
Comparative Analysis of Public RNA-Sequencing Data from Human Intestinal Enteroid (HIEs) Infected with Enteric RNA Viruses Identifies Universal and Virus-Specific Epithelial Responses


Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines and Virus Infection
2.2. Selection of GEO and EMBL-EBI Data
2.3. Differential Expression Analysis
2.4. Over-Representation Analysis
2.5. Transcription Factor Enrichment Analysis (TFEA)
2.6. Code Availability
3. Results
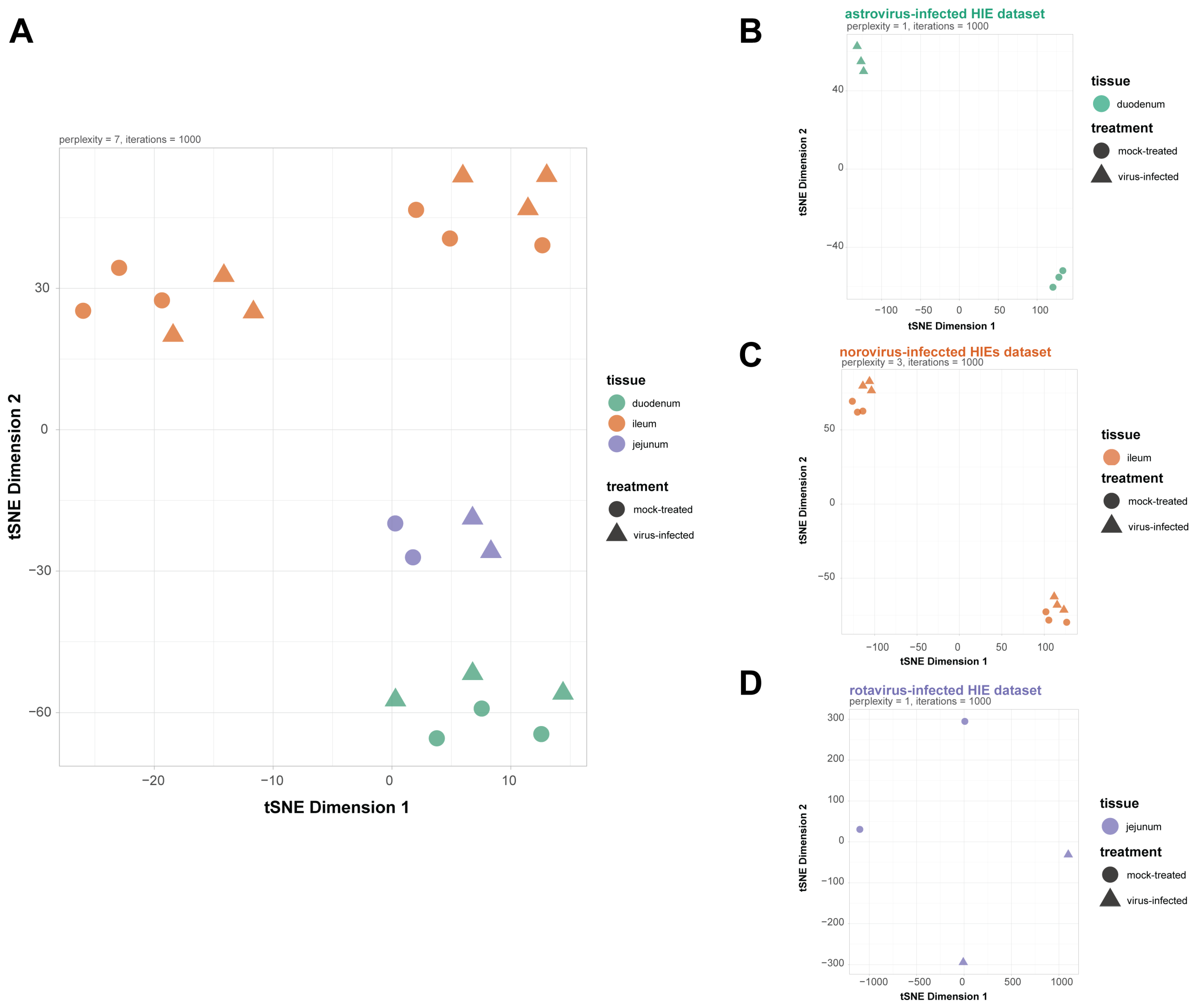
3.1. Samples Across Datasets Showed Divergence Due to Tissue Origin of the HIEs Line and Viral Infection
3.2. Most Non-Weakly Expressed Genes are Shared Across Datasets Including a Number of Innate Immune Response Genes
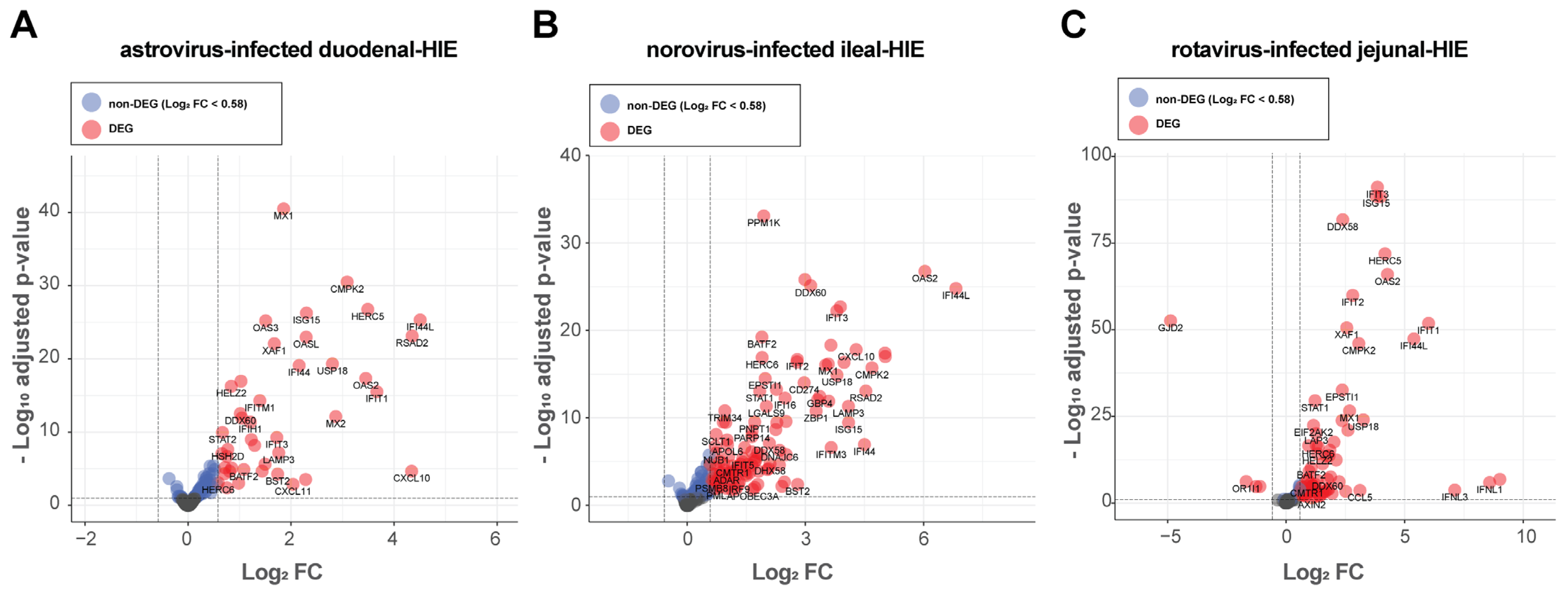
3.3. Differential Expression Analysis Showed That HIE Responses Against Astrovirus, Norovirus and Rotavirus Is Primarily Characterized by an Up-Regulation of Interferon-Stimulated Genes (ISGs)
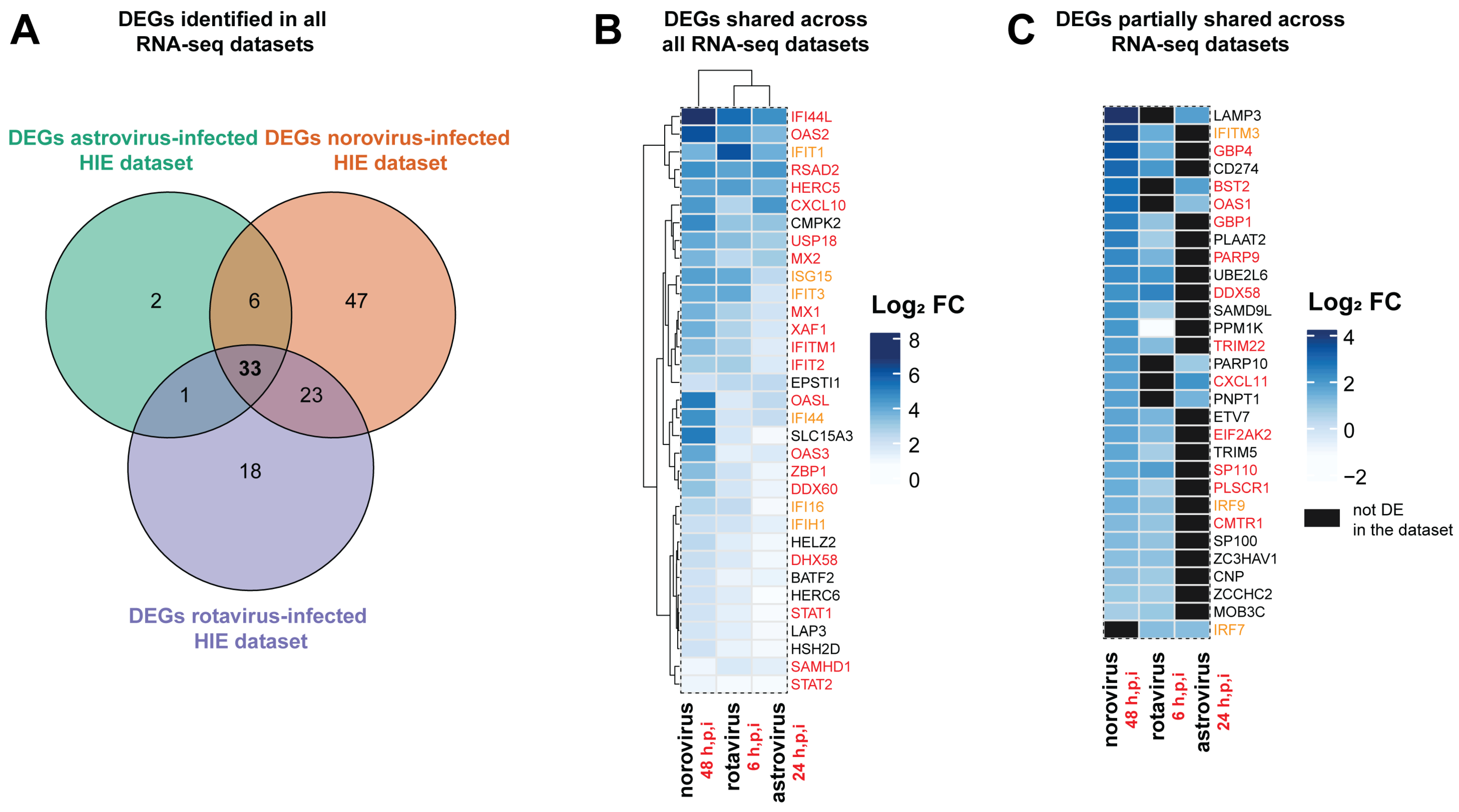
3.4. Antiviral Defense and IFN Signaling Represent a Conserved Response of the Epithelium to Viral Infection
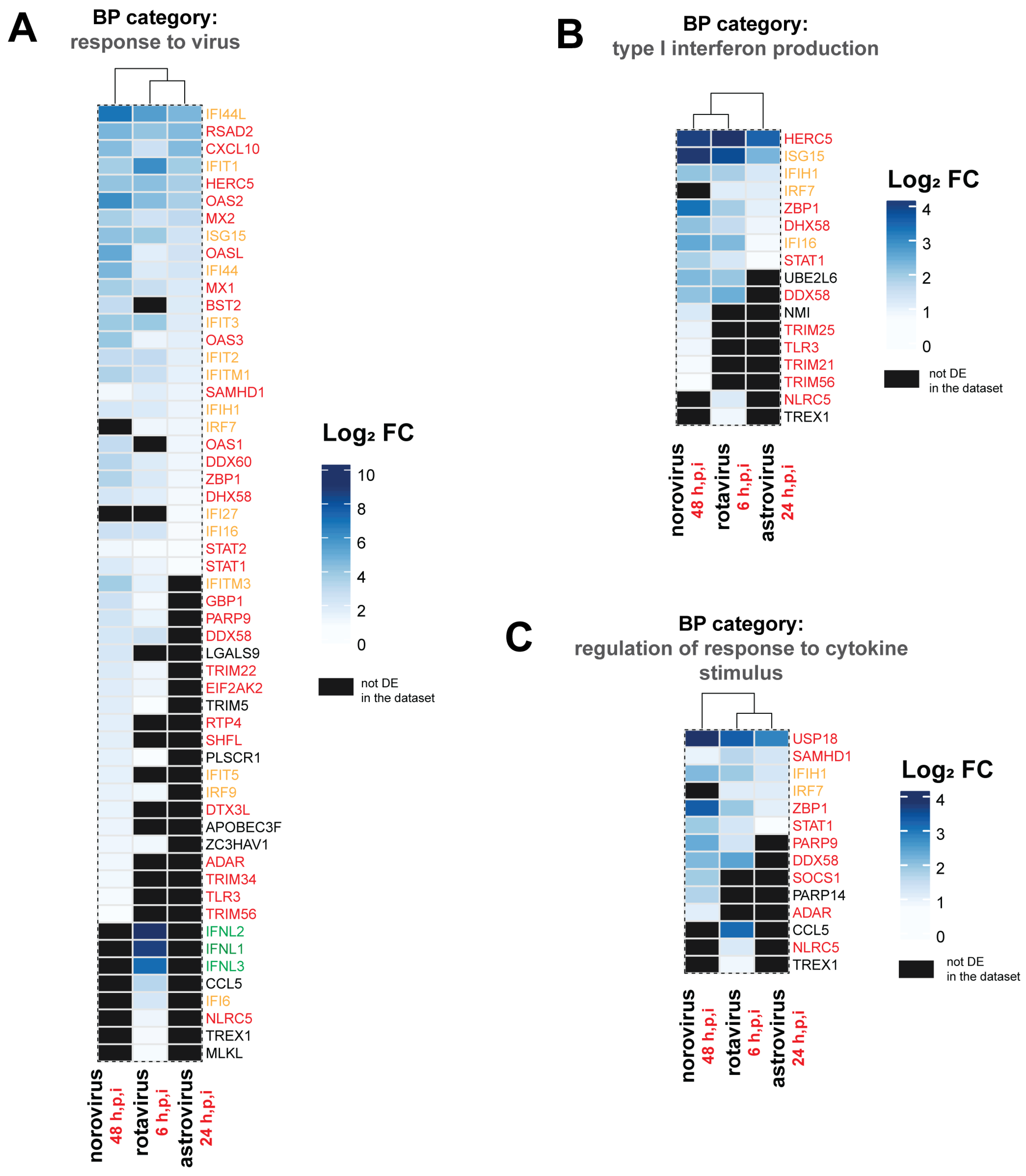
3.5. Distinct DEGs and Biological Processes (BP) were Also Noted for Each Enteric Virus
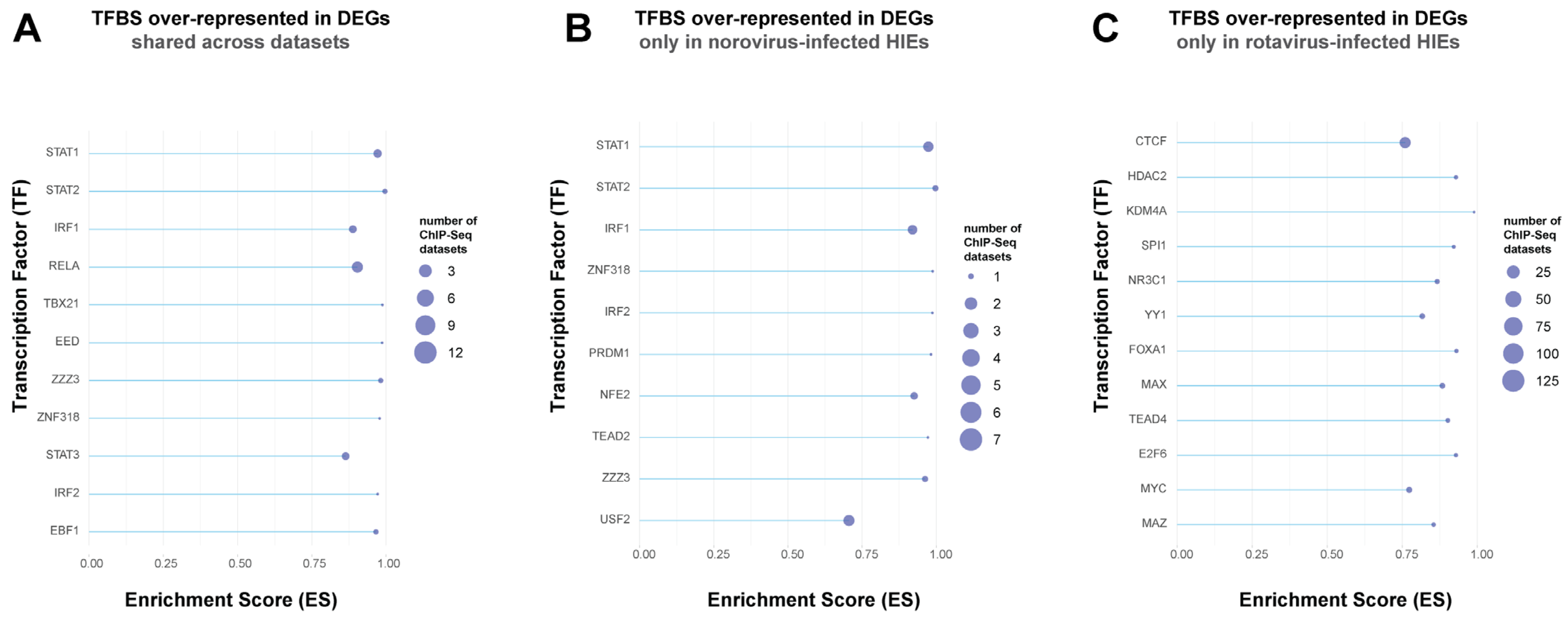
3.6. Transcription Factor Enrichment Analysis (TFEA) Revealed That the Conserved Intestinal Epithelial Response against the Three Enteric Viruses is Driven by STAT1/STAT2, IRF1, and NFkB
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Oude Munnink, B.B.; van der Hoek, L. Viruses Causing Gastroenteritis: The Known, The New and Those Beyond. Viruses 2016, 8, 42. [Google Scholar] [CrossRef]
- Hall, A.J.; Rosenthal, M.; Gregoricus, N.; Greene, S.A.; Ferguson, J.; Henao, O.L.; Vinjé, J.; Lopman, B.A.; Parashar, U.D.; Widdowson, M.-A. Incidence of Acute Gastroenteritis and Role of Norovirus, Georgia, USA, 2004–2005. Emerg. Infect. Dis. 2011, 17, 1381–1388. [Google Scholar] [CrossRef]
- Ahmed, S.M.; Hall, A.J.; Robinson, A.E.; Verhoef, L.; Premkumar, P.; Parashar, U.D.; Koopmans, M.; Lopman, B.A. Global prevalence of norovirus in cases of gastroenteritis: A systematic review and meta-analysis. Lancet Infect. Dis. 2014, 14, 725–730. [Google Scholar] [CrossRef] [Green Version]
- Crawford, S.E.; Ramani, S.; Tate, J.E.; Parashar, U.D.; Svensson, L.; Hagbom, M.; Franco, M.A.; Greenberg, H.B.; O’Ryan, M.; Kang, G.; et al. Rotavirus infection. Nat. Rev. Dis. Primers 2017, 3, 17083. [Google Scholar] [CrossRef] [Green Version]
- Vu, D.-L.; Bosch, A.; Pintó, R.M.; Guix, S. Epidemiology of Classic and Novel Human Astrovirus: Gastroenteritis and Beyond. Viruses 2017, 9, 33. [Google Scholar] [CrossRef] [PubMed]
