
Broad Impact of Exchange Protein Directly Activated by cAMP 2 (EPAC2) on Respiratory Viral Infections

 and
and 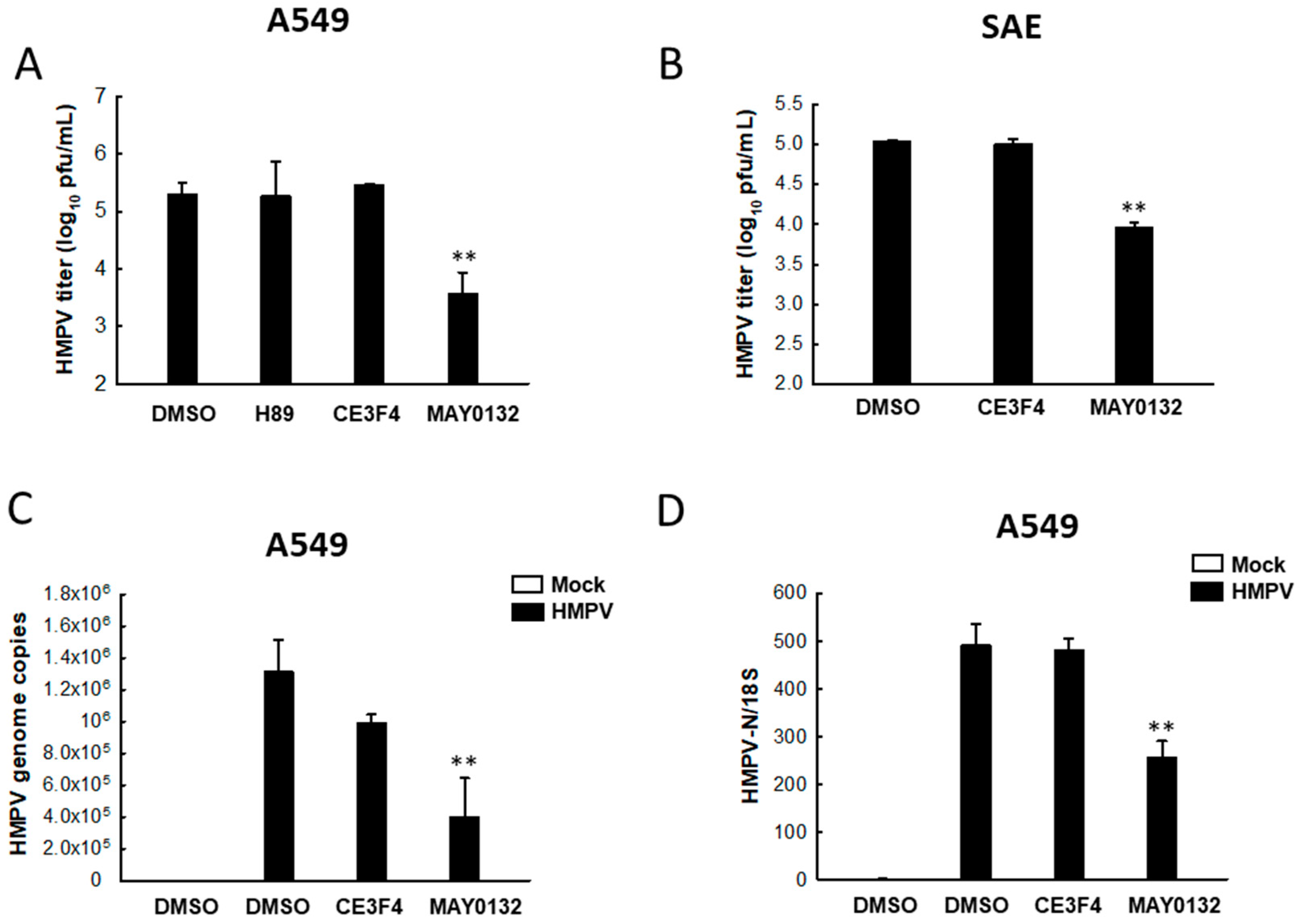{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells, Virus, and Reagents
2.2. Cytokine and Chemokine Quantification
2.3. Quantitative Real-Time PCR (qRT-PCR)
