Nicolaou, H.; Reiter, A.; Shohdi, W.M.; Šálek, M.; Řeřucha, Š.; et al. Reproductive seasonality of the Egyptian fruit bat (Rousettus aegyptiacus) at the northern limits of its distribution. J. Mammal. 2014, 95, 1036–1042. [Google Scholar] [CrossRef] [Green Version]
- Jacobsen, N.H.G.; Du Plessis, E. Observations on the ecology and biology of the Cape fruit bat Rousettus aegyptiacus leachi in the Eastern Transvaal. S. Afr. J. Sci. 1976, 72, 270–273. [Google Scholar]
- Gloza-Rausch, F.; Ipsen, A.; Seebens, A.; Göttsche, M.; Panning, M.; Felix Drexler, J.; Petersen, N.; Annan, A.; Grywna, K.; Müller, M.; et al. Detection and prevalence patterns of group I coronaviruses in bats, northern Germany. Emerg. Infect. Dis. 2008, 14, 626–631. [Google Scholar] [CrossRef]
- Meyer, B.; Drosten, C.; Müller, M.A. Serological assays for emerging coronaviruses: Challenges and pitfalls. Virus Res. 2014, 194, 175–183. [Google Scholar] [CrossRef] [PubMed]
- Chu, D.K.W.; Hui, K.P.Y.; Perera, R.A.P.M.; Miguel, E.; Niemeyer, D.; Zhao, J.; Channappanavar, R.; Dudas, G.; Oladipo, J.O.; Traoré, A.; et al. MERS coronaviruses from camels in Africa exhibit region-dependent genetic diversity. Proc. Natl. Acad. Sci. USA 2018, 115, 3144–3149. [Google Scholar] [CrossRef] [Green Version]
- Younan, M.; Bornstein, S.; Gluecks, I.V. MERS and the dromedary camel trade between Africa and the Middle East. Trop. Anim. Health Prod. 2016, 48, 1277–1282. [Google Scholar] [CrossRef]
- Liljander, A.; Meyer, B.; Jores, J.; Müller, M.A.; Lattwein, E.; Njeru, I.; Bett, B.; Drosten, C.; Corman, V.M. MERS-CoV antibodies in humans, Africa, 2013–2014. Emerg. Infect. Dis. 2016, 22, 1086–1089. [Google Scholar] [CrossRef] [Green Version]
- Chu, D.K.; Oladipo, J.O.; Perera, R.A.; Kuranga, S.A.; Chan, S.M.; Poon, L.L.; Peiris, M. Middle East respiratory syndrome coronavirus (MERS-CoV) in dromedary camels in Nigeria, 2015. Eurosurveillance 2015, 20, 30086. [Google Scholar] [CrossRef] [Green Version]
- Miguel, E.; Chevalier, V.; Ayelet, G.; Ben Bencheikh, M.N.; Boussini, H.; Chu, D.K.; El Berbri, I.; Fassi-Fihri, O.; Faye, B.; Fekadu, G.; et al. Risk factors for MERS coronavirus infection in dromedary camels in Burkina Faso, Ethiopia, and Morocco, 2015. Eurosurveillance 2017, 22, 30498. [Google Scholar] [CrossRef]
- Chen, Y.-N.; Su, B.-G.; Chen, H.-C.; Chou, C.-H.; Cheng, H.-C. Detection of specific antibodies to the nucleocapsid protein fragments of Severe Acute Respiratory Syndrome-coronavirus and scotophilus bat coronavirus-512 in three insectivorous bat species. Taiwan Vet. J. 2018, 44, 179–188. [Google Scholar] [CrossRef]
- Chang, F.Y.; Chen, H.C.; Chen, P.J.; Ho, M.S.; Hsieh, S.L.; Lin, J.C.; Liu, F.T.; Sytwu, H.K. Immunologic aspects of characteristics, diagnosis, and treatment of coronavirus disease 2019 (COVID-19). J. Biomed. Sci. 2020, 27, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Müller, M.A.; Paweska, J.T.; Leman, P.A.; Drosten, C.; Grywna, K.; Kemp, A.; Braack, L.; Sonnenberg, K.; Niedrig, M.; Swanepoel, R. Coronavirus antibodies in African bat species. Emerg. Infect. Dis. 2007, 13, 1367–1370. [Google Scholar] [CrossRef] [PubMed]
