
The Pro-Inflammatory Chemokines CXCL9, CXCL10 and CXCL11 Are Upregulated Following SARS-CoV-2 Infection in an AKT-Dependent Manner

 , ,
, ,  ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. SARS-CoV-2 Virus and In Vitro Infections
2.3. RNA Isolation and RT-qPCR
2.4. In Vivo hACE2 Mouse Studies
2.5. Viral Plaque Assay
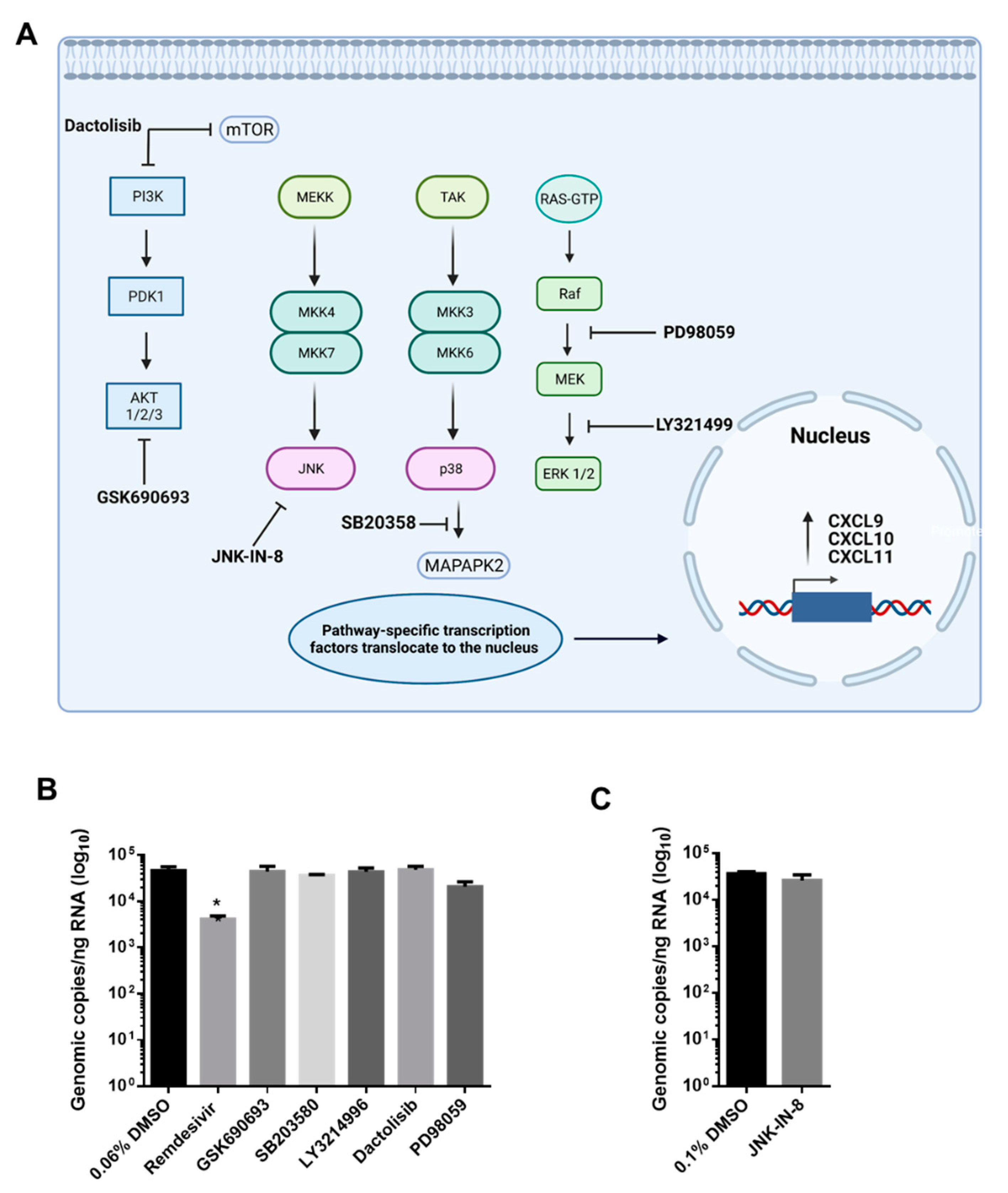
2.6. Kinase Inhibitor Treatment
2.7. Human Specimen Collection and Preparation
2.8. Statistics
3. Results
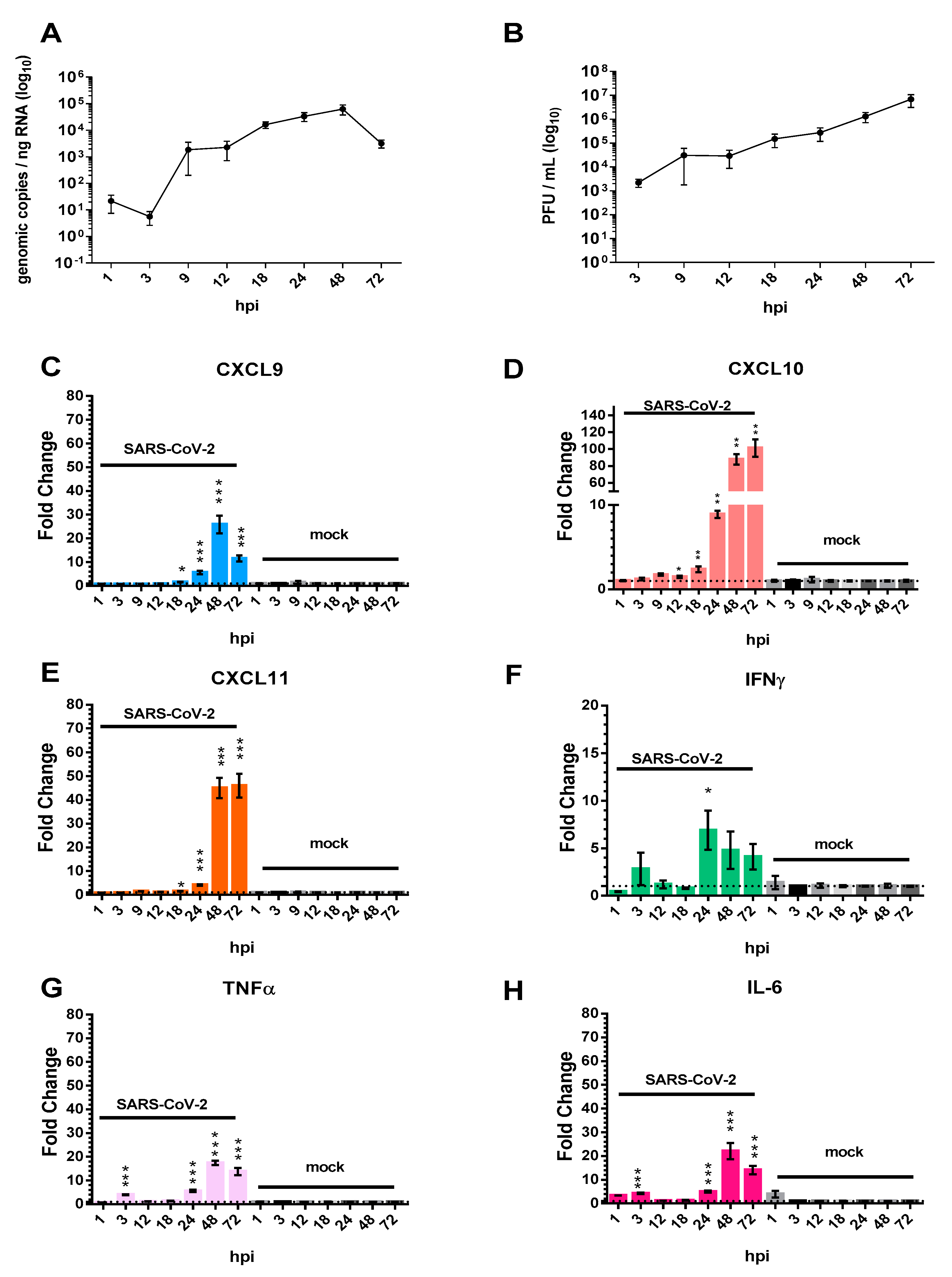
3.1. The Pro-Inflammatory Chemokines CXCL9, CXCL10, and CXCL11 Are Upregulated in Calu-3 Lung Epithelial Cells Following SARS-CoV-2 Infection
3.2. Chemokine/Cytokine Induction in the Lungs of SARS-CoV-2-Infected hACE2 Mice
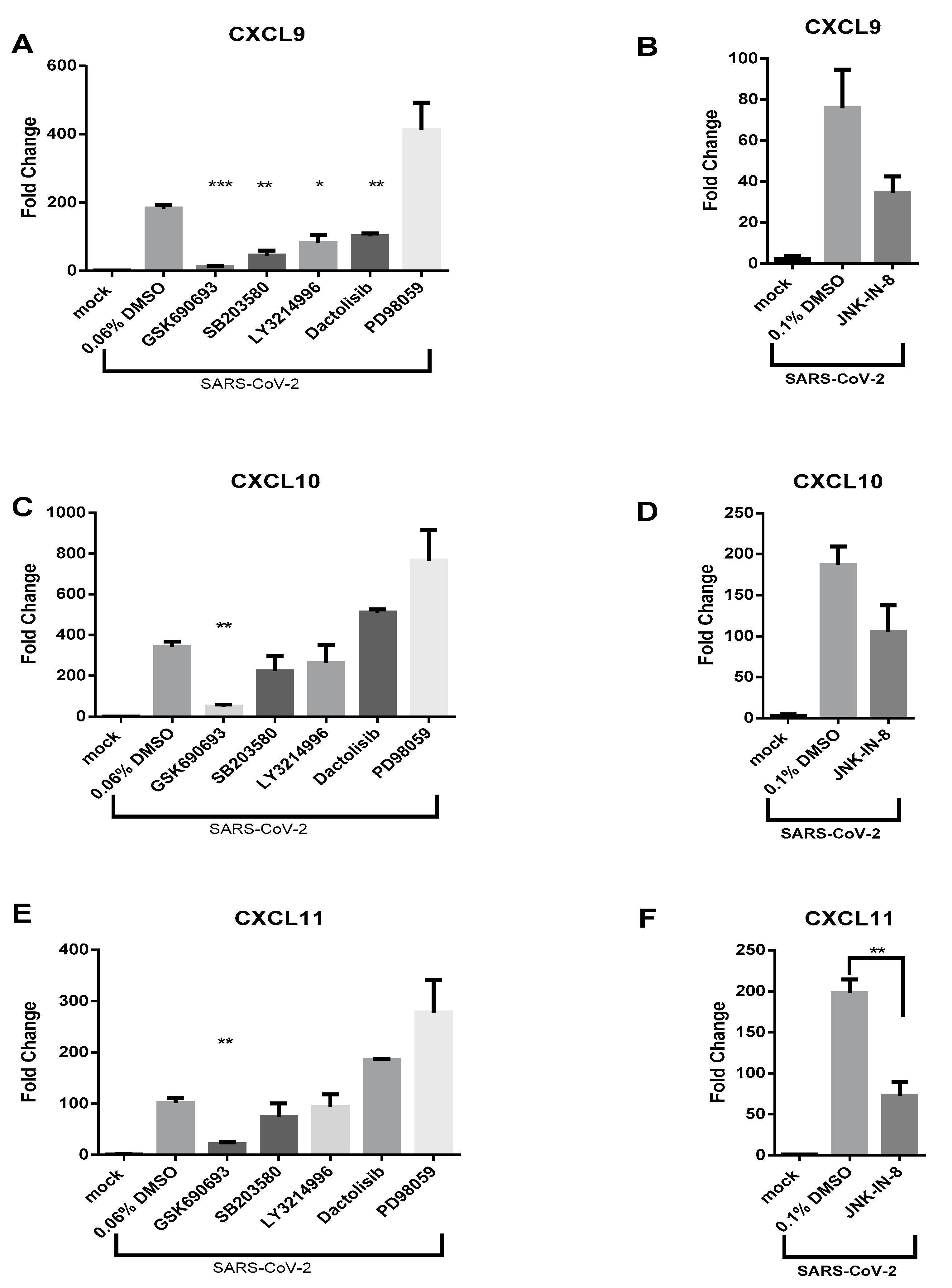
3.3. AKT Inhibitor, GSK690693, Prevents the Induction of CXCL9, CXCL10 and CXCL11 by SARS-CoV-2 in Calu-3 Cells
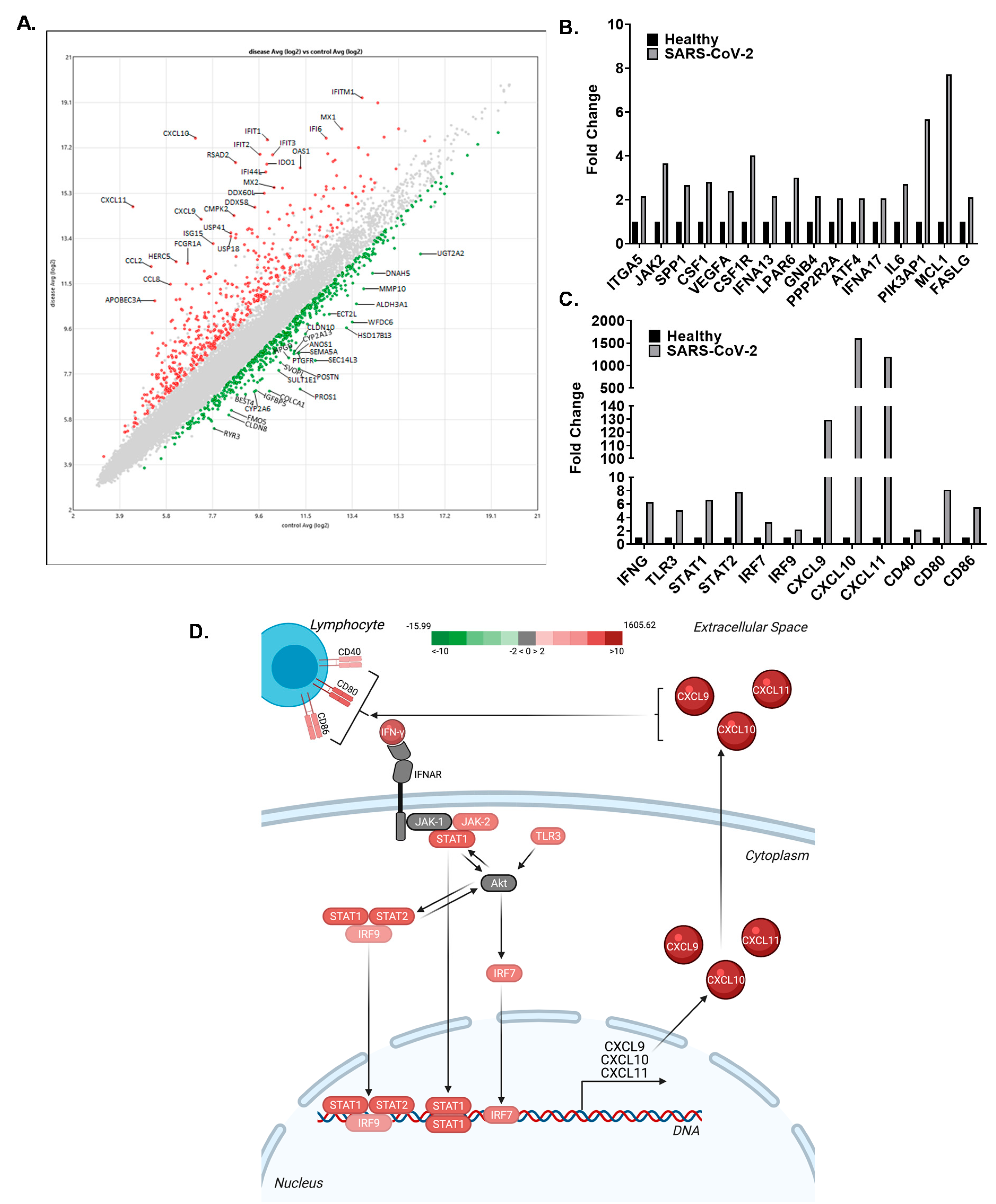
3.4. The CXCL9/10/11 Axis Is Significantly Upregulated in SARS-CoV-2 Positive Human Subjects
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jin, Y.; Yang, H.; Ji, W.; Wu, W.; Chen, S.; Zhang, W.; Duan, G. Virology, Epidemiology, Pathogenesis, and Control of COVID-19. Viruses 2020, 12, 372. [Google Scholar] [CrossRef] [Green Version]
- Zheng, J. SARS-CoV-2: An Emerging Coronavirus that Causes a Global Threat. Int. J. Biol. Sci. 2020, 16, 1678–1685. [Google Scholar] [CrossRef] [Green Version]