2.4. Reporter Gene Assay
2.5. Statistical Analysis
3. Results
3.1. EPAC2 Promotes HMPV Replication
3.2. EPAC2 Regulates HMPV-Induced Cytokines/Chemokines
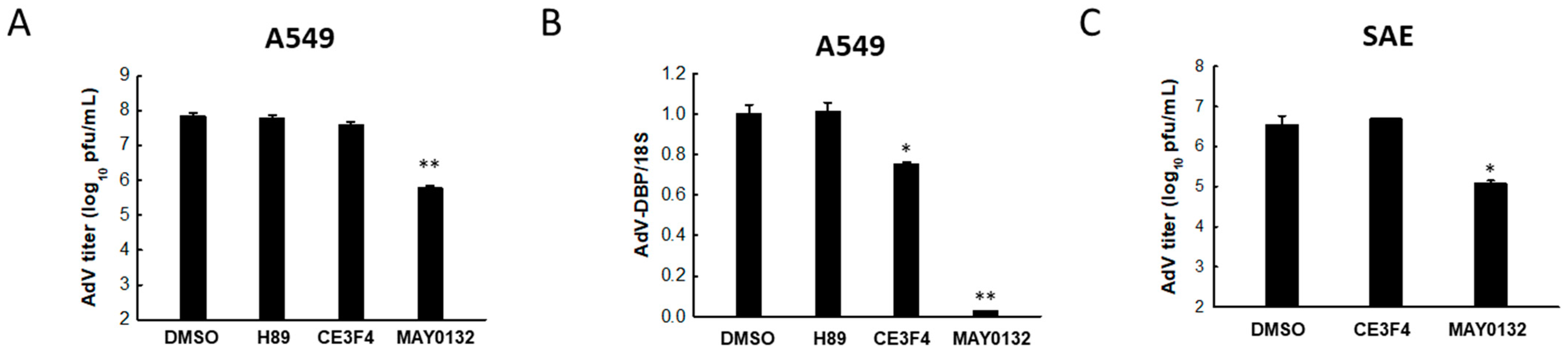
3.3. EPAC2 Is Responsible for AdV Replication
3.4. EPAC2 Regulates AdV-Induced Cytokines/Chemokines