- Tsuda, S.; Watanabe, S.; Masangkay, J.S.; Mizutani, T.; Alviola, P.; Ueda, N.; Iha, K.; Taniguchi, S.; Fujii, H.; Kato, K.; et al. Genomic and serological detection of bat coronavirus from bats in the Philippines. Arch. Virol. 2012, 157, 2349–2355. [Google Scholar] [CrossRef] [PubMed]
- McDermid, K.R.; Snyman, A.; Verreynne, F.J.; Carroll, J.P.; Penzhorn, B.L.; Yabsley, M.J. Surveillance for viral and parasitic pathogens in a vulnerable African lion (Panthera leo) population in the northern Tuli game reserve, Botswana. J. Wildl. Dis. 2017, 53, 54–61. [Google Scholar] [CrossRef] [Green Version]
- Evermann, J.F.; Laurenson, M.K.; McKeirnan, A.J.; Caro, T.M. Infectious disease surveillance in captive and free-living cheetahs: An integral part of the species survival plan. Zoo Biol. 1993, 12, 125–133. [Google Scholar] [CrossRef]
- Heeney, J.L.; Evermann, J.F.; McKeirnan, A.J.; Marker-Kraus, L.; Roelke, M.E.; Bush, M.; Wildt, D.E.; Meltzer, D.G.; Colly, L.; Lukas, J. Prevalence and implications of feline coronavirus infections of captive and free-ranging cheetahs (Acinonyx jubatus). J. Virol. 1990, 64, 1964–1972. [Google Scholar] [CrossRef] [Green Version]
- Wang, N.; Li, S.Y.; Yang, X.L.; Huang, H.M.; Zhang, Y.J.; Guo, H.; Luo, C.M.; Miller, M.; Zhu, G.; Chmura, A.A.; et al. Serological evidence of bat SARS-related coronavirus infection in humans, China. Virol. Sin. 2018, 33, 104–107. [Google Scholar] [CrossRef] [Green Version]
- Wolfe, N.D.; Daszak, P.; Kilpatrick, A.M.; Burke, D.S. Bushmeat hunting, deforestation, and prediction of zoonotic disease. Emerg. Infect. Dis. 2005, 11, 1822–1827. [Google Scholar] [CrossRef] [PubMed]
- Pernet, O.; Schneider, B.S.; Beaty, S.M.; LeBreton, M.; Yun, T.E.; Park, A.; Zachariah, T.T.; Bowden, T.A.; Hitchens, P.; Ramirez, C.M.; et al. Evidence for henipavirus spillover into human populations in Africa. Nat. Commun. 2014, 5, 5342. [Google Scholar] [CrossRef] [Green Version]
- Wacharapluesadee, S.; Sintunawa, C.; Kaewpom, T.; Khongnomnan, K.; Olival, K.J.; Epstein, J.H.; Rodpan, A.; Paiboon Sangsri, N.I.; Chindamporn, A.; Suksawa, K.; et al. Betacoronavirus in bat guano fertilizer, Thailand. Emerg. Infect. Dis. 2013, 19, 2012–2014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plowright, R.K.; Parrish, C.R.; McCallum, H.; Hudson, P.J.; Ko, A.I.; Graham, A.L.; Lloyd-Smith, J.O. Pathways to zoonotic spillover. Nat. Rev. Microbiol. 2017, 15, 502–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonwitt, J.; Kandeh, M.; Dawson, M.; Ansumana, R.; Sahr, F.; Kelly, A.H.; Brown, H. Participation of women and children in hunting activities in Sierra Leone and implications for control of zoonotic infections. PLoS Negl. Trop. Dis. 2017, 11, e0005699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mickleburgh, S.; Waylen, K.; Racey, P. Bats as bushmeat: A global review. Oryx 2009, 43, 217–234. [Google Scholar] [CrossRef] [Green Version]