- Zhou, P.; Yang, X.-L.; Wang, X.-G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.-R.; Zhu, Y.; Li, B.; Huang, C.-L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrison, A.G.; Lin, T.; Wang, P. Mechanisms of SARS-CoV-2 Transmission and Pathogenesis. Trends Immunol. 2020, 41, 1100–1115. [Google Scholar] [CrossRef]
- Martines, R.B. Pathology and Pathogenesis of SARS-CoV-2 Associated with Fatal Coronavirus Disease, United States. Emerg. Infect. Dis. 2020, 26, 2005–2015. [Google Scholar] [CrossRef]
- Dhama, K. Coronavirus Disease 2019-COVID-19. Clin. Microbiol. Rev. 2020, 33, e00028-20. [Google Scholar] [CrossRef]
- Khalaf, K. SARS-CoV-2: Pathogenesis, and Advancements in Diagnostics and Treatment. Front. Immunol. 2020, 11, 570927. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Wu, T.; Liu, Q.; Yang, Z. The SARS-CoV-2 outbreak: What we know. Int. J. Infect. Dis. 2020, 94, 44–48. [Google Scholar] [CrossRef]
- Felsenstein, S.; Herbert, J.A.; McNamara, P.S.; Hedrich, C.M. COVID-19: Immunology and treatment options. Clin. Immunol. 2020, 215, 108448. [Google Scholar] [CrossRef]
- Dhama, K. An update on SARS-CoV-2/COVID-19 with particular reference to its clinical pathology, pathogenesis, immunopathology and mitigation strategies. Travel Med. Infect. Dis. 2020, 37, 101755. [Google Scholar] [CrossRef] [PubMed]
- Chen, N. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: A descriptive study. Lancet 2020, 395, 507–513. [Google Scholar] [CrossRef] [Green Version]
- Ragab, D.; Eldin, H.S.; Taeimah, M.; Khattab, R.; Salem, R. The COVID-19 Cytokine Storm; What We Know So Far. Front. Immunol. 2020, 11, 1446. [Google Scholar] [CrossRef]