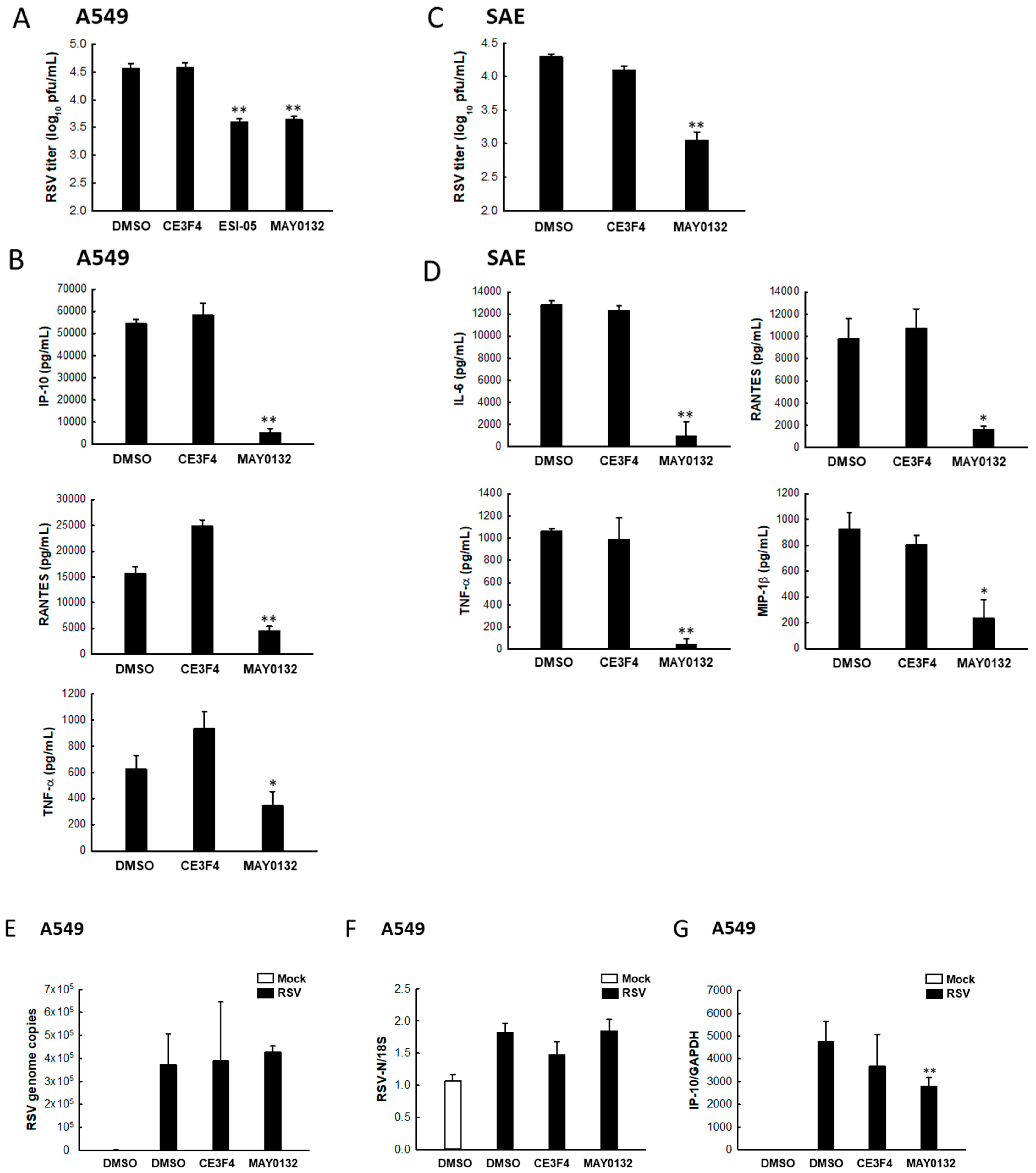
3.5. EPAC2 Regulates RSV Replication and Virus-Induced Cytokines/Chemokines
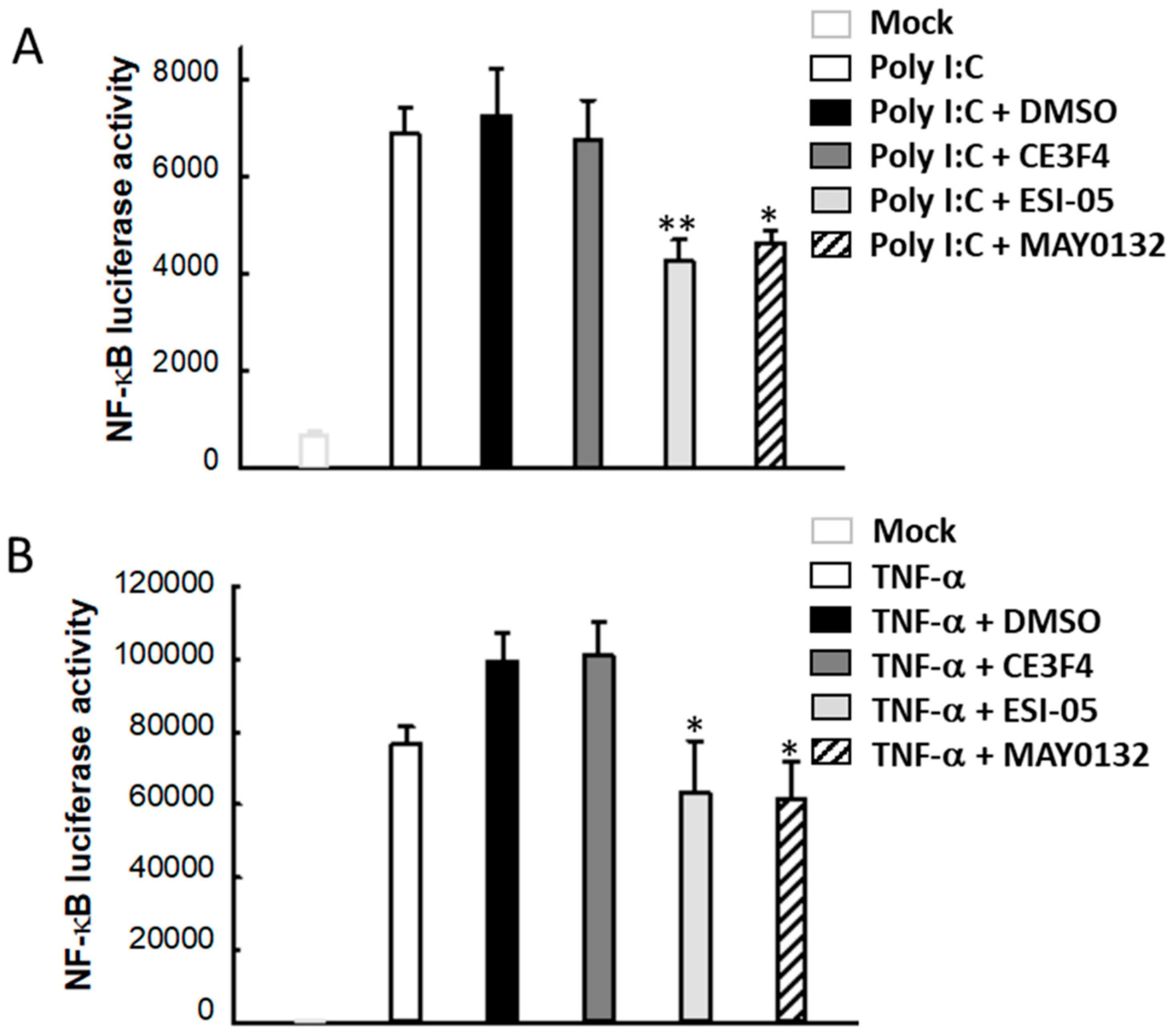
3.6. EPAC2 Regulates Poly I:C- and TNF-α-Induced Inflammatory Signaling Pathways
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Forum of International Respiratory Societies. The Global Impact of Respiratory Disease; European Respiratory Society: Lausanne, Switzerland, 2017. [Google Scholar]
- Wardlaw, T.M.; Johansson, E.W.; Hodge, M.; World Health Organization; UNICEF. Pneumonia: The Forgotten Killer of Children; UNICEF: New York, NY, USA; WHO: Geneva, Switzerland, 2006; p. 42.