- Holloway, E.M.; Czerwinski, M.; Tsai, Y.-H.; Wu, J.H.; Wu, A.; Childs, C.J.; Walton, K.D.; Sweet, C.W.; Yu, Q.; Glass, I.; et al. Mapping Development of the Human Intestinal Niche at Single-Cell Resolution. Cell Stem Cell 2021, 28, 568–580. [Google Scholar] [CrossRef] [PubMed]
- Lauder, E.K.K.; Schmidt, T.M.; Golob, J.L. Organoid-derived adult human colonic epithelium responds to co-culture with a probiotic strain of Bifidobacterium longum. bioRxiv 2020. [Google Scholar] [CrossRef]
- Blutt, S.E.; Crawford, S.E.; Ramani, S.; Zou, W.Y.; Estes, M.K. Engineered Human Gastrointestinal Cultures to Study the Microbiome and Infectious Diseases. Cell. Mol. Gastroenterol. Hepatol. 2018, 5, 241–251. [Google Scholar] [CrossRef] [Green Version]
- Kolawole, A.O.; Wobus, C.E. Gastrointestinal organoid technology advances studies of enteric virus biology. PLoS Pathog. 2020, 16, e1008212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolawole, A.O.; Mirabelli, C.; Hill, D.R.; Svoboda, S.A.; Janowski, A.B.; Passalacqua, K.D.; Rodriguez, B.N.; Dame, M.K.; Freiden, P.; Berger, R.P.; et al. Astrovirus replication in human intestinal enteroids reveals multi-cellular tropism and an intricate host innate immune landscape. PLoS Pathog. 2019, 15, e1008057. [Google Scholar] [CrossRef] [Green Version]
- Hosmillo, M.; Chaudhry, Y.; Nayak, K.; Sorgeloos, F.; Koo, B.-K.; Merenda, A.; Lillestol, R.; Drumright, L.; Zilbauer, M.; Goodfellow, I. Norovirus Replication in Human Intestinal Epithelial Cells Is Restricted by the Interferon-Induced JAK/STAT Signaling Pathway and RNA Polymerase II-Mediated Transcriptional Responses. MBio 2020, 11, e00215-20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saxena, K.; Simon, L.M.; Zeng, X.-L.; Blutt, S.E.; Crawford, S.E.; Sastri, N.P.; Karandikar, U.C.; Ajami, N.J.; Zachos, N.C.; Kovbasnjuk, O.; et al. A paradox of transcriptional and functional innate interferon responses of human intestinal enteroids to enteric virus infection. Proc. Natl. Acad. Sci. USA 2017, 114, E570–E579. [Google Scholar] [CrossRef] [Green Version]
- Sudmant, P.H.; Alexis, M.S.; Burge, C.B. Meta-analysis of RNA-seq expression data across species, tissues and studies. Genome Biol. 2015, 16, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Fasterius, E.; Szigyarto, C.A.-K. Analysis of public RNA-sequencing data reveals biological consequences of genetic heterogeneity in cell line populations. Sci. Rep. 2018, 8, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Bray, N.L.; Pimentel, H.; Melsted, P.; Pachter, L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 2016, 34, 525–527. [Google Scholar] [CrossRef] [PubMed]