- Mildenstein, T.; Tanshi, I.; Paul, A. Racey exploitation of bats for bushmeat and medicine. In Bats in the Anthropocene: Conservation of Bats in a Changing World; Voigt, C.C., Kingston, T., Eds.; Springer International Publishing: Cham, Switzerland, 2016; pp. 325–376. ISBN 978-3-319-25218-6. [Google Scholar]
- Nature Food. Exploring wet markets. Nat. Food 2020. [Google Scholar] [CrossRef]
- Jenkins, R.; Racey, P. Bats as bushmeat in Madagascar. Madag. Conserv. Dev. 2009, 3. [Google Scholar] [CrossRef] [Green Version]
- Weiss, S.; Nowak, K.; Fahr, J.; Wibbelt, G.; Mombouli, J.-V.; Parra, H.J.; Wolfe, N.D.; Schneider, B.S.; Leendertz, F.H. Henipavirus-related sequences in fruit bat bushmeat, Republic of Congo. Emerg. Infect. Dis. 2012, 18, 1536–1537. [Google Scholar] [CrossRef]
- Anti, P.; Owusu, M.; Agbenyega, O.; Annan, A.; Badu, E.K.; Nkrumah, E.E.; Tschapka, M.; Oppong, S.; Adu-Sarkodie, Y.; Drosten, C. Human–bat interactions in rural West Africa. Emerg. Infect. Dis. 2015, 21, 1418–1421. [Google Scholar] [CrossRef] [Green Version]
- Vora, N.M.; Osinubi, M.; Wallace, R.M.; Aman-Oloniyo, A.; Gbadegesin, Y.H.; Yennan, K.S.; Saliman, O.A.; Niezgoda, M.; Davis, L.; Recuenco, S. Assessment of potential zoonotic disease exposure and illness related to an annual bat festival—Idanre, Nigeria. MMWR Morb. Mortal Wkly Rep. 2014, 82, 3–6. [Google Scholar]
- Samuel, S. The Coronavirus Likely Came from China’s Wet Markets. They’re Reopening Anyway. Available online: https://www.vox.com/future-perfect/2020/4/15/21219222/coronavirus-china-ban-wet-markets-reopening (accessed on 11 June 2020).
- Daly, N. Chinese Citizens Push to Abolish Wildlife Trade as Coronavirus Persists. Available online: https://www.nationalgeographic.com/animals/2020/01/china-bans-wildlife-trade-after-coronavirus-outbreak/ (accessed on 11 June 2020).
- Goodman, S.M. Hunting of Microchiroptera in south-western Madagascar. Oryx 2006, 40, 225–228. [Google Scholar] [CrossRef] [Green Version]
- Gbogbo, F.; Kyei, M.O. Knowledge, perceptions and attitude of a community living around a colony of straw-coloured fruit bats (Eidolon helvum) in Ghana after Ebola virus disease outbreak in West Africa. Zoonoses Public Health 2017, 64, 628–635. [Google Scholar] [CrossRef]
- Lawson, E.T.; Ohemeng, F.; Ayivor, J.; Leach, M.; Waldman, L.; Ntiamoa-Baidu, Y. Understanding framings and perceptions of spillover. Disaster Prev. Manag. An. Int. J. 2017, 26, 396–411. [Google Scholar] [CrossRef]
- Ohemeng, F.; Lawson, E.T.; Ayivor, J.; Leach, M.; Waldman, L.; Ntiamoa-Baidu, Y. Socio-cultural determinants of human–bat interactions in rural Ghana. Anthrozoos 2017, 30, 181–194. [Google Scholar] [CrossRef]