- Kasper, D.; Fauci, A.; Hauser, S.; Longo, D.; Jameson, J.L.; Loscalzo, J. Infections Due to DNA and RNA Respiratory Viruses. In Harrison’s Principles of Internal Medicine, 19th ed.; McGraw-Hill Education: New York, NY, USA, 2014. [Google Scholar]
- Lu, R.; Zhao, X.; Li, J.; Niu, P.; Yang, B.; Wu, H.; Wang, W.; Song, H.; Huang, B.; Zhu, N.; et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: Implications for virus origins and receptor binding. Lancet 2020, 395, 565–574. [Google Scholar] [CrossRef] [Green Version]
- World Health Organization. Disease Outbreak News. Novel Coronavirus—China; WHO: Geneva, Switzerland, 2020. [Google Scholar]
- World Health Organization. WHO Coronavirus (COVID-19) Dashboard; WHO: Geneva, Switzerland, 2020. [Google Scholar]
- Singh, K.; Kondal, D.; Mohan, S.; Jaganathan, S.; Deepa, M.; Venkateshmurthy, N.S.; Jarhyan, P.; Anjana, R.M.; Narayan, K.M.V.; Mohan, V.; et al. Health, psychosocial, and economic impacts of the COVID-19 pandemic on people with chronic conditions in India: A mixed methods study. BMC Public Health 2021, 21, 1–15. [Google Scholar] [CrossRef]
- Falsey, A.R.; Erdman, D.; Anderson, L.J.; Walsh, E.E. Human Metapneumovirus Infections in Young and Elderly Adults. J. Infect. Dis. 2003, 187, 785–790. [Google Scholar] [CrossRef] [PubMed]
- Madhi, S.A.; Ludewick, H.; Abed, Y.; Klugman, K.P.; Boivin, G. Human Metapneumovirus-Associated Lower Respiratory Tract Infections among Hospitalized Human Immunodeficiency Virus Type 1 (HIV-1)-Infected and HIV-1-Uninfected African Infants. Clin. Infect. Dis. 2003, 37, 1705–1710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viazov, S.; Ratjen, F.; Scheidhauer, R.; Fiedler, M.; Roggendorf, M. High Prevalence of Human Metapneumovirus Infection in Young Children and Genetic Heterogeneity of the Viral Isolates. J. Clin. Microbiol. 2003, 41, 3043–3045. [Google Scholar] [CrossRef] [Green Version]
- Crowe, J.E. Human Metapneumovirus as a Major Cause of Human Respiratory Tract Disease. Pediatr. Infect. Dis. J. 2004, 23, S215–S221. [Google Scholar] [CrossRef] [PubMed]
- Edwards, K.M.; Zhu, Y.; Griffin, M.R.; Weinberg, G.A.; Hall, C.B.; Szilagyi, P.G.; Staat, M.A.; Iwane, M.; Prill, M.M.; Williams, J.V. Burden of Human Metapneumovirus Infection in Young Children. N. Engl. J. Med. 2013, 368, 633–643. [Google Scholar] [CrossRef] [Green Version]
- Hall, C.B.; Weinberg, G.A.; Iwane, M.K.; Blumkin, A.K.; Edwards, K.M.; Staat, M.A.; Auinger, P.; Griffin, M.R.; Poehling, K.A.; Erdman, D.; et al. The Burden of Respiratory Syncytial Virus Infection in Young Children. N. Engl. J. Med. 2009, 360, 588–598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Pillai, P.; Miyake, F.; Nair, H. The role of viral co-infections in the severity of acute respiratory infections among children infected with respiratory syncytial virus (RSV): A systematic review and meta-analysis. J. Glob. Health 2020, 10, 010426. [Google Scholar] [CrossRef]
- Semple, M.G.; Cowell, A.; Dove, W.F.; Greensill, J.; McNamara, P.; Halfhide, C.P.; Shears, P.; Smyth, R.L.; Hart, C.A. Dual Infection of Infants by Human Metapneumovirus and Human Respiratory Syncytial Virus Is Strongly Associated with Severe Bronchiolitis. J. Infect. Dis. 2005, 191, 382–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, S.; Lin, Z.; Zhang, Y.; Lv, F.; Li, H.; Zhang, X.; Lin, L.; Zhu, H.-H.; Xu, Z.; Li, C.; et al. The Epidemiology, Molecular, and Clinical of Human Adenoviruses in Children Hospitalized with Acute Respiratory Infections. Front. Microbiol. 2021, 12, 629971. [Google Scholar] [CrossRef]