- Soneson, C.; Love, M.I.; Robinson, M.D. Differential analyses for RNA-seq: Transcript-level estimates improve gene-level inferences. F1000Res. 2015, 4, 1521. [Google Scholar] [CrossRef] [PubMed]
- Rau, A.; Gallopin, M.; Celeux, G.; Jaffrézic, F. Data-based filtering for replicated high-throughput transcriptome sequencing experiments. Bioinformatics 2013, 29, 2146–2152. [Google Scholar] [CrossRef] [Green Version]
- Gibney, G.; Baxevanis, A.D. Searching NCBI Databases Using Entrez. Curr. Protoc. Bioinform. 2011, 34, 1–3. [Google Scholar] [CrossRef]
- Naming human genes. Nat. Genet. 2020, 52, 751. [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Stephens, M. False discovery rates: A new deal. Biostatistics 2016, 18, 275–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blighe, K.; Rana, S.; Lewis, M. EnhancedVolcano: Publication-Ready Volcano Plots with Enhanced Colouring and Labeling. 2018. Available online: https://github.com/kevinblighe/EnhancedVolcano.
- Simmons, S.; Peng, J.; Bienkowska, J.; Berger, B. Discovering what Dimensionality Reduction Really Tells Us about RNA-Seq Data. J. Comput. Biol. 2015, 22, 715–728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. clusterProfiler: An R Package for Comparing Biological Themes Among Gene Clusters. OMICS A J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Gu, Z.; Eils, R.; Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puente-Santamaria, L.; Wasserman, W.W.; Del Peso, L. TFEA.ChIP: A tool kit for transcription factor binding site enrichment analysis capitalizing on ChIP-seq datasets. Bioinformatics 2019, 35, 5339–5340. [Google Scholar] [CrossRef]
- Consortium, E.P. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar] [CrossRef]
- Lin, S.-C.; Qu, L.; Ettayebi, K.; Crawford, S.E.; Blutt, S.E.; Robertson, M.J.; Zeng, X.-L.; Tenge, V.R.; Ayyar, B.V.; Karandikar, U.C.; et al. Human norovirus exhibits strain-specific sensitivity to host interferon pathways in human intestinal enteroids. Proc. Natl. Acad. Sci. USA 2020, 117, 23782–23793. [Google Scholar] [CrossRef] [PubMed]
- Baccarella, A.; Williams, C.R.; Parrish, J.Z.; Kim, C.C. Empirical assessment of the impact of sample number and read depth on RNA-Seq analysis workflow performance. BMC Bioinform. 2018, 19, 1–12. [Google Scholar] [CrossRef]
- He, L.; Wang, B.; Li, Y.; Zhu, L.; Li, P.; Zou, F.; Bin, L. The Solute Carrier Transporter SLC15A3 Participates in Antiviral Innate Immune Responses against Herpes Simplex Virus-1. J. Immunol. Res. 2018, 2018, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McNab, F.W.; Mayerbarber, K.D.; Sher, A.; Wack, A.; O’Garra, A. Type I interferons in infectious disease. Nat. Rev. Immunol. 2015, 15, 87–103. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.K.; Tomida, J.; Wood, R.D. Disruption of DNA polymerase zeta engages an innate immune response. Cell Rep. 2021, 34, 108775. [Google Scholar] [CrossRef]
