
A Comprehensive Molecular Epidemiological Analysis of SARS-CoV-2 Infection in Cyprus from April 2020 to January 2021: Evidence of a Highly Polyphyletic and Evolving Epidemic


 , , , , ,
, , , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Participants
2.2. RNA Extraction and SARS-CoV-2 Real-Time RT-PCR
2.2.1. BMV UCY Cohort
2.2.2. NIPD Cohort
2.2.3. FGH Cohort
2.3. Next Generation Sequencing (NGS)
2.3.1. Library Preparation
2.3.2. Processing of Raw Sequencing Data
2.4. Bioinformatic Analysis
2.4.1. Lineage Classification
2.4.2. Mutation Calling
2.4.3. Phylogenetic, Phylodynamic and Phylogeographic Inferences
3. Results
3.1. The Appearance of SARS-CoV-2 Lineages in Cyprus
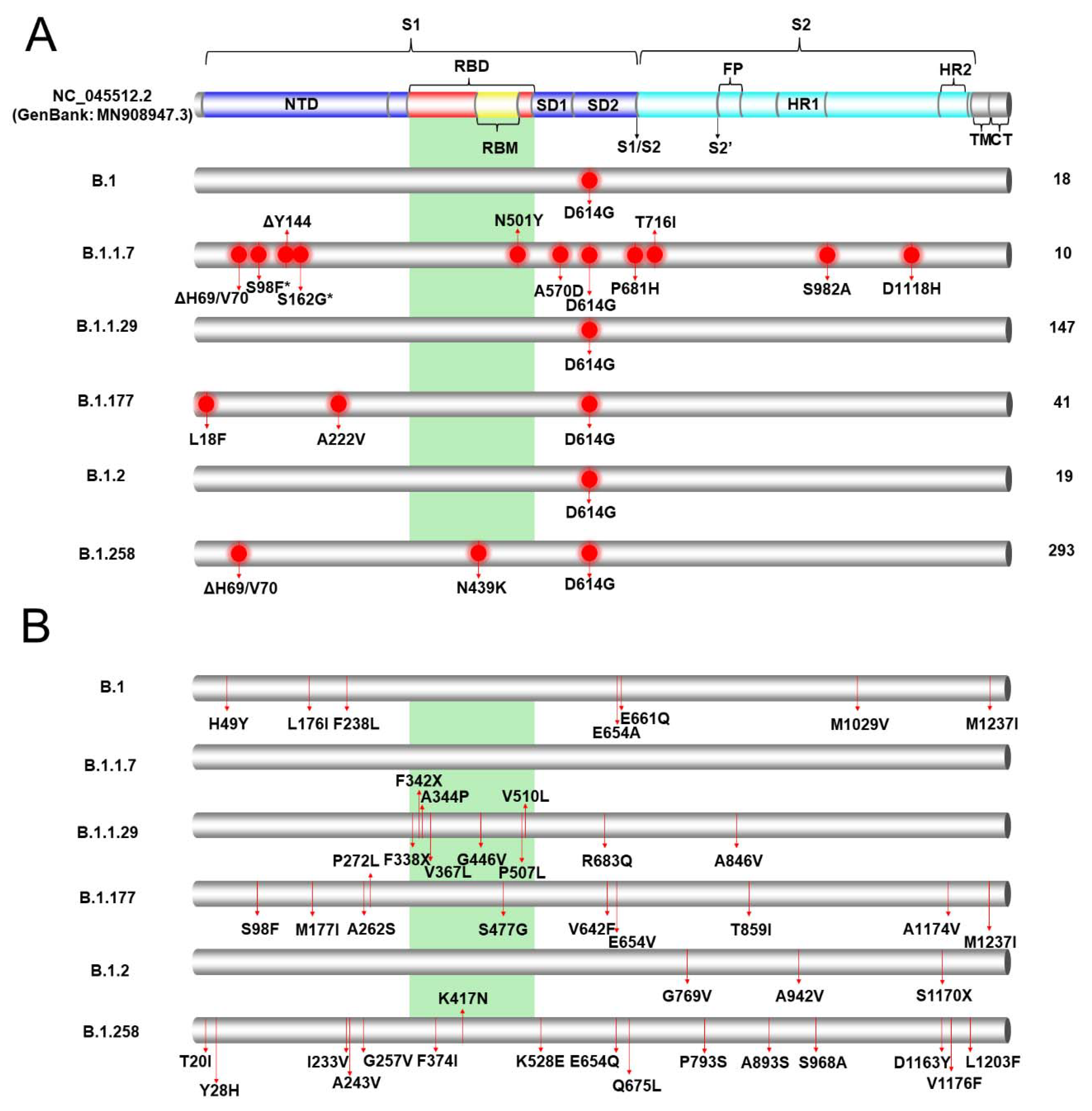
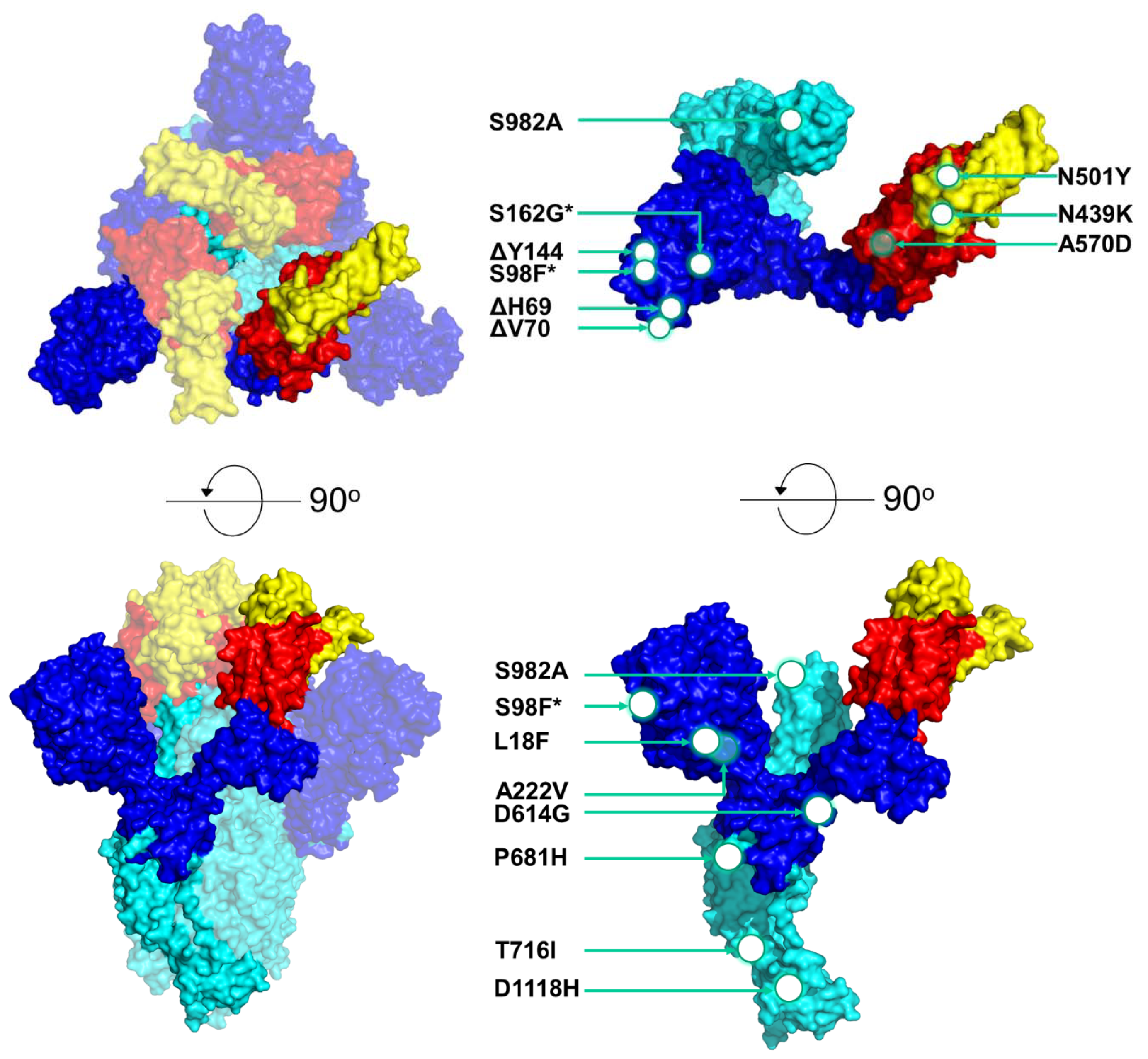
3.2. Mutational Analysis of the S Protein to Identify the Most Prevalent Lineages in Cyprus
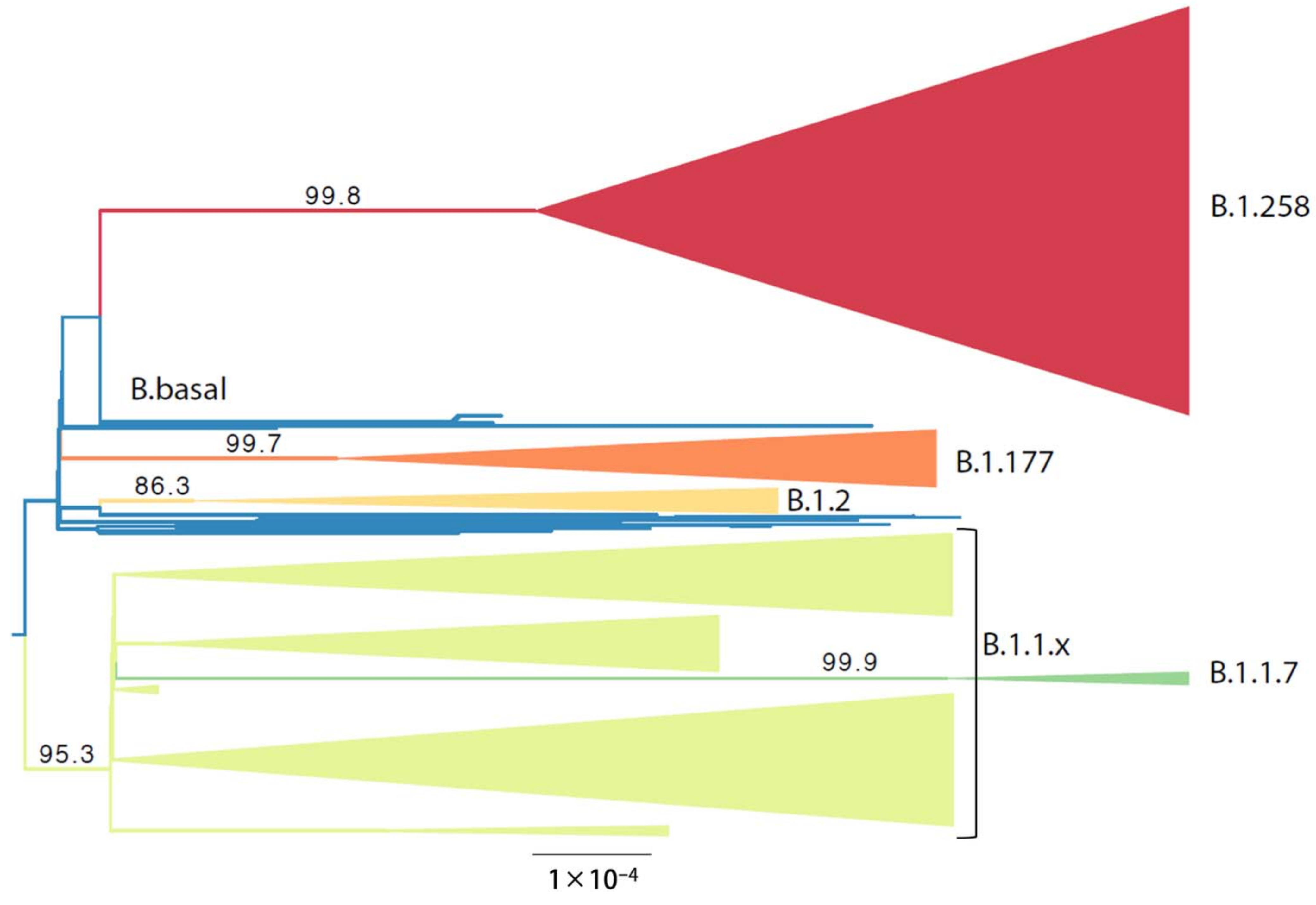
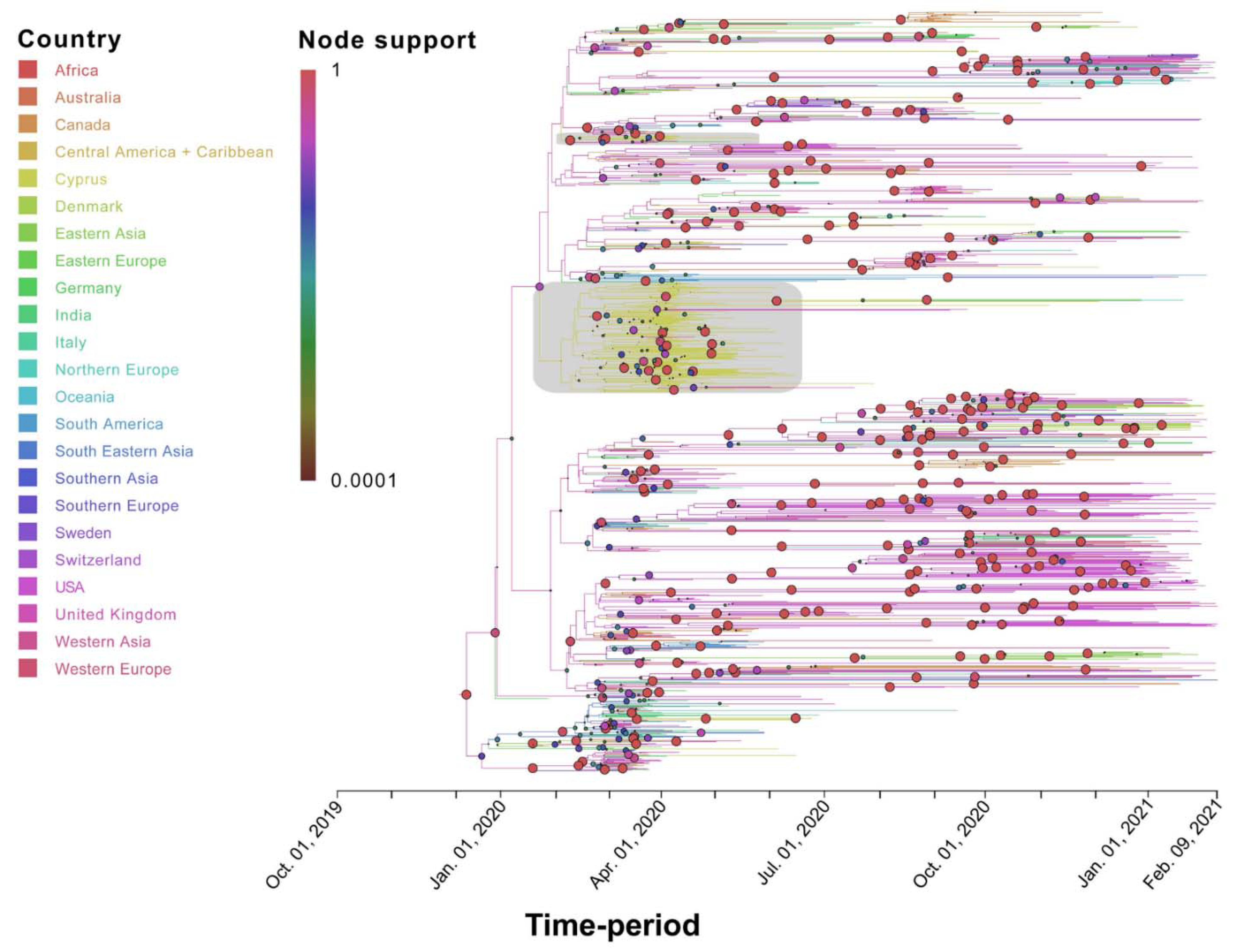
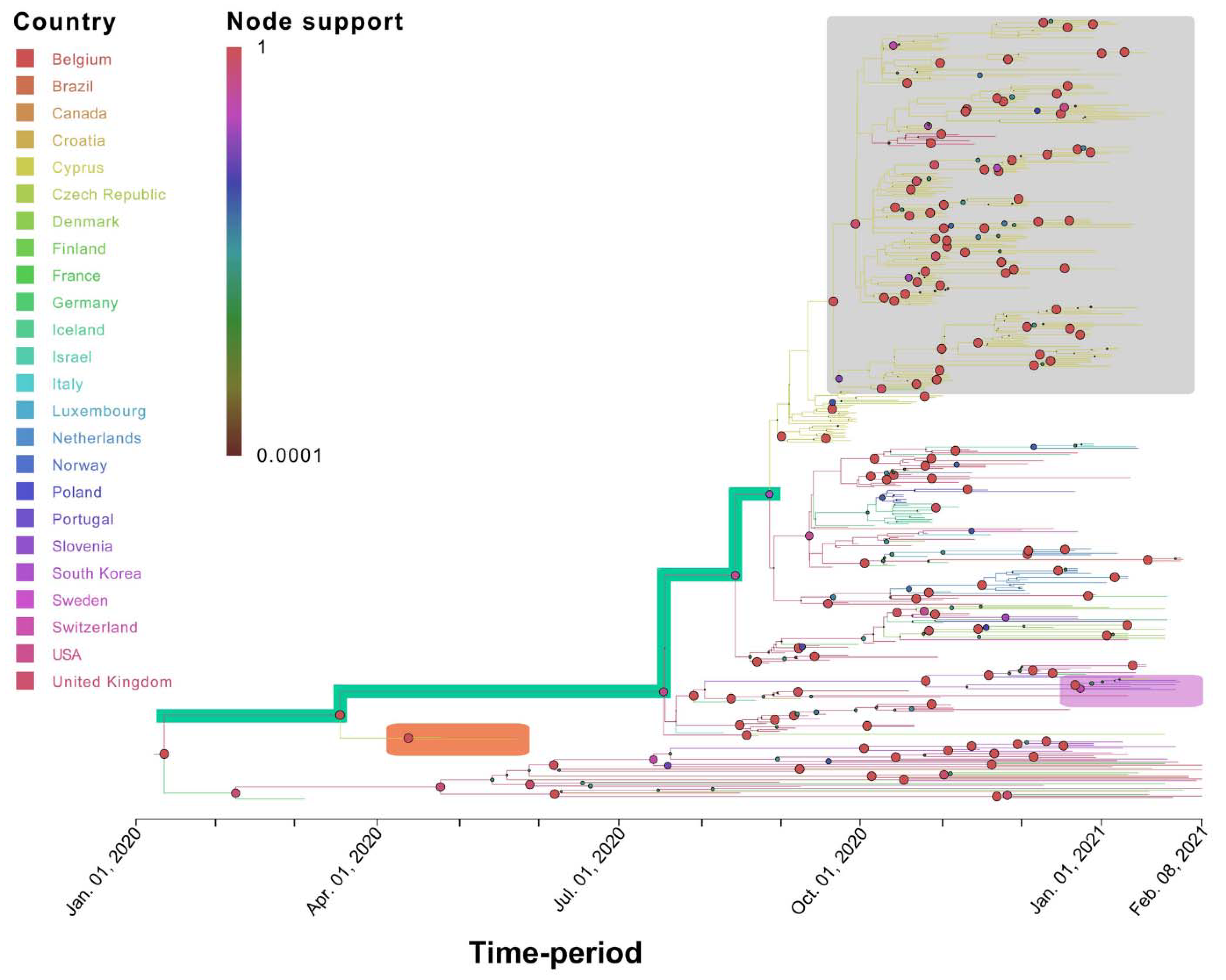
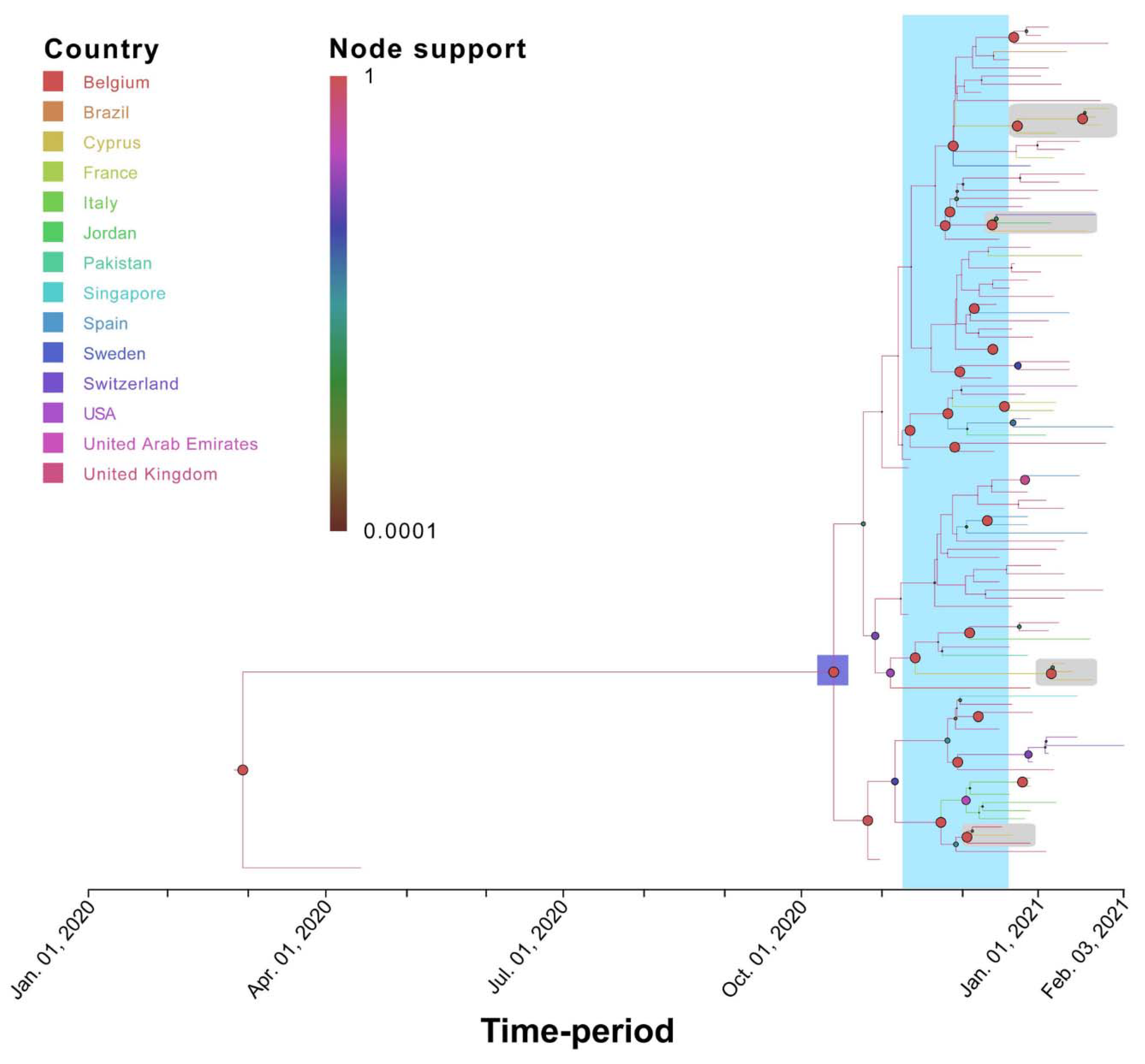
3.3. Phylogenetic Analysis of Cypriot SARS-CoV-2-Infected Individuals