- Scott, M.K.; Chommanard, C.; Lu, X.; Appelgate, D.; Grenz, L.; Schneider, E.; Gerber, S.I.; Erdman, D.D.; Thomas, A. Human Adenovirus Associated with Severe Respiratory Infection, Oregon, USA, 2013–2014. Emerg. Infect. Dis. 2016, 22, 1044–1051. [Google Scholar] [CrossRef] [Green Version]
- Alharbi, S.; Van Caeseele, P.; Consunji-Araneta, R.; Zoubeidi, T.; Fanella, S.; Souid, A.-K.; Alsuwaidi, A.R. Epidemiology of severe pediatric adenovirus lower respiratory tract infections in Manitoba, Canada, 1991–2005. BMC Infect. Dis. 2012, 12, 55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gray, G.C.; McCarthy, T.; Lebeck, M.G.; Schnurr, D.P.; Russell, K.L.; Kajon, A.E.; Landry, M.L.; Leland, D.S.; Storch, G.A.; Ginocchio, C.C.; et al. Genotype prevalence and risk factors for severe clinical adenovirus infection, United States 2004–2006. Clin. Infect. Dis. 2007, 45, 1120–1131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Echavarría, M. Adenoviruses in Immunocompromised Hosts. Clin. Microbiol. Rev. 2008, 21, 704–715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Resch, B. Product review on the monoclonal antibody palivizumab for prevention of respiratory syncytial virus infection. Hum. Vaccines Immunother. 2017, 13, 2138–2149. [Google Scholar] [CrossRef] [Green Version]
- De Rooij, J.; Zwartkruis, F.J.; Verheijen, M.H.; Cool, R.H.; Nijman, S.M.; Wittinghofer, A.; Bos, J.L. Epac is a Rap1 gua-nine-nucleotide-exchange factor directly activated by cyclic AMP. Nature 1998, 396, 474–477. [Google Scholar] [CrossRef]
- Kawasaki, H.; Springett, G.M.; Mochizuki, N.; Toki, S.; Nakaya, M.; Matsuda, M.; Housman, D.E.; Graybiel, A.M. A family of cAMP-binding proteins that directly activate Rap1. Science 1998, 282, 2275–2279. [Google Scholar] [CrossRef] [Green Version]
- Metrich, M.; Lucas, A.; Gastineau, M.; Samuel, J.L.; Heymes, C.; Morel, E.; Lezoualc’’h, F. Epac mediates beta-adrenergic receptor-induced cardiomyocyte hypertrophy. Circ. Res. 2008, 102, 959–965. [Google Scholar] [CrossRef] [Green Version]
- Kumar, N.; Gupta, S.; Dabral, S.; Singh, S.; Sehrawat, S. Role of exchange protein directly activated by cAMP (EPAC1) in breast cancer cell migration and apoptosis. Mol. Cell. Biochem. 2017, 430, 115–125. [Google Scholar] [CrossRef]
- Suzuki, S.; Yokoyama, U.; Abe, T.; Kiyonari, H.; Yamashita, N.; Kato, Y.; Kurotani, R.; Sato, M.; Okumura, S.; Ishikawa, Y. Differential Roles of Epac in Regulating Cell Death in Neuronal and Myocardial Cells. J. Biol. Chem. 2010, 285, 24248–24259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griggs, R.B.; Santos, D.F.; Laird, D.E.; Doolen, S.; Donahue, R.R.; Wessel, C.R.; Fu, W.; Sinha, G.P.; Wang, P.; Zhou, J.; et al. Methylglyoxal and a spinal TRPA1-AC1-Epac cascade facilitate pain in the db/db mouse model of type 2 diabetes. Neurobiol. Dis. 2019, 127, 76–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao, X.; Mei, F.; Agrawal, A.; Peters, C.J.; Ksiazek, T.G.; Cheng, X.; Tseng, C.