- Chiang, H.-S.; Liu, H.M. The Molecular Basis of Viral Inhibition of IRF- and STAT-Dependent Immune Responses. Front. Immunol. 2019, 9, 3086. [Google Scholar] [CrossRef] [Green Version]
- Schoggins, J.W.; Wilson, S.J.; Panis, M.; Murphy, M.Y.; Jones, C.T.; Bieniasz, P.; Rice, C.M. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 2011, 472, 481–485. [Google Scholar] [CrossRef]
- Dang, W.; Xu, L.; Yin, Y.; Chen, S.; Wang, W.; Hakim, M.S.; Chang, K.-O.; Peppelenbosch, M.P.; Pan, Q. IRF-1, RIG-I and MDA5 display potent antiviral activities against norovirus coordinately induced by different types of interferons. Antivir. Res. 2018, 155, 48–59. [Google Scholar] [CrossRef]
- Maloney, N.S.; Thackray, L.B.; Goel, G.; Hwang, S.; Duan, E.; Vachharajani, P.; Xavier, R.; Virgin, H.W. Essential cell-autonomous role for interferon (IFN) regulatory factor 1 in IFN-gamma-mediated inhibition of norovirus replication in macrophages. J. Virol. 2012, 86, 12655–12664. [Google Scholar] [CrossRef] [Green Version]
- Iaconis, G.; Jackson, B.; Childs, K.; Boyce, M.; Goodbourn, S.; Blake, N.; Iturriza-Gomara, M.; Seago, J. Rotavirus NSP1 Inhibits Type I and Type III Interferon Induction. Viruses 2021, 13, 589. [Google Scholar] [CrossRef]
- Basagoudanavar, S.H.; Thapa, R.J.; Nogusa, S.; Wang, J.; Beg, A.A.; Balachandran, S. Distinct roles for the NF-kappa B RelA subunit during antiviral innate immune responses. J. Virol. 2011, 85, 2599–2610. [Google Scholar] [CrossRef] [Green Version]
- Ren, G.; Cui, K.; Zhang, Z.; Zhao, K. Division of labor between IRF1 and IRF2 in regulating different stages of transcriptional activation in cellular antiviral activities. Cell Biosci. 2015, 5, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pentland, I.; Parish, J.L. Targeting CTCF to Control Virus Gene Expression: A Common Theme amongst Diverse DNA Viruses. Viruses 2015, 7, 3574–3585. [Google Scholar] [CrossRef] [Green Version]
- Pervolaraki, K.; Stanifer, M.L.; Münchau, S.; Renn, L.A.; Albrecht, D.; Kurzhals, S.; Senís, E.; Grimm, D.; Schröder-Braunstein, J.; Rabin, R.L.; et al. Type I and Type III Interferons Display Different Dependency on Mitogen-Activated Protein Kinases to Mount an Antiviral State in the Human Gut. Front. Immunol. 2017, 8, 459. [Google Scholar] [CrossRef] [PubMed]
- Su, R.; Shereen, M.A.; Zeng, X.; Liang, Y.; Li, W.; Ruan, Z.; Li, Y.; Liu, W.; Liu, Y.; Wu, K.; et al. The TLR3/IRF1/Type III IFN Axis Facilitates Antiviral Responses against Enterovirus Infections in the Intestine. MBio 2020, 11, e02540-20. [Google Scholar] [CrossRef]