- Kamins, A.O.; Rowcliffe, J.M.; Ntiamoa-Baidu, Y.; Cunningham, A.A.; Wood, J.L.N.; Restif, O. Characteristics and risk perceptions of Ghanaians potentially exposed to bat-borne zoonoses through bushmeat. Ecohealth 2015, 12, 104–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Musila, S.; Prokop, P.; Gichuki, N. Knowledge and perceptions of, and attitudes to, bats by people living around Arabuko-Sokoke Forest, Malindi-Kenya. Anthrozoos 2018, 31, 247–262. [Google Scholar] [CrossRef]
- Lawson, E.T.; Ayivor, J.S.; Ohemeng, F.; Ntiamoa-Baidu, Y. Avoiding bites and scratches? Understanding the public health implication of human-bat interactions in Ghana. Zoonoses Public Health 2018, 66, 108–116. [Google Scholar] [CrossRef] [PubMed]
- Holbrook, M.G.; Anthony, S.J.; Navarrete-Macias, I.; Bestebroer, T.; Munster, V.J.; van Doremalen, N. Updated and validated pan-coronavirus PCR assay to detect all coronavirus genera. Viruses 2021, 13, 599. [Google Scholar] [CrossRef]
- Chen, L.; Liu, B.; Yang, J.; Jin, Q. DBatVir: The database of bat-associated viruses. Database 2014, 2014, bau021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilbert, A.T.; Fooks, A.R.; Hayman, D.T.S.; Horton, D.L.; Müller, T.; Plowright, R.; Peel, A.J.; Bowen, R.; Wood, J.L.N.; Mills, J.; et al. Deciphering serology to understand the ecology of infectious diseases in wildlife. Ecohealth 2013, 10, 298–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, T. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and high- performance computing. Nat. Methods 2015, 9, 6–9. [Google Scholar] [CrossRef]
- Suchard, M.A.; Lemey, P.; Baele, G.; Ayres, D.L.; Drummond, A.J.; Rambaut, A. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 2018, 4, 1–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, M.A.; Pfeiffer, W.; Schwartz, T. Creating the CIPRES science gateway for inference of large phylogenetic trees. In Proceedings of the Gateway Computing Environments Workshop (GCE), New Orleans, LA, USA, 14 November 2010; pp. 1–8. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Search criteria: | Google scholar searches with keywords: “bat, bats, fruit bats, insectivorous bats, animal, mammal, livestock, domestic, domesticated, wildlife, coronavirus, coronaviruses, detections, Africa, Sub-Saharan, Southern Africa, Eastern Africa, nucleic acid, molecular detection, serology, serological, surveillance, survey” were used to search for peer-reviewed publications documenting surveys for coronaviruses in mammals from Africa (mainland Africa as well as islands associated with Africa such as Madagascar, Reunion Island, Seychelles). |
| Selection criteria: | For a suitably thorough synopsis of the findings, publications were limited to research available until the end of December 2020 and excluded dissertations, theses, or non-peer-reviewed publications. Sequences included in phylogenetic analyses in this review also excluded sequences from dissertations, theses, or unpublished sequences on GenBank that are not linked to available publications. However, PREDICT surveillance data (‘PREDICT 1 and 2 surveillance and test data’) linked to a 2017 publication was accessed online from Healthmap.org [56] and included both surveillance among bats and other wildlife and livestock. |