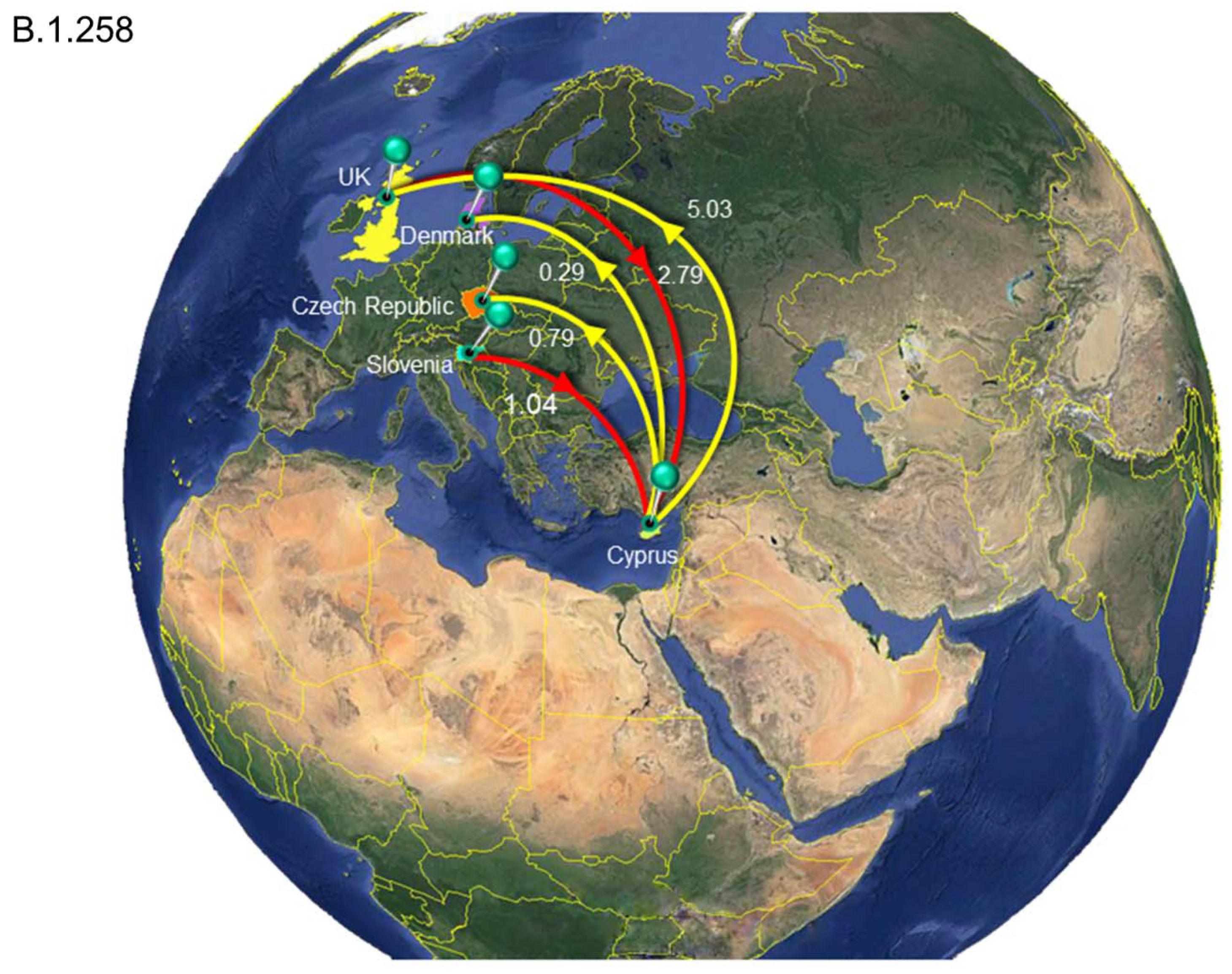
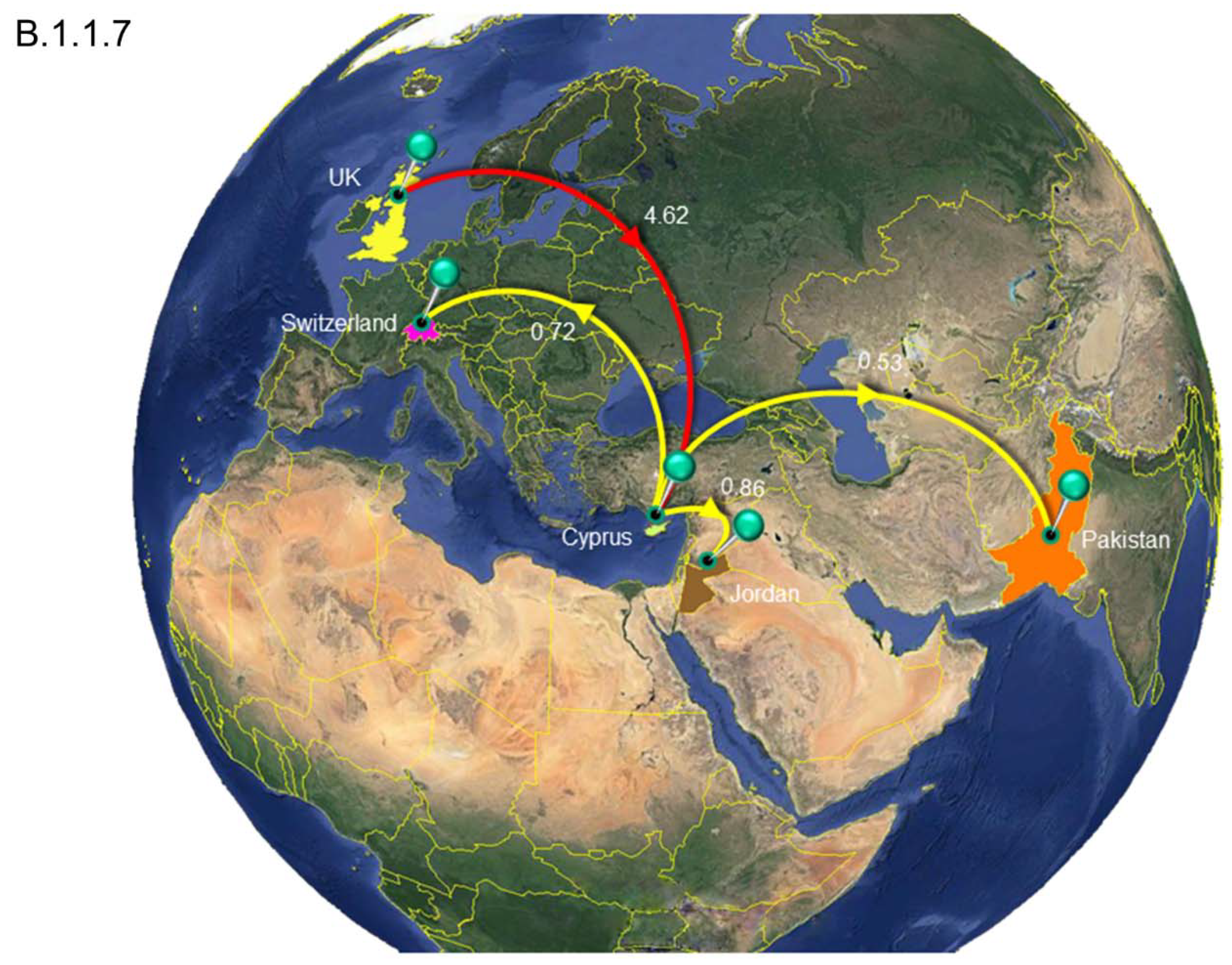
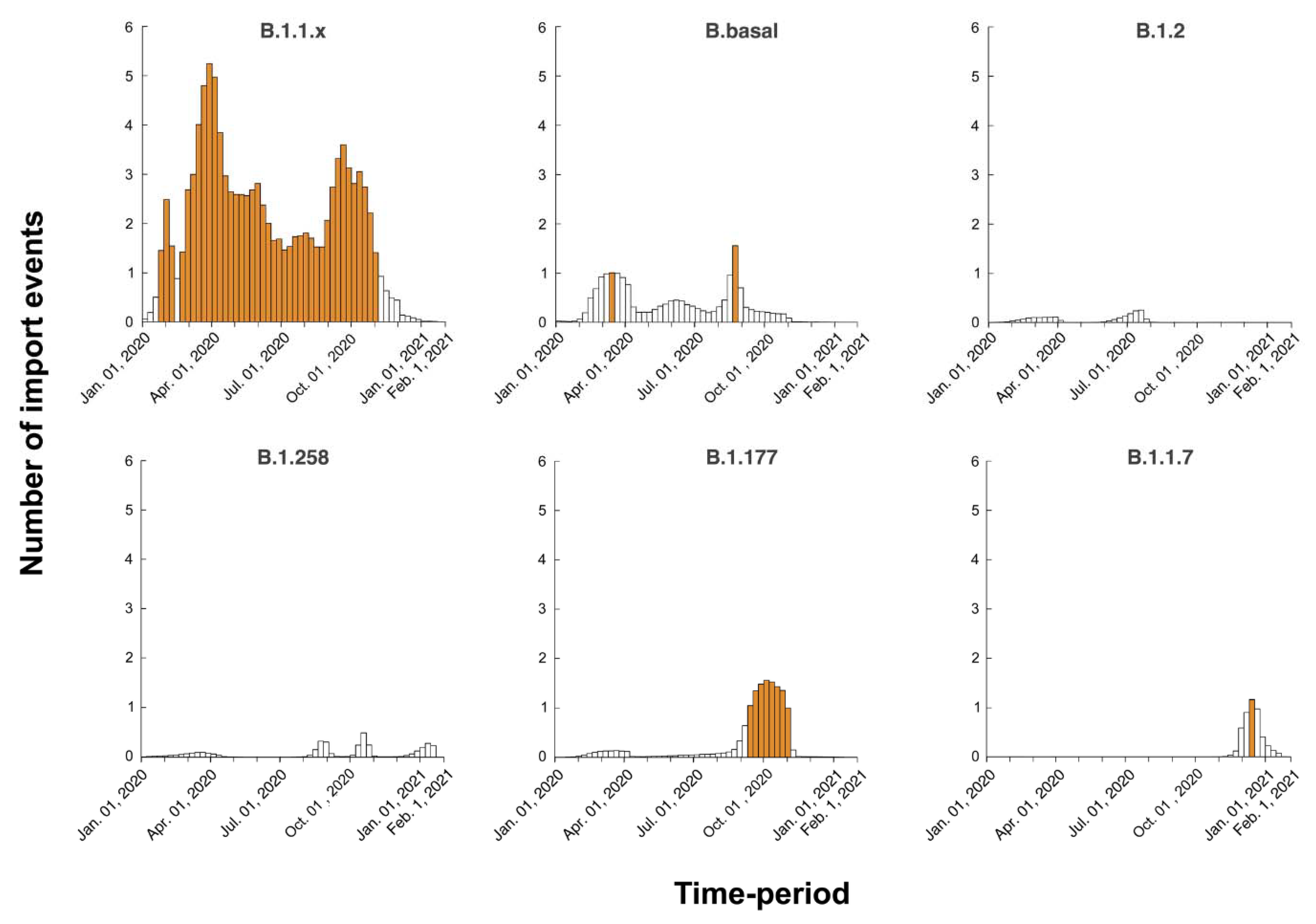
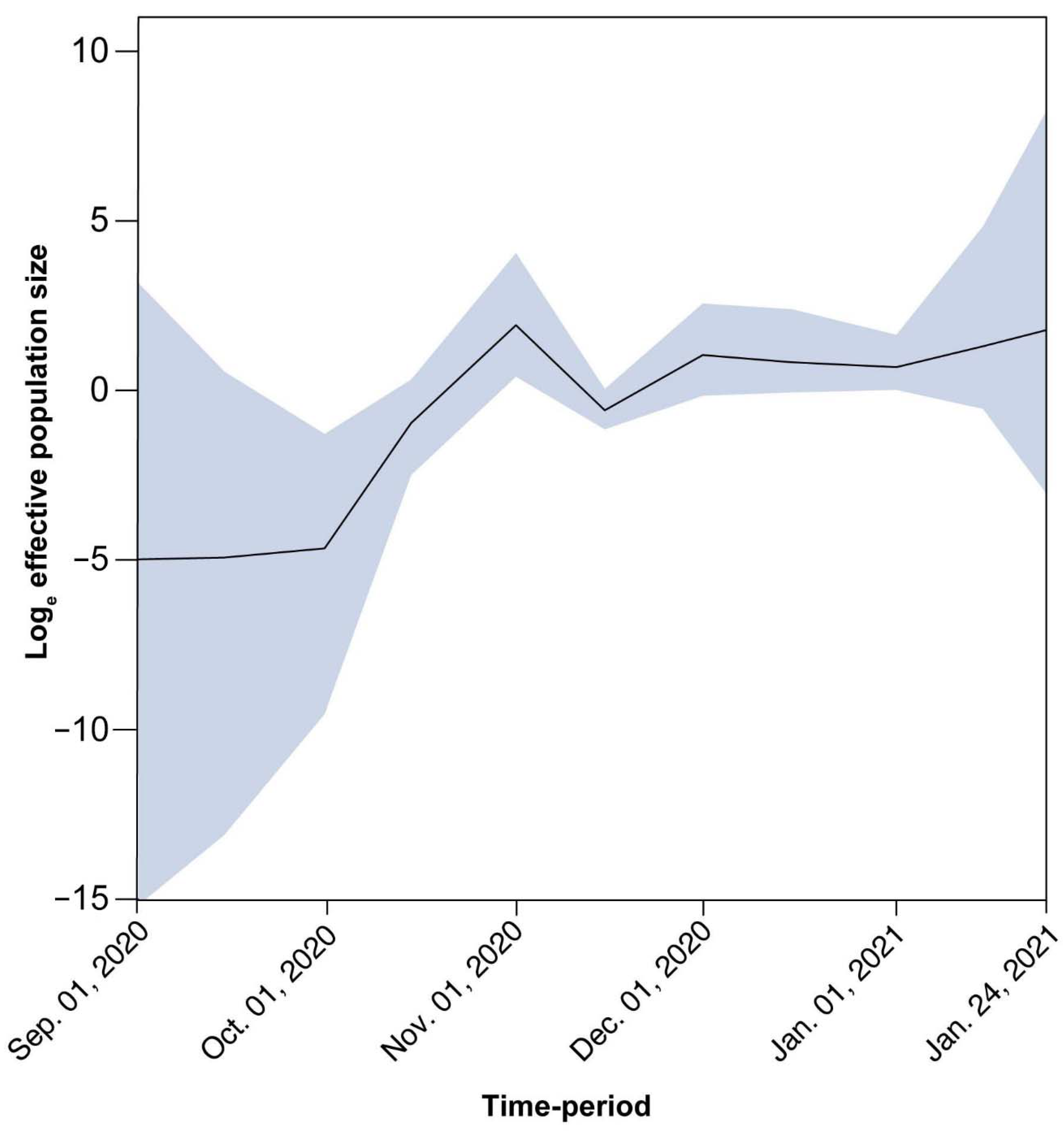
3.4. Timed Migration Histories
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lu, R.; Zhao, X.; Li, J.; Niu, P.; Yang, B.; Wu, H.; Wang, W.; Song, H.; Huang, B.; Zhu, N.; et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: Implications for virus origins and receptor binding. Lancet 2020, 395, 565–574. [Google Scholar] [CrossRef] [Green Version]
- Peiris, J.S.M.; Yuen, K.Y.; Osterhaus, A.D.M.E.; Stöhr, K. The Severe Acute Respiratory Syndrome. N. Engl. J. Med. 2003, 349, 2431–2441. [Google Scholar] [CrossRef] [Green Version]
- Zaki, A.M.; Van Boheemen, S.; Bestebroer, T.M.; Osterhaus, A.D.M.E.; Fouchier, R.A.M. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N. Engl. J. Med. 2012, 367, 1814–1820. [Google Scholar] [CrossRef]
- GISAID. GISAID Initiative. Available online: https://www.gisaid.org/ (accessed on 24 February 2021).