- Wong, C.K.; Lam, C.W.K.; Wu, A.K.L.; Ip, W.K.; Lee, N.L.S.; Chan, I.H.S.; Lit, L.C.W.; Hui, D.S.C.; Chan, M.H.M.; Chung, S.S.C.; et al. Plasma inflammatory cytokines and chemokines in severe acute respiratory syndrome. Clin. Exp. Immunol. 2004, 136, 95–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahallawi, W.H.; Khabour, O.F.; Zhang, Q.; Makhdoum, H.M.; Suliman, B.A. MERS-CoV infection in humans is associated with a pro-inflammatory Th1 and Th17 cytokine profile. Cytokine 2018, 104, 8–13. [Google Scholar] [CrossRef]
- Huang, C. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Liang, Y.; Wang, M.-L.; Chien, C.-S.; Yarmishyn, A.A.; Yang, Y.-P.; Lai, W.-Y.; Luo, Y.-H.; Lin, Y.-T.; Chen, Y.-J.; Chang, P.-C.; et al. Highlight of Immune Pathogenic Response and Hematopathologic Effect in SARS-CoV, MERS-CoV, and SARS-Cov-2 Infection. Front. Immunol. 2020, 11, 1022. [Google Scholar] [CrossRef]
- Xiong, Y.; Liu, Y.; Cao, L.; Wang, D.; Guo, M.; Jiang, A.; Guo, D.; Hu, W.; Yang, J.; Tang, Z.; et al. Transcriptomic characteristics of bronchoalveolar lavage fluid and peripheral blood mononuclear cells in COVID-19 patients. Emerg. Microbes Infect. 2020, 9, 761–770. [Google Scholar] [CrossRef]
- Ichikawa, A.; Kuba, K.; Morita, M.; Chida, S.; Tezuka, H.; Hara, H.; Sasaki, T.; Ohteki, T.; Ranieri, V.M.; Dos Santos, C.C.; et al. CXCL10-CXCR3 Enhances the Development of Neutrophil-mediated Fulminant Lung Injury of Viral and Nonviral Origin. Am. J. Respir. Crit. Care Med. 2013, 187, 65–77. [Google Scholar] [CrossRef] [PubMed]