| Criteria for ‘primary surveillance reports’: | Reports containing a description of the collection and testing of samples from animals for coronavirus surveillance. For bat surveillance, we focused on surveillance strategies using nucleic acid detection methodologies such as family-wide consensus PCR analysis or unbiased high throughput metagenomic sequencing. This includes re-testing samples from an earlier report with a different assay and reporting additional coronaviruses detected. Primary surveillance reports may contain varying levels of characterization for detected viruses. We expanded this criterion for livestock and non-bat wildlife to include both nucleic acid and serological surveillance. |
| Criteria for ‘secondary characterization reports’: | Refers specifically to studies based on a primary surveillance report that does not describe new sample collection but a detailed characterization of viral sequences identified in a previous publication or more in-depth analysis of data obtained from primary surveillance reports. |
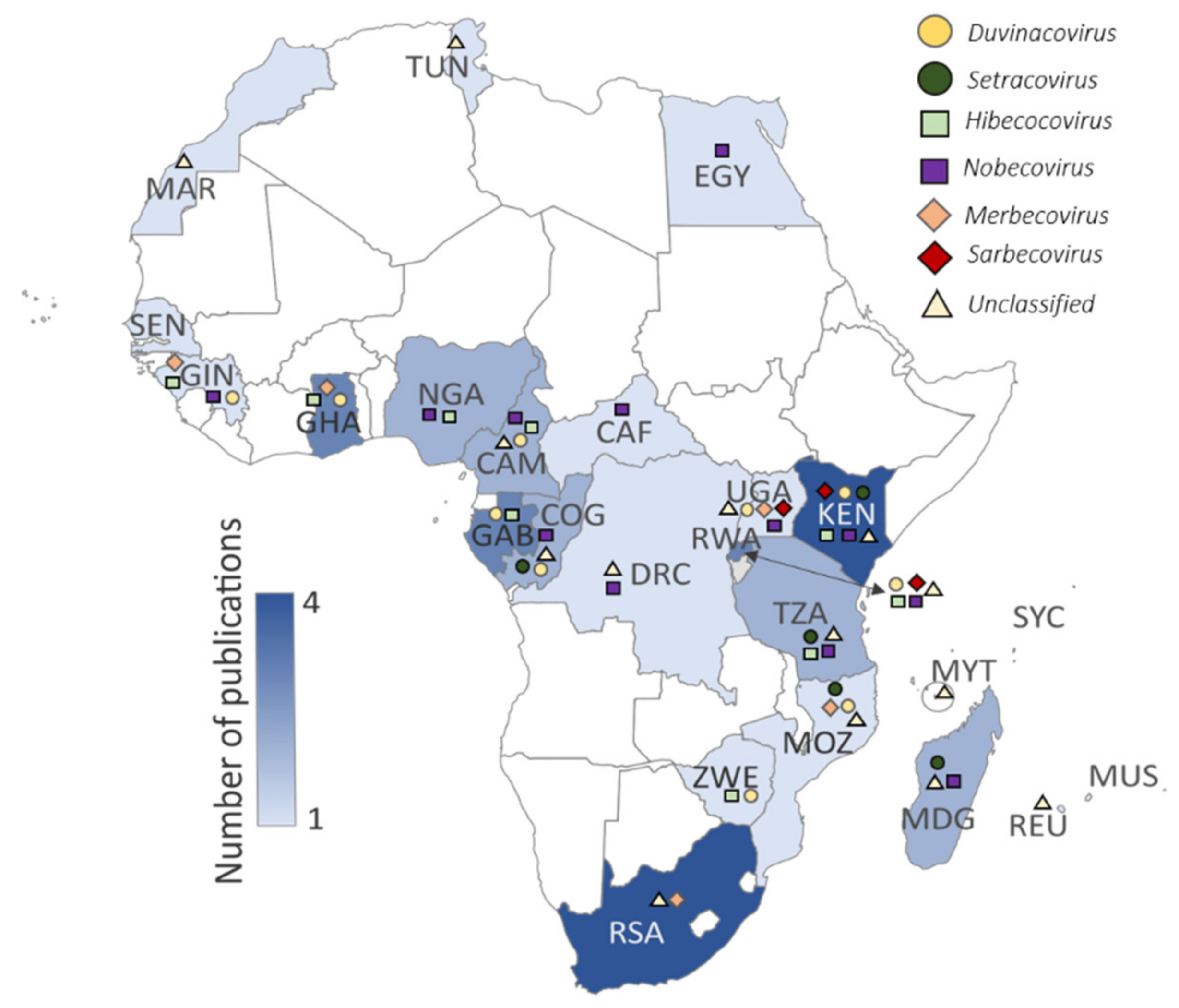
| Country (3 Letter Country Code) | References [Primary Surveillance]/(Characterization Report) * |
|---|---|
| Cameroon | [30,34] |
| Central African Republic (CAF) | [45] |
| Democratic Republic of the Congo (DRC) | [30] |
| Egypt (EGY) | [27] |
| Gabon (GAB) | [30,40,45] |
| Ghana (GHA) | [37,44,46] |
| Guinea (GIN) | [39] |
| Kenya (KEN) | [19,26,29]/([57]) |
| Madagascar (MDG) | [38,47] |
| Mauritius (MUS) | [38] |
| Mayotte (MYT) | [38] |
| Morocco (MAR) | [38] |
| Mozambique (MOZ) | [38] |
| Nigeria (NGA) | [28,41] |
| Republic of the Congo (COG) | [30,45] |
| Reunion Island (REU) | [38] |
| Rwanda (RWA) | [30,35,36]/([60]) |
| Senegal (SEN) | [45] |
| Seychelles (SYC) | [38] |
| South Africa (RSA) | [32,42,43]/([58]) |
| Tanzania (TZA) | [30]/([60]) |
| Tunisia (TUN) | [33] |
| Uganda (UGA) | [30]/([59,60]) |
| Zimbabwe (ZWE) | [31] |
| Bat Families Tested | Number of Species | Species Tested | Bat Species Positive | Number of Individuals Tested Per Family * | Positive Individuals # |
|---|---|---|---|---|---|
| Pteropodidae | 44 | 22 | 14 | 10,851 | 881 (8.1%) |
| Hipposideridae | 21 | 10 | 8 | 8563 | 257 (3%) |
| Molossidae | 44 | 16 | 8 | 2144 | 286 (13.3%) |
| Miniopteridae | 22 | 12 | 5 | 1464 | 120 (8.2%) |
| Vespertilionidae | 114 | 37 | 9 | 918 | 41 (4.5%) |
| Rhinolophidae | 38 | 14 | 9 | 728 | 68 (9.3%) |
| Emballonuridae | 11 | 4 | 0 | 678 | 0 |
| Nycteridae | 15 | 6 | 3 | 299 | 51 (17.1%) |
| Rhinonycteridae | 6 | 3 | 2 | 250 | 74 (29.6%) |
| Megadermatidae | 2 | 2 | 1 | 25 | 3 (12%) |
| Rhinopomatidae | 3 | 1 | 0 | 1 | 0 |