- Hannah, R.; Esteban, O.; Diana, B.; Edouard, M.; Joe, H.; Bobbie, M.; Charlie, G.; Cameron, A. Coronavirus (COVID-19) Cases-Our World in Data. Available online: https://ourworldindata.org/covid-cases?country=IND~USA~GBR~CAN~DEU~FRA#confirmed-cases (accessed on 24 February 2021).
- McGrail, D.J.; Dai, J.; McAndrews, K.M.; Kalluri, R. Enacting national social distancing policies corresponds with dramatic reduction in COVID19 infection rates. PLoS ONE 2020, 15, e0236619. [Google Scholar] [CrossRef]
- Mukherjee, R. Global efforts on vaccines for COVID-19: Since, sooner or later, we all will catch the coronavirus. J. Biosci. 2020, 45, 1–10. [Google Scholar] [CrossRef]
- Liu, Y.; Morgenstern, C.; Kelly, J.; Lowe, R.; Jit, M. The impact of non-pharmaceutical interventions on SARS-CoV-2 transmission across 130 countries and territories. MedRxiv 2020, 19, 1–2. [Google Scholar]
- Leung, K.; Wu, J.T.; Liu, D.; Leung, G.M. First-wave COVID-19 transmissibility and severity in China outside Hubei after control measures, and second-wave scenario planning: A modelling impact assessment. Lancet 2020, 395, 1382–1393. [Google Scholar] [CrossRef]