- Coperchini, F.; Chiovato, L.; Ricci, G.; Croce, L.; Magri, F.; Rotondi, M. The cytokine storm in COVID-19: Further advances in our understanding the role of specific chemokines involved. Cytokine Growth Factor Rev. 2021, 58, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Coperchini, F.; Chiovato, L.; Rotondi, M. Interleukin-6, CXCL10 and Infiltrating Macrophages in COVID-19-Related Cytokine Storm: Not One for All But All for One! Front. Immunol. 2021, 12, 668507. [Google Scholar] [CrossRef]
- Baer, A.; Kehn-Hall, K. Viral Concentration Determination Through Plaque Assays: Using Traditional and Novel Overlay Systems. J. Vis. Exp. 2014, 2014, e52065. [Google Scholar] [CrossRef]
- Chu, H.; Chan, J.; Yuen, T.; Shuai, H.; Yip, C.; Tsang, J.; Huang, X.; Chai, Y.; Yang, D.; Hou, Y.; et al. Comparative tropism, replication kinetics, and cell damage profiling of SARS-CoV-2 and SARS-CoV with implications for clinical manifestations, transmissibility, and laboratory studies of COVID-19: An observational study. Lancet Microbe 2020, 1, e14–e23. [Google Scholar] [CrossRef]
- Bestle, D. TMPRSS2 and furin are both essential for proteolytic activation of SARS-CoV-2 in human airway cells. Life Sci. Alliance 2020, 3, e202000786. [Google Scholar] [CrossRef] [PubMed]
- Tokunaga, R.; Zhang, W.; Naseem, M.; Puccini, A.; Berger, M.D.; Soni, S.; McSkane, M.; Baba, H.; Lenz, H.-J. CXCL9, CXCL10, CXCL11/CXCR3 axis for immune activation—A target for novel cancer therapy. Cancer Treat. Rev. 2018, 63, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Alomar, S.Y. IL-1beta (interleukin-1beta) stimulates the production and release of multiple cytokines and chemokines by human preadipocytes. Arch. Physiol. Biochem. 2016, 122, 117–122. [Google Scholar] [CrossRef] [PubMed]