- Ioannidis, I.; Ye, F.; McNally, B.; Willette, M.; Flaño, E. Toll-Like Receptor Expression and Induction of Type I and Type III Interferons in Primary Airway Epithelial Cells. J. Virol. 2013, 87, 3261–3270. [Google Scholar] [CrossRef] [Green Version]
- McCartney, S.A.; Thackray, L.B.; Gitlin, L.; Gilfillan, S.; Virgin, H.W.; Colonna, M. MDA-5 recognition of a murine norovirus. PLoS Pathog. 2008, 4, e1000108. [Google Scholar] [CrossRef]
- Perng, Y.-C.; Lenschow, D.J. ISG15 in antiviral immunity and beyond. Nat. Rev. Genet. 2018, 16, 423–439. [Google Scholar] [CrossRef] [PubMed]
- Stanifer, M.L.; Guo, C.; Doldan, P.; Boulant, S. Importance of Type I and III Interferons at Respiratory and Intestinal Barrier Surfaces. Front. Immunol. 2020, 11, 608645. [Google Scholar] [CrossRef]
- Ding, S.; Zhu, S.; Ren, L.; Feng, N.; Song, Y.; Ge, X.; Li, B.; Flavell, R.A.; Greenberg, H.B. Rotavirus VP3 targets MAVS for degradation to inhibit type III interferon expression in intestinal epithelial cells. Elife 2018, 7, e39494. [Google Scholar] [CrossRef]
- Sen, A.; Sharma, A.; Greenberg, H. Rotavirus Degrades Multiple Interferon (IFN) Type Receptors to Inhibit IFN Signaling and Protects against Mortality from Endotoxin in Suckling Mice. J. Virol. 2017, 92, e01394-17. [Google Scholar] [CrossRef] [Green Version]
- Graff, J.W.; Ettayebi, K.; Hardy, M.E. Rotavirus NSP1 inhibits NFkappaB activation by inducing proteasome-dependent degradation of beta-TrCP: A novel mechanism of IFN antagonism. PLoS Pathog. 2009, 5, e1000280. [Google Scholar] [CrossRef] [Green Version]
- Di Fiore, I.J.; Pane, J.A.; Holloway, G.; Coulson, B.S. NSP1 of human rotaviruses commonly inhibits NF-kappaB signalling by inducing beta-TrCP degradation. J. Gen. Virol. 2015, 96, 1768–1776. [Google Scholar] [CrossRef]
- Morelli, M.; Dennis, A.F.; Patton, J.T. Putative E3 ubiquitin ligase of human rotavirus inhibits NF-kappaB activation by using molecular mimicry to target beta-TrCP. MBio 2015, 6, e02490-14. [Google Scholar] [CrossRef] [Green Version]
- Ding, S.; Mooney, N.; Li, B.; Kelly, M.R.; Feng, N.; Loktev, A.V.; Sen, A.; Patton, J.T.; Jackson, P.K.; Greenberg, H.B. Comparative Proteomics Reveals Strain-Specific beta-TrCP Degradation via Rotavirus NSP1 Hijacking a Host Cullin-3-Rbx1 Complex. PLoS Pathog. 2016, 12, e1005929. [Google Scholar] [CrossRef] [PubMed]
- Roth, A.N.; Karst, S.M. Norovirus mechanisms of immune antagonism. Curr. Opin. Virol. 2016, 16, 24–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Differential Expression Analysis Summary | |||||