| Myzopodidae | 2 | 0 | 0 | 0 | - |
| Cistugonidae | 2 | 0 | 0 | 0 | - |
| Totals | 324 | 127 | 59 | 25,921 | 1779 (6.9%) |
| Animals Groups | Birds 1 and Poultry/ Other Fowl | Carivores 2 | Cattle/ Buffalo 3 | Dogs 4 | Goats/ Sheep 4 | Non-Human Primates | Pangolins 5 | Rodents/ Shrews | Swine 4 | Ungulates 7 | Other 6 | Grand Total |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Cameroon | - | 67 | - | - | - | 3475 | 79 | 4653 | - | 144 | 16 | 8434 |
| DR Congo | 7 | 6 | 10 | - | 16 | 1574 | 3 | 1848 | 1 | 15 | 2 | 3482 |
| Ethiopia | - | - | - | - | - | 454 | - | - | - | - | - | 454 |
| Gabon | 1 | 11 | - | - | - | 82 | 18 | 1141 | - | 548 | 37 | 1838 |
| Ghana | - | - | 1230 | - | 2194 | 496 | - | 532 | 716 | 108 | - | 5276 |
| Guinea | - | - | - | 6 | 321 | - | - | 904 | 8 | - | - | 1239 |
| Ivory Coast | 12 | - | - | - | - | 59 | - | 293 | - | - | - | 364 |
| Kenya | - | - | - | - | - | 334 | - | 369 | - | 514 | - | 1217 |
| Liberia | - | - | - | - | - | - | - | 205 | - | - | - | 205 |
| Republic of Congo | - | 2 | - | - | - | 352 | - | 461 | - | 14 | - | 829 |
| Rwanda | - | - | - | - | - | 762 | - | 708 | - | - | - | 1470 |
| Senegal | - | - | - | - | - | 253 | - | 263 | - | - | - | 516 |
| Sierra Leone | - | 5 | - | 318 | 938 | 15 | - | 369 | 1012 | - | - | 2657 |
| South Sudan | - | - | - | - | - | - | - | 46 | - | - | - | 46 |
| Tanzania | - | 8 | 53 | 120 | 105 | 444 | - | 1513 | 95 | 39 | 1 | 2378 |
| Uganda | - | - | - | - | 13 | 1238 | - | 762 | 1 | 83 | - | 2097 |
| Grand Total | 20 | 99 | 1293 | 444 | 3587 | 9538 | 100 | 14,067 | 1833 | 1465 | 56 | 32,502 |
| Coronavirus nucleic acid | - | 1 | - | - | 14 | - | 13 | - | 1 | - | 29 |
 | |
|---|---|
| Consideration | Activity |
| Formulate a strong research question around the aim of the research to be conducted. | Scope of the surveillance—only coronaviruses or broader surveillance. What will the primary focus of the project be? Assessment of risk for settlements near known colonies? Review the literature and determine important species to target. |
| Assemble an interdisciplinary team | Collaborate with experts in virology, taxonomists, field biologists, veterinarians, ecologists, specific community leaders, social sciences, and policy-makers. A large interdisciplinary team is essential for accurate long-term surveillance. |
| Identify high-risk species or animal populations based on a predetermined research question | As a starting point, collaborations can assist in identifying accessible locations of interests, such as specific roosts (day or maternity roosts, etc.) for bat host species considered higher risk (from literature). The roosts can be assessed for population presence over time to enable longitudinal surveillance planning. The region must be assessed for nearby human settlements and the occurrence of animals (farmed, free-roaming, or other wildlife). |