- McLoughlin, R.; Jenkins, B.J.; Grail, D.; Williams, A.S.; Fielding, C.; Parker, C.R.; Ernst, M.; Topley, N.; Jones, S.A. IL-6 trans-signaling via STAT3 directs T cell infiltration in acute inflammation. Proc. Natl. Acad. Sci. USA 2005, 102, 9589–9594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffmann, M. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280. [Google Scholar] [CrossRef]
- McCray, P.B., Jr.; Pewe, L.; Wohlford-Lenane, C.; Hickey, M.; Manzel, L.; Shi, L.; Netland, J.; Jia, H.P.; Halabi, C.; Sigmund, C.D.; et al. Lethal Infection of K18-hACE2 Mice Infected with Severe Acute Respiratory Syndrome Coronavirus. J. Virol. 2007, 81, 813–821. [Google Scholar] [CrossRef] [Green Version]
- Winkler, E.S. SARS-CoV-2 infection of human ACE2-transgenic mice causes severe lung inflammation and impaired function. Nat. Immunol. 2020, 21, 1327–1335. [Google Scholar] [CrossRef]
- Qi, Z.; Wang, J.; Han, X.; Yang, J.; Zhao, G.; Cao, Y. Listr1 locus regulates innate immunity against Listeria monocytogenes infection in the mouse liver possibly through Cxcl11 polymorphism. Immunogenetics 2014, 66, 231–242. [Google Scholar] [CrossRef]
- Burke, S.J. Synergistic expression of the CXCL10 gene in response to IL-1beta and IFN-gamma involves NF-kappaB, phosphorylation of STAT1 at Tyr701, and acetylation of histones H3 and H4. J. Immunol. 2013, 191, 323–336. [Google Scholar] [CrossRef] [Green Version]