|---|---|---|---|---|---|
| Dataset | HIEs | # Genes Analyzed | DEGs | Up-Regulated-DEGs | Down-Regulated DEGs |
| astrovirus-infected | duodenal | 11,824 | 42 | 42 | 0 |
| norovirus-infected | ileal | 12,917 | 109 | 109 | 0 |
| rotavirus-infected | jejunal | 12,416 | 75 | 71 | 4 |
| Shared Differentially Expressed Genes (DEGs) and IFN Responses | ||||||
|---|---|---|---|---|---|---|
| HGNC Symbol | Ensemble Gene ID | Entrez Gene ID | Entrez Gene Description | Type I IFN | Type II IFN | Type III IFN |
| BATF2 | ENSG00000168062 | 116071 | basic leucine zipper ATF-like transcription factor 2 | yes | yes | yes |
| CMPK2 | ENSG00000134326 | 129607 | cytidine/uridine monophosphate kinase 2 | yes | yes | no |
| CXCL10 | ENSG00000169245 | 3627 | C-X-C motif chemokine ligand 10 | yes | yes | no |
| DDX58 | ENSG00000107201 | 23586 | DExD/H-box helicase 58 | yes | yes | yes |
| DDX60 | ENSG00000137628 | 55601 | DExD/H-box helicase 60 | yes | yes | no |
| DHX58 | ENSG00000108771 | 79132 | DExH-box helicase 58 | yes | yes | no |
| EPSTI1 | ENSG00000133106 | 94240 | epithelial stromal interaction 1 | yes | yes | yes |
| HELZ2 | ENSG00000130589 | 85441 | helicase with zinc finger 2 | yes | yes | yes |
| HERC5 | ENSG00000138646 | 51191 | HECT and RLD domain containing E3 ubiquitin protein ligase 5 | yes | yes | no |
| HSH2D | ENSG00000196684 | 84941 | hematopoietic SH2 domain containing | yes | yes | no |
| IFI16 | ENSG00000163565 | 3428 | interferon gamma inducible protein 16 | yes | yes | no |
| IFIH1 | ENSG00000115267 | 64135 | interferon induced with helicase C domain 1 | yes | yes | yes |
| IFIT1 | ENSG00000185745 | 3434 | interferon induced protein with tetratricopeptide repeats 1 | yes | yes | yes |
| IFIT2 | ENSG00000119922 | 3433 | interferon induced protein with tetratricopeptide repeats 2 | yes | yes | yes |
| IFIT3 | ENSG00000119917 | 3437 | interferon induced protein with tetratricopeptide repeats 3 | yes | yes | yes |
| IFITM1 | ENSG00000185885 | 8519 | interferon induced transmembrane protein 1 | yes | yes | no |
| ISG15 | ENSG00000187608 | 9636 | ISG15 ubiquitin like modifier | yes | yes | yes |
| LAMP3 | ENSG00000078081 | 27074 | lysosomal associated membrane protein 3 | yes | yes | no |
| LAP3 | ENSG00000002549 | 51056 | leucine aminopeptidase 3 | yes | yes | no |
| MX1 | ENSG00000157601 | 4599 | MX dynamin like GTPase 1 | yes | yes | yes |
| MX2 | ENSG00000183486 | 4600 | MX dynamin like GTPase 2 | yes | yes | no |
| OAS2 | ENSG00000111335 | 4939 | 2′-5′-oligoadenylate synthetase 2 | yes | yes | no |
| OAS3 | ENSG00000111331 | 4940 | 2′-5′-oligoadenylate synthetase 3 | yes | yes | yes |