- Rabaan, A.A.; Al-Ahmed, S.H.; Sah, R.; Al-Tawfiq, J.A.; Haque, S.; Harapan, H.; Arteaga-Livias, K.; Aldana, D.K.B.; Kumar, P.; Dhama, K.; et al. Genomic Epidemiology and Recent Update on Nucleic Acid–Based Diagnostics for COVID-19. Curr. Trop. Med. Rep. 2020, 7, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Cento, V.; Alteri, C.; Merli, M.; Di Ruscio, F.; Tartaglione, L.; Rossotti, R.; Travi, G.; Vecchi, M.; Raimondi, A.; Nava, A.; et al. Effectiveness of infection-containment measures on SARS-CoV-2 seroprevalence and circulation from May to July 2020, in Milan, Italy. PLoS ONE 2020, 15, e0242765. [Google Scholar] [CrossRef]
- Parczewski, M.; Ciechanowicz, A. Molecular epidemiology of SARS-CoV-2: A review of current data on genetic variability of the virus. Pol. Arch. Intern. Med. 2021, 131, 63–69. [Google Scholar] [CrossRef]
- Pachetti, M.; Marini, B.; Benedetti, F.; Giudici, F.; Mauro, E.; Storici, P.; Masciovecchio, C.; Angeletti, S.; Ciccozzi, M.; Gallo, R.C.; et al. Emerging SARS-CoV-2 mutation hot spots include a novel RNA-dependent-RNA polymerase variant. J. Transl. Med. 2020, 18, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, S.; Zhang, Z.; He, F. The phylogenetic relationship within SARS-CoV-2s: An expanding basal clade. Mol. Phylogenet. Evol. 2021, 157, 107017. [Google Scholar] [CrossRef]