- Vazirinejad, R.; Ahmadi, Z.; Arababadi, M.K.; Hassanshahi, G.; Kennedy, H. The Biological Functions, Structure and Sources of CXCL10 and Its Outstanding Part in the Pathophysiology of Multiple Sclerosis. Neuroimmunomodulation 2014, 21, 322–330. [Google Scholar] [CrossRef]
- Liu, M.; Guo, S.; Hibbert, J.M.; Jain, V.; Singh, N.; Wilson, N.O.; Stiles, J.K. CXCL10/IP-10 in infectious diseases pathogenesis and potential therapeutic implications. Cytokine Growth Factor Rev. 2011, 22, 121–130. [Google Scholar] [CrossRef]
- Fenwick, P.S.; Macedo, P.; Kilty, I.C.; Barnes, P.J.; Donnelly, L.E. Effect of JAK Inhibitors on Release of CXCL9, CXCL10 and CXCL11 from Human Airway Epithelial Cells. PLoS ONE 2015, 10, e0128757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choy, K.T. Remdesivir, lopinavir, emetine, and homoharringtonine inhibit SARS-CoV-2 replication in vitro. Antivir. Res. 2020, 178, 104786. [Google Scholar] [CrossRef] [PubMed]
- Blanco-Melo, D. Imbalanced Host Response to SARS-CoV-2 Drives Development of COVID-19. Cell 2020, 181, 1036–1045.e9. [Google Scholar] [CrossRef] [PubMed]
- Metzemaekers, M.; Vanheule, V.; Janssens, R.; Struyf, S.; Proost, P. Overview of the Mechanisms that May Contribute to the Non-Redundant Activities of Interferon-Inducible CXC Chemokine Receptor 3 Ligands. Front. Immunol. 2018, 8, 1970. [Google Scholar] [CrossRef]
- Woodland, D.L.; Scott, I. T Cell Memory in the Lung Airways. Proc. Am. Thorac. Soc. 2005, 2, 126–131. [Google Scholar] [CrossRef] [Green Version]
- Edward Arthur Steinhaus, 1914–1969. Annu. Rev. Entomol. 1971, 16, 10–11.