| OASL | ENSG00000135114 | 8638 | 2′-5′-oligoadenylate synthetase like | yes | yes | yes |
| PARP10 | ENSG00000178685 | 84875 | poly(ADP-ribose) polymerase family member 10 | yes | yes | yes |
| RSAD2 | ENSG00000134321 | 91543 | radical S-adenosyl methionine domain containing 2 | yes | yes | no |
| SAMHD1 | ENSG00000101347 | 25939 | SAM and HD domain containing deoxynucleoside triphosphate triphosphohydrolase 1 | yes | yes | no |
| STAT1 | ENSG00000115415 | 6772 | signal transducer and activator of transcription 1 | yes | yes | yes |
| STAT2 | ENSG00000170581 | 6773 | signal transducer and activator of transcription 2 | yes | yes | yes |
| USP18 | ENSG00000184979 | 11274 | ubiquitin specific peptidase 18 | yes | yes | yes |
| XAF1 | ENSG00000132530 | 54739 | XIAP associated factor 1 | yes | yes | no |
| ZBP1 | ENSG00000124256 | 81030 | Z-DNA binding protein 1 | yes | yes | no |
| Gene Ontology (GO) Over-Representation Analysis Summary | ||||
|---|---|---|---|---|
| Dataset | HIE | # DEGs | # DEGs Assigned to BP Categories | # BP Categories Over-Represented |
| astrovirus-infected | duodenal | 42 | 41 | 18 |
| norovirus-infected | ileal | 109 | 103 | 26 |
| rotavirus-infected | jejunal | 75 | 69 | 23 |
| Uniquely Over-Represented Biological Processes (BPs) for each Dataset Analyzed | |||||||
|---|---|---|---|---|---|---|---|
| Dataset | HIEs | GO Term ID | GO Term Description | Gene Ratio | Gene Count | adj. p-Value | Gene ID |
| astrovirus-infected | duodenal | GO:0050688 | regulation of defense response to virus | 0.12 | 5 | 0.00012 | IFIT1,HERC5,DDX60,DHX58,STAT1 |
| norovirus-infected | ileal | GO:1903900 | regulation of viral life cycle | 0.27 | 28 | 0.00000 | OAS2,OASL,RSAD2,ISG15,LAMP3,OAS3,IFITM3,IFIT1,MX1, IFITM1,BST2,OAS1,IFI16,LGALS9,TRIM22,PARP10,EIF2AK2, TRIM5,SHFL,PLSCR1,IFIT5,APOBEC3F,ZC3HAV1,ADAR, TRIM34,TRIM25,TRIM14,TRIM21 |
| norovirus-infected | ileal | GO:0019058 | viral life cycle | 0.27 | 28 | 0.00000 | OAS2,OASL,RSAD2,ISG15,LAMP3,OAS3,IFITM3,IFIT1,MX1, IFITM1,BST2,OAS1,IFI16,LGALS9,TRIM22,PARP10,EIF2AK2, TRIM5,SHFL,PLSCR1,IFIT5,APOBEC3F,ZC3HAV1,ADAR, TRIM34,TRIM25,TRIM14,TRIM21 |
| norovirus-infected | ileal | GO:0034340 | response to type I interferon | 0.26 | 27 | 0.00000 | OAS2,OASL,RSAD2,ISG15,OAS3,USP18,IFIT3,IFITM3,XAF1, IFIT1,MX1,MX2,IFITM1,ZBP1,BST2,IFIT2,OAS1,STAT1,IFI35, SHFL,IRF9,SP100,ADAR,STAT2,SAMHD1,TRIM56,PSMB8 |
| norovirus-infected | ileal | GO:0000209 | protein polyubiquitination | 0.15 | 15 | 0.00000 | HERC5,UBE2L6,HERC6,TRIM22,PARP10,TRIM5,DTX3L,NMI, RNF213,TRIM34,PSMB9,TRIM14,TRIM21,TRIM56,PSMB8 |
| norovirus-infected | ileal | GO:0002683 | negative regulation of immune system process | 0.12 | 12 | 0.00020 | CD274,BST2,GBP1,IFI16,DHX58,LGALS9,SOCS1,PARP14,NMI, ADAR,SAMHD1,TLR3 |