- Wang, R.; Hozumi, Y.; Yin, C.; Wei, G.W. Decoding SARS-CoV-2 Transmission and Evolution and Ramifications for COVID-19 Diagnosis, Vaccine, and Medicine. J. Chem. Inf. Model. 2020, 60, 5853–5865. [Google Scholar] [CrossRef] [PubMed]
- Ghadimi-Moghadam, A.; Haghani, M.; Bevelacqua, J.J.; Jafarzadeh, A.; Kaveh-Ahangar, A.; Mortazavi, S.M.J.; Ghadimi-Moghadam, A.; Mortazavi, S.A.R. Covid-19 tragic pandemic: Concerns over unintentional “directed accelerated evolution” of novel coronavirus (sars-cov-2) and introducing a modified treatment method for ards. J. Biomed. Phys. Eng. 2020, 10, 241–246. [Google Scholar] [CrossRef]
- Rambaut, A.; Holmes, E.C.; O’Toole, Á.; Hill, V.; McCrone, J.T.; Ruis, C.; du Plessis, L.; Pybus, O.G. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat. Microbiol. 2020, 5, 1403–1407. [Google Scholar] [CrossRef] [PubMed]
- Nextstrain Nextstrain. Available online: https://nextstrain.org/ (accessed on 24 February 2021).