- Chu, H. Comparative Replication and Immune Activation Profiles of SARS-CoV-2 and SARS-CoV in Human Lungs: An Ex Vivo Study With Implications for the Pathogenesis of COVID-19. Clin. Infect. Dis. 2020, 71, 1400–1409. [Google Scholar] [CrossRef] [Green Version]
- Yang, D. Attenuated Interferon and Proinflammatory Response in SARS-CoV-2-Infected Human Dendritic Cells Is Associated With Viral Antagonism of STAT1 Phosphorylation. J. Infect. Dis. 2020, 222, 734–745. [Google Scholar] [CrossRef]
- Conti, P. Coronavirus-19 (SARS-CoV-2) induces acute severe lung inflammation via IL-1 causing cytokine storm in COVID-19: A promising inhibitory strategy. J. Biol. Regul. Homeost. Agents 2020, 34, 1971–1975. [Google Scholar]
- Conti, P.; Caraffa, A.; Gallenga, C.; Kritas, S.K.; Frydas, I.; Younes, A.; Di Emidio, P.; Tetè, G.; Pregliasco, F.; Ronconi, G. The British variant of the new coronavirus-19 (Sars-Cov-2) should not create a vaccine problem. J. Biol. Regul. Homeost. Agents 2021, 35, 1–4. [Google Scholar]
- Oladunni, F.S.; Park, J.-G.; Pino, P.A.; Gonzalez, O.; Akhter, A.; Allue-Guardia, A.; Olmo-Fontanez, A.; Gautam, S.; Garcia-Vilanova, A.; Ye, C.; et al. Lethality of SARS-CoV-2 infection in K18 human angiotensin-converting enzyme 2 transgenic mice. Nat. Commun. 2020, 11, 6122. [Google Scholar] [CrossRef]
- Costela-Ruiz, V.J. SARS-CoV-2 infection: The role of cytokines in COVID-19 disease. Cytokine Growth Factor Rev. 2020, 54, 62–75. [Google Scholar] [CrossRef]
- DeDiego, M.L. Inhibition of NF-kappaB-mediated inflammation in severe acute respiratory syndrome coronavirus-infected mice increases survival. J. Virol. 2014, 88, 913–924. [Google Scholar] [CrossRef] [Green Version]
- Nieto-Torres, J.L.; DeDiego, M.L.; Verdiá-Báguena, C.; Guardeno, J.M.J.; Regla-Nava, J.A.; Fernandez-Delgado, R.; Castaño-Rodriguez, C.; Alcaraz, A.; Torres, J.; Aguilella, V.M.; et al. Severe Acute Respiratory Syndrome Coronavirus Envelope Protein Ion Channel Activity Promotes Virus Fitness and Pathogenesis. PLoS Pathog. 2014, 10, e1004077. [Google Scholar] [CrossRef]
- Siu, K.-L.; Yuen, K.-S.; Castano-Rodriguez, C.; Ye, Z.-W.; Yeung, M.-L.; Fung, S.-Y.; Yuan, S.; Chan, C.-P.; Yuen, K.-Y.; Enjuanes, L.; et al. Severe acute respiratory syndrome Coronavirus ORF3a protein activates the NLRP3 inflammasome by promoting TRAF3-dependent ubiquitination of ASC. FASEB J. 2019, 33, 8865–8877. [Google Scholar] [CrossRef] [PubMed]
- Conti, P.; Ronconf, G.; Caraffa, A.; Gallenga, C.E.; Ross, R.; Frydas, I.; Kritas, S.K. Induction of pro-inflammatory cytokines (IL-1 and IL-6) and lung inflammation by Coronavirus-19 (COVI-19 or SARS-CoV-2): Anti-inflammatory strategies. J. Biol. Regul. Homeost. Agents 2020, 34, 327–331. [Google Scholar] [PubMed]
- Chen, J. Cellular immune responses to severe acute respiratory syndrome coronavirus (SARS-CoV) infection in senescent BALB/c mice: CD4+ T cells are important in control of SARS-CoV infection. J. Virol. 2010, 84, 1289–1301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahajan, K.; Mahajan, N.P. PI3K-independent AKT activation in cancers: A treasure trove for novel therapeutics. J. Cell. Physiol. 2012, 227, 3178–3184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahajan, K.; Coppola, D.; Challa, S.; Fang, B.; Chen, Y.A.; Zhu, W.; Lopez, A.S.; Koomen, J.; Engelman, R.W.; Rivera, C.; et al. Ack1 Mediated AKT/PKB Tyrosine 176 Phosphorylation Regulates Its Activation. PLoS ONE 2010, 5, e9646. [Google Scholar] [CrossRef] [PubMed]