| norovirus-infected | ileal | GO:0051701 | interaction with host | 0.11 | 11 | 0.00002 | IFITM3,IFIT1,IFITM1,LGALS9,TRIM22,EIF2AK2,TRIM5,TRIM34, TRIM25,TRIM14,TRIM21 |
| norovirus-infected | ileal | GO:0043123 | positive regulation of I-kappaB kinase/NF-kappaB signaling | 0.10 | 10 | 0.00006 | BST2,LGALS9,TRIM22,TRIM5,IFIT5,TRIM34,TRIM25,TLR3, TRIM14,TRIM21 |
| norovirus-infected | ileal | GO:0046718 | viral entry into host cell | 0.09 | 9 | 0.00001 | IFITM3,IFITM1,LGALS9,TRIM22,TRIM5,TRIM34,TRIM25, TRIM14,TRIM21 |
| norovirus-infected | ileal | GO:0051092 | positive regulation of NF-kappaB transcription factor activity | 0.09 | 9 | 0.00007 | LGALS9,TRIM22,EIF2AK2,TRIM5,TRIM34,TRIM25,TLR3, TRIM14,TRIM21 |
| norovirus-infected | ileal | GO:0071360 | cellular response to exogenous dsRNA | 0.04 | 4 | 0.00029 | IFIT1,DDX58,IFIH1,TLR3 |
| rotavirus-infected | jejunal | GO:0048525 | negative regulation of viral process | 0.25 | 17 | 0.00000 | IFNL3,IFIT1,OAS2,RSAD2,ISG15,CCL5,MX1,IFITM1,IFI16, OASL,IFITM3,OAS3,STAT1,EIF2AK2,ZC3HAV1,TRIM5,PLSCR1 |
| rotavirus-infected | jejunal | GO:0050778 | positive regulation of immune response | 0.23 | 16 | 0.00000 | IFNL2,IFNL1,IFNL3,RSAD2,CCL5,DDX58,IFI16,CD274,ZBP1, PARP9,NLRC5,IRF7,GBP1,TREX1,TRIM5,PLSCR1 |
| rotavirus-infected | jejunal | GO:0007259 | receptor signaling pathway via JAK-STAT | 0.10 | 7 | 0.00002 | IFNL2,IFNL1,IFNL3,CCL5,PARP9,STAT1,STAT2 |
| rotavirus-infected | jejunal | GO:0002753 | cytoplasmic pattern recognition receptor signaling pathway | 0.07 | 5 | 0.00110 | DDX58,IFIH1,DDX60,DHX58,IRF7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cieza, R.J.; Golob, J.L.; Colacino, J.A.; Wobus, C.E. Comparative Analysis of Public RNA-Sequencing Data from Human Intestinal Enteroid (HIEs) Infected with Enteric RNA Viruses Identifies Universal and Virus-Specific Epithelial Responses. Viruses 2021, 13, 1059. https://0-doi-org.brum.beds.ac.uk/10.3390/v13061059
Cieza RJ, Golob JL, Colacino JA, Wobus CE. Comparative Analysis of Public RNA-Sequencing Data from Human Intestinal Enteroid (HIEs) Infected with Enteric RNA Viruses Identifies Universal and Virus-Specific Epithelial Responses. Viruses. 2021; 13(6):1059. https://0-doi-org.brum.beds.ac.uk/10.3390/v13061059
Chicago/Turabian StyleCieza, Roberto J., Jonathan L. Golob, Justin A. Colacino, and Christiane E. Wobus. 2021. "Comparative Analysis of Public RNA-Sequencing Data from Human Intestinal Enteroid (HIEs) Infected with Enteric RNA Viruses Identifies Universal and Virus-Specific Epithelial Responses" Viruses 13, no. 6: 1059. https://0-doi-org.brum.beds.ac.uk/10.3390/v13061059