| Perform initial surveillance targeting either large roosts or multiple smaller roosts | Assess viral presence and diversity with once-off or seasonal surveillance (statistically significant). Population-level sampling of excreted samples such as fecal collection (beneath roosting bats) is simple and non-invasive. Proper species identification should be conducted with both barcodes and morphological identification. |
| Nucleic acid testing with a suitable assay | Review the literature and use a recently updated assay to ensure detection of all available diversity. Test the assay sensitivity for comparisons. Based on the scope of the project and resource conservation—consider a specific or randomly primed approach. |
| Plan longitudinal surveillance (duration, types of samples collected, measurement, and ecological data collection). Plan to survey animal species in the region preferably concurrently or sequentially following bat surveillance. | Based on initial findings, plan for longitudinal surveillance according to specified intervals (based on bat presence at roosts or species movements): seasonal or periodic (monthly). Sampling must occur across different reproductive stages. Surveillance can be done at the population-level (overall) and individual-level (to determine demographics of infection prevalence). |
| Serological surveillance | Review options for serological assays (commercial or developed assays). Collaboration with experts may be critical. Serological testing (bats, non-bat animals, and humans) is important to understand coronavirus antibody responses, duration of protection, and exposure—optimize suitable assays. |
| Viral characterization | Recover complete genomes of selected viruses for classification and functional studies. Assessing possible zoonotic potential with pathogenesis studies and protein modeling. Collaborate with specialists that can assist and help develop local capacity. |
| Investigate human-animal interactions | Perform observational and behavioural studies to assess human-wildlife-livestock interactions. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Geldenhuys, M.; Mortlock, M.; Epstein, J.H.; Pawęska, J.T.; Weyer, J.; Markotter, W. Overview of Bat and Wildlife Coronavirus Surveillance in Africa: A Framework for Global Investigations. Viruses 2021, 13, 936. https://0-doi-org.brum.beds.ac.uk/10.3390/v13050936
Geldenhuys M, Mortlock M, Epstein JH, Pawęska JT, Weyer J, Markotter W. Overview of Bat and Wildlife Coronavirus Surveillance in Africa: A Framework for Global Investigations. Viruses. 2021; 13(5):936. https://0-doi-org.brum.beds.ac.uk/10.3390/v13050936
Chicago/Turabian StyleGeldenhuys, Marike, Marinda Mortlock, Jonathan H. Epstein, Janusz T. Pawęska, Jacqueline Weyer, and Wanda Markotter. 2021. "Overview of Bat and Wildlife Coronavirus Surveillance in Africa: A Framework for Global Investigations" Viruses 13, no. 5: 936. https://0-doi-org.brum.beds.ac.uk/10.3390/v13050936