- Kostaki, E.G.; Pavlopoulos, G.A.; Verrou, K.-M.; Ampatziadis-Michailidis, G.; Harokopos, V.; Hatzis, P.; Moulos, P.; Siafakas, N.; Pournaras, S.; Hadjichristodoulou, C.; et al. Molecular epidemiology of SARS-CoV-2 in Greece reveals low rates of onward virus transmission after lifting of travel restrictions based on risk assessment during summer 2020. MedRxiv 2021. [Google Scholar] [CrossRef]
- Lorenzo-Redondo, R.; Ozer, E.A.; Achenbach, C.J.; D’Aquila, R.T.; Hultquist, J.F. Molecular epidemiology in the HIV and SARS-CoV-2 pandemics. Curr. Opin. HIV AIDS 2021, 16, 11–24. [Google Scholar] [CrossRef]
- Tang, X.; Wu, C.; Li, X.; Song, Y.; Yao, X.; Wu, X.; Duan, Y.; Zhang, H.; Wang, Y.; Qian, Z.; et al. On the origin and continuing evolution of SARS-CoV-2. Natl. Sci. Rev. 2020, 7, 1012–1023. [Google Scholar] [CrossRef] [Green Version]
- Plante, J.A.; Liu, Y.; Liu, J.; Xia, H.; Johnson, B.A.; Lokugamage, K.G.; Zhang, X.; Muruato, A.E.; Zou, J.; Fontes-Garfias, C.R.; et al. Spike mutation D614G alters SARS-CoV-2 fitness. Nature 2020, 592, 116–121. [Google Scholar] [CrossRef] [PubMed]
- Gómez, C.E.; Perdiguero, B.; Esteban, M. Emerging sars-cov-2 variants and impact in global vaccination programs against sars-cov-2/covid-19. Vaccines 2021, 9, 1–13. [Google Scholar] [CrossRef]
- Quattrocchi, A.; Mamais, I.; Tsioutis, C.; Christaki, E.; Constantinou, C.; Koliou, M.; Pana, Z.-D.; Silvestros, V.; Theophanous, F.; Haralambous, C.; et al. Extensive Testing and Public Health Interventions for the Control of COVID-19 in the Republic of Cyprus between March and May 2020. J. Clin. Med. 2020, 9, 3598. [Google Scholar] [CrossRef]
- Illumina Inc. bcl2fastq and bcl2fastq2 Conversion Software. Available online: https://emea.support.illumina.com/sequencing/sequencing_software/bcl2fastq-conversion-software.html (accessed on 3 March 2021).
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Grubaugh, N.D.; Gangavarapu, K.; Quick, J.; Matteson, N.L.; De Jesus, J.G.; Main, B.J.; Tan, A.L.; Paul, L.M.; Brackney, D.E.; Grewal, S.; et al. An amplicon-based sequencing framework for accurately measuring intrahost virus diversity using PrimalSeq and iVar. Genome Biol. 2019, 20, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, K.; Rozewicki, J.; Yamada, K.D. MAFFT online service: Multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform. 2019, 20, 1160–1166. [Google Scholar] [CrossRef] [Green Version]
- Larsson, A. AliView: A fast and lightweight alignment viewer and editor for large datasets. Bioinformatics 2014, 30, 3276–3278. [Google Scholar] [CrossRef]
- De Maio, N.; Walker, C.; Borges, R.; Weilguny, L.; Slodkowicz, G.; Goldman, N. Masking Strategies for SARS-CoV-2 Alignments 2020. Available online: https://virological.org/t/masking-strategies-for-sars-cov-2-alignments/480 (accessed on 15 February 2021).
- Nguyen, L.T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, M.; Zaretskaya, I.; Raytselis, Y.; Merezhuk, Y.; McGinnis, S.; Madden, T.L. NCBI BLAST: A better web interface. Nucleic Acids Res. 2008, 36, W5–W9. [Google Scholar] [CrossRef]
- O’Toole, I.; Scher, E.; Underwood, A.; Jackson, B.; Hill, V.; McCrone, J.T.; Ruis, C.; Abu-Dahab, K.; Taylor, B.; Yeats, C.; et al. PANGO lineages. Available online: https://cov-lineages.org/index.html (accessed on 3 March 2021).
- Rambaut, A.; Lam, T.T.; Carvalho, L.M.; Pybus, O.G. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol. 2016, 2, vew007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suchard, M.A.; Lemey, P.; Baele, G.; Ayres, D.L.; Drummond, A.J.; Rambaut, A. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 2018, 4, vey016. [Google Scholar] [CrossRef] [Green Version]
- Shapiro, B.; Ho, S.Y.W.; Drummond, A.J.; Suchard, M.A.; Pybus, O.G.; Rambaut, A. A bayesian phylogenetic method to estimate unknown sequence ages. Mol. Biol. Evol. 2011, 28, 879–887. [Google Scholar] [CrossRef] [Green Version]
- Gill, M.S.; Lemey, P.; Faria, N.R.; Rambaut, A.; Shapiro, B.; Suchard, M.A. Improving bayesian population dynamics inference: A coalescent-based model for multiple loci. Mol. Biol. Evol. 2013, 30, 713–724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemey, P.; Rambaut, A.; Drummond, A.J.; Suchard, M.A. Bayesian phylogeography finds its roots. PLoS Comput. Biol. 2009, 5, e1000520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edwards, C.J.; Suchard, M.A.; Lemey, P.; Welch, J.J.; Barnes, I.; Fulton, T.L.; Barnett, R.; O’Connell, T.C.; Coxon, P.; Monaghan, N.; et al. Ancient hybridization and an irish origin for the modern polar bear matriline. Curr. Biol. 2011, 21, 1251–1258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minin, V.N.; Suchard, M.A. Fast, accurate and simulation-free stochastic mapping. Philos. Trans. R. Soc. B Biol. Sci. 2008, 363, 3985–3995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemey, P.; Rambaut, A.; Bedford, T.; Faria, N.; Bielejec, F.; Baele, G.; Russell, C.A.; Smith, D.J.; Pybus, O.G.; Brockmann, D.; et al. Unifying Viral Genetics and Human Transportation Data to Predict the Global Transmission Dynamics of Human Influenza H3N2. PLoS Pathog. 2014, 10, e1003932. [Google Scholar] [CrossRef] [Green Version]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior summarization in Bayesian phylogenetics using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef] [Green Version]
- Krammer, F. SARS-CoV-2 vaccines in development. Nature 2020, 586, 516–527. [Google Scholar] [CrossRef] [PubMed]
- Tegally, H.; Wilkinson, E.; Giovanetti, M.; Iranzadeh, A.; Fonseca, V.; Giandhari, J.; Doolabh, D.; Pillay, S.; San, E.J.; Msomi, N.; et al. Emergence and rapid spread of a new severe acute respiratory syndrome-related coronavirus 2 (SARS-CoV-2) lineage with multiple spike mutations in South Africa. MedRxiv 2020. [Google Scholar] [CrossRef]
- Villoutreix, B.; Calvez, V.; Marcelin, A.-G.; Khatib, A.-M. In Silico Investigation of the New UK (B.1.1.7) and South African (501Y.V2) SARS-CoV-2 Variants with a Focus at the ACE2–Spike RBD Interface. Int. J. Mol. Sci. 2021, 22, 1695. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Wang, P.; Nair, M.S.; Yu, J.; Rapp, M.; Wang, Q.; Luo, Y.; Chan, J.F.W.; Sahi, V.; Figueroa, A.; et al. Potent neutralizing antibodies against multiple epitopes on SARS-CoV-2 spike. Nature 2020, 584, 450–456. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, M.; Uemura, K.; Sato, A.; Toba, S.; Sanaki, T.; Maenaka, K.; Hall, W.W.; Orba, Y.; Sawa, H. SARS-CoV-2 variants with mutations at the S1/ S2 cleavage site are generated in vitro during propagation in TMPRSS2-deficient cells. PLoS Pathog. 2021, 17, e1009233. [Google Scholar] [CrossRef] [PubMed]
- Gobeil, S.M.C.; Janowska, K.; McDowell, S.; Mansouri, K.; Parks, R.; Manne, K.; Stalls, V.; Kopp, M.F.; Henderson, R.; Edwards, R.J.; et al. D614G Mutation Alters SARS-CoV-2 Spike Conformation and Enhances Protease Cleavage at the S1/S2 Junction. Cell Rep. 2021, 34, 108630. [Google Scholar] [CrossRef] [PubMed]
- Bateman, A.; Martin, M.J.; O’Donovan, C.; Magrane, M.; Alpi, E.; Antunes, R.; Bely, B.; Bingley, M.; Bonilla, C.; Britto, R.; et al. UniProt: The universal protein knowledgebase. Nucleic Acids Res. 2017, 45, D158–D169. [Google Scholar] [CrossRef] [Green Version]
- Xia, X. Domains and Functions of Spike Protein in Sars-Cov-2 in the Context of Vaccine Design. Viruses 2021, 13, 109. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Yang, C.; Xu, X.F.; Xu, W.; Liu, S. wen Structural and functional properties of SARS-CoV-2 spike protein: Potential antivirus drug development for COVID-19. Acta Pharmacol. Sin. 2020, 41, 1141–1149. [Google Scholar] [CrossRef]
- Mittal, A.; Manjunath, K.; Ranjan, R.K.; Kaushik, S.; Kumar, S.; Verma, V. COVID-19 pandemic: Insights into structure, function, and hACE2 receptor recognition by SARS-CoV-2. PLoS Pathog. 2020, 16, e1008762. [Google Scholar] [CrossRef]
- Kim, S.; Lee, J.H.; Lee, S.; Shim, S.; Nguyen, T.T.; Hwang, J.; Kim, H.; Choi, Y.O.; Hong, J.; Bae, S.; et al. The progression of sars coronavirus 2 (Sars-cov2): Mutation in the receptor binding domain of spike gene. Immune Netw. 2020, 20, 1–11. [Google Scholar] [CrossRef]
- Wang, P.; Nair, M.S.; Liu, L.; Iketani, S.; Luo, Y.; Guo, Y.; Wang, M.; Yu, J.; Zhang, B.; Kwong, P.D.; et al. Antibody Resistance of SARS-CoV-2 Variants B.1.351 and B.1.1.7. Nature 2021, 593, 130–135. [Google Scholar] [CrossRef] [PubMed]
- Khelashvili, G.; Plante, A.; Doktorova, M.; Weinstein, H. Ca(2+)-dependent mechanism of membrane insertion and destabilization by the SARS-CoV-2 fusion peptide. Biophys. J. 2021, 120, 1105–1119. [Google Scholar] [CrossRef]
- Wang, Q.; Ma, J.; Acevedo, A. High-Potency Polypeptide-based Interference for Coronavirus Spike Glycoproteins. BioRxiv 2021. [Google Scholar] [CrossRef]
- Ghafari, M.; du Plessis, L.; Pybus, O.G.; Katzourakis, A.; Time Dependence of SARS-CoV-2 Substitution Rates. Virological 2020. Available online: https://virological.org/t/time-dependence-of-sars-cov-2-substitution-rates/542 (accessed on 15 February 2021).