- Joung, S.M.; Park, Z.-Y.; Rani, S.; Takeuchi, O.; Akira, S.; Lee, J.Y. Akt Contributes to Activation of the TRIF-Dependent Signaling Pathways of TLRs by Interacting with TANK-Binding Kinase 1. J. Immunol. 2010, 186, 499–507. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Kim, O.; Yang, J.; Sato, K.; Eisenmann, K.M.; McCarthy, J.; Chen, H.; Qiu, Y. Regulation of Akt/PKB Activation by Tyrosine Phosphorylation. J. Biol. Chem. 2001, 276, 31858–31862. [Google Scholar] [CrossRef] [Green Version]
- Jiang, T.; Qiu, Y. Interaction between Src and a C-terminal Proline-rich Motif of Akt Is Required for Akt Activation. J. Biol. Chem. 2003, 278, 15789–15793. [Google Scholar] [CrossRef] [Green Version]
- Reddy, N.M.; Potteti, H.R.; Vegiraju, S.; Chen, H.-J.; Tamatam, C.M.; Reddy, S.P. PI3K-AKT Signaling via Nrf2 Protects against Hyperoxia-Induced Acute Lung Injury, but Promotes Inflammation Post-Injury Independent of Nrf2 in Mice. PLoS ONE 2015, 10, e0129676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.-F.; Liu, Y.-Y.; Yang, C.-T.; Chien, Y.; Twu, N.-F.; Wang, M.-L.; Wang, C.-Y.; Huang, C.-C.; Kao, K.-C.; Hsu, H.-S.; et al. Improvement of ventilator-induced lung injury by IPS cell-derived conditioned medium via inhibition of PI3K/Akt pathway and IP-10-dependent paracrine regulation. Biomaterials 2013, 34, 78–91. [Google Scholar] [CrossRef]
- Kalil, A.C.; Patterson, T.F.; Mehta, A.K.; Tomashek, K.M.; Wolfe, C.R.; Ghazaryan, V.; Marconi, V.C.; Ruiz-Palacios, G.M.; Hsieh, L.; Kline, S.; et al. Baricitinib plus Remdesivir for Hospitalized Adults with Covid-19. N. Engl. J. Med. 2021, 384, 795–807. [Google Scholar] [CrossRef]
- Song, M.; Bode, A.M.; Dong, Z.; Lee, M.-H. AKT as a Therapeutic Target for Cancer. Cancer Res. 2019, 79, 1019–1031. [Google Scholar] [CrossRef] [Green Version]
- Dorlo, T.P.C.; Balasegaram, M.; Beijnen, J.H.; De Vries, P.J. Miltefosine: A review of its pharmacology and therapeutic efficacy in the treatment of leishmaniasis. J. Antimicrob. Chemother. 2012, 67, 2576–2597. [Google Scholar] [CrossRef]
- National Research Council (U.S.); Committee for the Update of the Guide for the Care and Use of Laboratory Animals; Institute for Laboratory Animal Research (U.S.); National Academies Press (U.S.). Guide for the Care and Use of Laboratory Animals; National Academies Press: Washington, DC, USA, 2011; pp. 25–220.






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Callahan, V.; Hawks, S.; Crawford, M.A.; Lehman, C.W.; Morrison, H.A.; Ivester, H.M.; Akhrymuk, I.; Boghdeh, N.; Flor, R.; Finkielstein, C.V.; et al. The Pro-Inflammatory Chemokines CXCL9, CXCL10 and CXCL11 Are Upregulated Following SARS-CoV-2 Infection in an AKT-Dependent Manner. Viruses 2021, 13, 1062. https://0-doi-org.brum.beds.ac.uk/10.3390/v13061062
Callahan V, Hawks S, Crawford MA, Lehman CW, Morrison HA, Ivester HM, Akhrymuk I, Boghdeh N, Flor R, Finkielstein CV, et al. The Pro-Inflammatory Chemokines CXCL9, CXCL10 and CXCL11 Are Upregulated Following SARS-CoV-2 Infection in an AKT-Dependent Manner. Viruses. 2021; 13(6):1062. https://0-doi-org.brum.beds.ac.uk/10.3390/v13061062
Chicago/Turabian StyleCallahan, Victoria, Seth Hawks, Matthew A. Crawford, Caitlin W. Lehman, Holly A. Morrison, Hannah M. Ivester, Ivan Akhrymuk, Niloufar Boghdeh, Rafaela Flor, Carla V. Finkielstein, and et al. 2021. "The Pro-Inflammatory Chemokines CXCL9, CXCL10 and CXCL11 Are Upregulated Following SARS-CoV-2 Infection in an AKT-Dependent Manner" Viruses 13, no. 6: 1062. https://0-doi-org.brum.beds.ac.uk/10.3390/v13061062