- Worobey, M.; Pekar, J.; Larsen, B.B.; Nelson, M.I.; Hill, V.; Joy, J.B.; Rambaut, A.; Suchard, M.A.; Wertheim, J.O.; Lemey, P. The emergence of SARS-CoV-2 in Europe and North America. Science 2020, 370, 564–570. [Google Scholar] [CrossRef] [PubMed]
- Davies, N.G.; Abbott, S.; Barnard, R.C.; Jarvis, C.I.; Kucharski, A.J.; Munday, J.D.; Pearson, C.A.B.; Russell, T.W.; Tully, D.C.; Washburne, A.D.; et al. Estimated transmissibility and impact of SARS-CoV-2 lineage B.1.1.7 in England. Science 2021, 372, eabg3055. [Google Scholar] [CrossRef]
- Li, R.; Ma, X.; Deng, J.; Chen, Q.; Liu, W.; Peng, Z.; Qiao, Y.; Lin, Y.; He, X.; Zhang, H. Differential efficiencies to neutralize the novel mutants B.1.1.7 and 501Y.V2 by collected sera from convalescent COVID-19 patients and RBD nanoparticle-vaccinated rhesus macaques. Cell. Mol. Immunol. 2021, 18, 1058–1060. [Google Scholar] [CrossRef]
- Engelbrecht, S.; Delaney, K.; Kleinhans, B.; Wilkinson, E.; Tegally, H.; Stander, T.; van Zyl, G.; Preiser, W.; de Oliveira, T. Multiple Early Introductions of SARS-CoV-2 to Cape Town, South Africa. Viruses 2021, 13, 526. [Google Scholar] [CrossRef] [PubMed]
- Republic of Cyprus Ministry of Health New Coronavirus Disease (COVID-19). Available online: https://www.pio.gov.cy/coronavirus/eng/categories/important-announcements (accessed on 24 March 2021).
- Korber, B.; Fischer, W.M.; Gnanakaran, S.; Yoon, H.; Theiler, J.; Abfalterer, W.; Hengartner, N.; Giorgi, E.E.; Bhattacharya, T.; Foley, B.; et al. Tracking Changes in SARS-CoV-2 Spike: Evidence that D614G Increases Infectivity of the COVID-19 Virus. Cell 2020, 182, 812–827. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Thi Nhu Thao, T.; Hoffmann, D.; Taddeo, A.; Ebert, N.; Labroussaa, F.; Pohlmann, A.; King, J.; Steiner, S.; Kelly, J.N.; et al. SARS-CoV-2 spike D614G change enhances replication and transmission. Nature 2021, 592, 122–127. [Google Scholar] [CrossRef] [PubMed]
- Greaney, A.J.; Loes, A.N.; Crawford, K.H.D.; Starr, T.N.; Malone, K.D.; Chu, H.Y.; Bloom, J.D. Comprehensive mapping of mutations in the SARS-CoV-2 receptor-binding domain that affect recognition by polyclonal human plasma antibodies. Cell Host Microbe 2021, 29, 463–476. [Google Scholar] [CrossRef]
- Baker, R.E.; Yang, W.; Vecchi, G.A.; Metcalf, C.J.E.; Grenfell, B.T. Susceptible supply limits the role of climate in the early SARS-CoV-2 pandemic. Science 2020, 369, 315–319. [Google Scholar] [CrossRef] [PubMed]
- U.S. Embassy in Cyprus Health Alert: Cyprus (June 3) Easing of Airport Restrictions and Available Departing Flight. Available online: https://cy.usembassy.gov/health-alert-cyprus-june-3-easing-of-airport-restrictions-and-available-departing-flight/ (accessed on 28 March 2021).
- Pater, A.A.; Bosmeny, M.S.; Barkau, C.L.; Ovington, K.N.; Chilamkurthy, R.; Parasrampuria, M.; Eddington, S.B.; Yinusa, A.O.; White, A.A.; Metz, P.E.; et al. Emergence and Evolution of a Prevalent New SARS-CoV-2 Variant in the United States. bioRxiv 2021. [Google Scholar] [CrossRef]
- Tegally, H.; Wilkinson, E.; Lessells, R.J.; Giandhari, J.; Pillay, S.; Msomi, N.; Mlisana, K.; Bhiman, J.N.; von Gottberg, A.; Walaza, S.; et al. Sixteen novel lineages of SARS-CoV-2 in South Africa. Nat. Med. 2021, 27, 440–446. [Google Scholar] [CrossRef]
- Brejová, B.; Hodorová, V.; Boršová, K.; Čabanová, V.; Reizigová, L.; Paul, E.D.; Čekan, P.; Klempa, B.; Nosek, J.; Vinař, T.B. 1.258∆, a SARS-CoV-2 Variant with ∆H69/∆V70 in the Spike Protein Circulating in the Czech Republic and Slovakia. arXiv 2021, arXiv:2102.04689. Available online: https://arxiv.org/abs/2102.04689 (accessed on 15 February 2021).
- Gupta, R.; Kemp, S.; Harvey, W.; Carabelli, A.; Robertson, D. Recurrent independent emergence and transmission of SARS-CoV-2 Spike amino acid H69/V70 deletions. Res. Sq. 2021. [Google Scholar] [CrossRef]
- Kemp, S.A.; Datir, R.P.; Collier, D.A.; Ferreira, I.A.T.M.; Carabelli, A.; Harvey, W.; Robertson, D.L.; Gupta, R.K. Recurrent emergence and transmission of a SARS-CoV-2 Spike deletion ∆H69/V70. BioRxiv 2020. [Google Scholar] [CrossRef]
- Thomson, E.C.; Rosen, L.E.; Shepherd, J.G.; Spreafico, R.; da Silva Filipe, A.; Wojcechowskyj, J.A.; Davis, C.; Piccoli, L.; Pascall, D.J.; Dillen, J.; et al. Circulating SARS-CoV-2 spike N439K variants maintain fitness while evading antibody-mediated immunity. Cell 2021, 184, 1171–1187. [Google Scholar] [CrossRef] [PubMed]
- Luan, B.; Huynh, T. Insights on SARS-CoV-2's Mutations for Evading Human Antibodies: Sacrifice and Survival. BioRxiv 2021. [Google Scholar] [CrossRef]
- Guthrie, J.L.; Teatero, S.; Zittermann, S.; Chen, Y.; Sullivan, A.; Rilkoff, H.; Joshi, E.; Sivaraman, K.; de Borja, R.; Sundaravadanam, Y.; et al. Detection of the novel SARS-CoV-2 European lineage B.1.177 in Ontario, Canada. MedRxiv 2020. [Google Scholar] [CrossRef]
- La Rosa, G.; Mancini, P.; Bonanno Ferraro, G.; Veneri, C.; Iaconelli, M.; Lucentini, L.; Bonadonna, L.; Brusaferro, S.; Brandtner, D.; Fasanella, A.; et al. Rapid Screening for SARS-CoV-2 Variants of Concern in Clinical and Environmental Samples Using Nested RT-PCR Assays Targeting Key Mutations of the Spike Protein. MedRxiv 2021. [Google Scholar] [CrossRef]
- Grabowski, F.; Kochanczyk, M.; Lipniacki, T. L18F substrain of SARS-CoV-2 VOC-202012/01 is rapidly spreading in England. MedRxiv 2021. [Google Scholar] [CrossRef]
- Bartolini, B.; Rueca, M.; Gruber, C.E.M.; Messina, F.; Giombini, E.; Ippolito, G.; Capobianchi, M.R.; Di Caro, A. The newly introduced SARS-CoV-2 variant A222V is rapidly spreading in Lazio region, Italy. MedRxiv 2020. [Google Scholar] [CrossRef]
- Singh, A.; Steinkellner, G.; Köchl, K.; Gruber, K.; Gruber, C.C. Serine 477 plays a crucial role in the interaction of the SARS-CoV-2 spike protein with the human receptor ACE2. Sci. Rep. 2021, 11, 4320. [Google Scholar] [CrossRef] [PubMed]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Time-Period | April–June 2020 | July–September 2020 | October–November 2020 | December 2020–January 2021 | Total |
|---|---|---|---|---|---|
| Lineage | Number of Sequences Per Lineage (%) | Number of Sequences Per Lineage (%) | Number of Sequences Per Lineage (%) | Number of Sequences Per Lineage (%) | Number of Sequences Per Lineage (%) |
| A | 1 (0.51) | - | - | - | 1 (0.17) |
| B | 5 (2.56) | - | - | - | 5 (0.84) |
| B.1 | 17 (8.72) | 1 (1.32) | - | - | 18 (3.02) |
| B.1.1.1 | - | 6 (7.89) | 1 (0.61) | - | 7 (1.17) |
| B.1.1.7 | - | - | - | 10 (6.17) | 10 (1.68) |
| B.1.1.29 | 144 (73.85) | - | 1 (0.61) | 2 (1.23) | 147 (24.66) |
| B.1.1.41 | 4 (2.05) | - | - | - | 4 (0.67) |
| B.1.1.67 | - | 1 (1.32) | - | - | 1 (0.17) |
| B.1.1.130 | 1 (0.51) | - | - | - | 1 (0.17) |
| B.1.1.131 | 2 (1.03) | - | - | - | 2 (0.34) |
| B.1.1.141 | - | - | - | 2 (1.23) | 2 (0.34) |
| B.1.1.153 | - | - | 3 (1.84) | - | 3 (0.5) |
| B.1.1.159 | 1 (0.51) | - | 1 (0.61) | - | 2 (0.34) |
| B.1.1.161 | 5 (2.56) | - | - | - | 5 (0.84) |
| B.1.1.192 | - | 2 (2.63) | - | - | 2 (0.34) |
| B.1.1.218 | - | 1 (1.32) | 1 (0.61) | - | 2 (0.34) |
| B.1.1.230 | - | 1 (1.32) | - | - | 1 (0.17) |
| B.1.1.251 | 4 (2.05) | - | - | - | 4 (0.67) |
| B.1.1.277 | 4 (2.05) | - | - | - | 4 (0.67) |
| B.1.1.288 | - | 2 (2.63) | - | - | 2 (0.34) |
| B.1.1.307 | - | 1 (1.32) | - | - | 1 (0.17) |
| B.1.1.315 | - | - | - | 1 (0.62) | 1 (0.17) |
| B.1.1.317 | - | 2 (2.63) | - | - | 2 (0.34) |
| B.1.2 | 1 (0.51) | 18 (23.68) | - | - | 19 (3.19) |
| B.1.36 | - | 2 (2.63) | - | - | 2 (0.34) |
| B.1.160 | - | 1 (1.32) | 1 (0.61) | - | 2 (0.34) |
| B.1.177 | 1 (0.51) | - | 30 (18.4) | 10 (6.17) | 41 (6.88) |
| B.1.177.8 | - | - | 1 (0.61) | - | 1 (0.17) |
| B.1.221.1 | - | - | 1 (0.61) | - | 1 (0.17) |
| B.1.236 | - | 6 (7.89) | - | - | 6 (1.01) |
| B.1.258 | 2 (1.03) | 32 (42.11) | 123 (75.46) | 136 (83.95) | 293 (49.16) |
| B.1.258.17 | - | - | - | 1 (0.62) | 1 (0.17) |
| B.1.313 | 2 (1.03) | - | - | - | 2 (0.34) |
| B.6 | 1 (0.51) | - | - | - | 1 (0.17) |
| Total | 195 | 76 | 163 | 162 | 596 |
| Clade.Lineage 1 | From 2 | To 3 | Average 4 | Lower 5 | Upper 6 |
|---|---|---|---|---|---|
| Clade.B.1.1.x | All | Cyprus | 106.38 | 34 | 161 |
| Eastern Europe | Cyprus | 2.44 | 0 | 8 | |
| Germany | Cyprus | 2.07 | 0 | 7 | |
| Italy | Cyprus | 1.39 | 0 | 5 | |
| South Eastern Asia | Cyprus | 1.36 | 0 | 5 | |
| Southern Europe | Cyprus | 6.48 | 0 | 13 | |
| United Kingdom | Cyprus | 92.64 | 27 | 142 | |
| Cyprus | All | 64.68 | 14 | 104 | |
| Cyprus | United Kingdom | 64.68 | 14 | 104 | |
| Clade.B.basal | All 7 | Cyprus | 17.75 | 8 | 25 |
| Germany | Cyprus | 1.95 | 0 | 6 | |
| United Kingdom | Cyprus | 15.8 | 6 | 24 | |
| Cyprus | All | 1.44 | 1 | 3 | |
| Cyprus | Brazil | 0.33 | 0 | 1 | |
| Cyprus | Chile | 1.1 | 0 | 2 | |
| Clade.B.1.2 | All | Cyprus | 1.94 | 1 | 3 |
| USA | Cyprus | 1.94 | 1 | 3 | |
| Cyprus | All | 1.74 | 1 | 5 | |
| Cyprus | USA | 1.74 | 1 | 5 | |
| Clade.B.1.258 | All | Cyprus | 3.84 | 1 | 5 |
| Slovenia | Cyprus | 1.04 | 0 | 2 | |
| United Kingdom | Cyprus | 2.79 | 0 | 4 | |
| Cyprus | All | 6.11 | 2 | 10 | |
| Cyprus | Czech Republic | 0.79 | 0 | 3 | |
| Cyprus | Denmark | 0.29 | 0 | 2 | |
| Cyprus | United Kingdom | 5.03 | 1 | 9 | |
| Clade.B.1.177 | All | Cyprus | 13.99 | 11 | 17 |
| United Kingdom | Cyprus | 13.99 | 11 | 17 | |
| Cyprus | All | 1.01 | 1 | 1 | |
| Cyprus | Finland | 1.01 | 1 | 1 | |
| Clade.B.1.1.7 | All | Cyprus | 4.62 | 3 | 7 |
| United Kingdom | Cyprus | 4.62 | 3 | 7 | |
| Cyprus | All | 2.11 | 1 | 4 | |
| Cyprus | Jordan | 0.86 | 0 | 2 | |
| Cyprus | Pakistan | 0.53 | 0 | 1 | |
| Cyprus | Switzerland | 0.72 | 0 | 2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chrysostomou, A.C.; Vrancken, B.; Koumbaris, G.; Themistokleous, G.; Aristokleous, A.; Masia, C.; Eleftheriou, C.; Iοannou, C.; Stylianou, D.C.; Ioannides, M.; et al. A Comprehensive Molecular Epidemiological Analysis of SARS-CoV-2 Infection in Cyprus from April 2020 to January 2021: Evidence of a Highly Polyphyletic and Evolving Epidemic. Viruses 2021, 13, 1098. https://0-doi-org.brum.beds.ac.uk/10.3390/v13061098
Chrysostomou AC, Vrancken B, Koumbaris G, Themistokleous G, Aristokleous A, Masia C, Eleftheriou C, Iοannou C, Stylianou DC, Ioannides M, et al. A Comprehensive Molecular Epidemiological Analysis of SARS-CoV-2 Infection in Cyprus from April 2020 to January 2021: Evidence of a Highly Polyphyletic and Evolving Epidemic. Viruses. 2021; 13(6):1098. https://0-doi-org.brum.beds.ac.uk/10.3390/v13061098
Chicago/Turabian StyleChrysostomou, Andreas C., Bram Vrancken, George Koumbaris, George Themistokleous, Antonia Aristokleous, Christina Masia, Christina Eleftheriou, Costakis Iοannou, Dora C. Stylianou, Marios Ioannides, and et al. 2021. "A Comprehensive Molecular Epidemiological Analysis of SARS-CoV-2 Infection in Cyprus from April 2020 to January 2021: Evidence of a Highly Polyphyletic and Evolving Epidemic" Viruses 13, no. 6: 1098. https://0-doi-org.brum.beds.ac.uk/10.3390/v13061098