-T.K. Blocking of Exchange Proteins Directly Activated by cAMP Leads to Reduced Replication of Middle East Respiratory Syndrome Coronavirus. J. Virol. 2014, 88, 3902–3910. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.-F.; Huang, G.; Wang, Y.-M.; Cheng, M.; Zhu, F.-F.; Zhong, J.-N.; Gao, Y.-D. Exchange protein directly activated by cAMP (Epac) protects against airway inflammation and airway remodeling in asthmatic mice. Respir. Res. 2019, 20, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Parnell, E.; Palmer, T.M.; Yarwood, S.J. The future of EPAC-targeted therapies: Agonism versus antagonism. Trends Pharmacol. Sci. 2015, 36, 203–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; Schwede, F.; Wienk, H.; Tengholm, A.; Rehmann, H. A Membrane Permeable Prodrug of S223 for Selective Epac2 Activation in Living Cells. Cells 2019, 8, 1589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lezoualc’H, F.; Fazal, L.; Laudette, M.; Conte, C. Cyclic AMP Sensor EPAC Proteins and Their Role in Cardiovascular Function and Disease. Circ. Res. 2016, 118, 881–897. [Google Scholar] [CrossRef]
- Choi, E.-J.; Ren, Y.; Chen, Y.; Liu, S.; Wu, W.; Ren, J.; Wang, P.; Garofalo, R.P.; Zhou, J.; Bao, X. Exchange Proteins Directly Activated by cAMP and Their Roles in Respiratory Syncytial Virus Infection. J. Virol. 2018, 92, 22. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Yan, Y.; He, H.; Wang, L.; Zhang, N.; Zhang, J.; Huang, H.; Wu, N.; Ren, H.; Qian, M.; et al. IFN-stimulated P2Y13 protects mice from viral infection by suppressing the cAMP/EPAC1 signaling pathway. J. Mol. Cell Biol. 2018, 11, 395–407. [Google Scholar] [CrossRef] [Green Version]
- Drelich, A.; Judy, B.; He, X.; Chang, Q.; Yu, S.; Li, X.; Lu, F.; Wakamiya, M.; Popov, V.; Zhou, J.; et al. Exchange Protein Directly Activated by cAMP Modulates Ebola Virus Uptake into Vascular Endothelial Cells. Viruses 2018, 10, 563. [Google Scholar] [CrossRef] [Green Version]
- Courilleau, D.; Bouyssou, P.; Fischmeister, R.; Lezoualc’H, F.; Blondeau, J.-P. The (R)-enantiomer of CE3F4 is a preferential inhibitor of human exchange protein directly activated by cyclic AMP isoform 1 (Epac1). Biochem. Biophys. Res. Commun. 2013, 440, 443–448. [Google Scholar] [CrossRef]
- Liu, Z.; Zhu, Y.; Chen, H.; Wang, P.; Mei, F.C.; Ye, N.; Cheng, X.; Zhou, J. Structure-activity relationships of 2-substituted phenyl-N-phenyl-2-oxoacetohydrazonoyl cyanides as novel antagonists of exchange proteins directly activated by cAMP (EPACs). Bioorganic Med. Chem. Lett. 2017, 27, 5163–5166. [Google Scholar] [CrossRef] [PubMed]
- Bao, X.; Liu, T.; Shan, Y.; Li, K.; Garofalo, R.P.; Casola, A. Human Metapneumovirus Glycoprotein G Inhibits Innate Immune Responses. PLoS Pathog. 2008, 4, e1000077. [Google Scholar] [CrossRef] [PubMed]
- Bao, X.; Sinha, M.; Liu, T.; Hong, C.; Luxon, B.A.; Garofalo, R.P.; Casola, A. Identification of human metapneu-movirus-induced gene networks in airway epithelial cells by microarray analysis. Virology 2008, 374, 114–127. [Google Scholar] [CrossRef] [Green Version]
- Bao, X.; Kolli, D.; Liu, T.; Shan, Y.; Garofalo, R.P.; Casola, A. Human metapneumovirus small hydrophobic protein inhibits NF-kappaB transcriptional activity. J. Virol. 2008, 82, 8224–8229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reyes, E.D.; Kulej, K.; Pancholi, N.J.; Akhtar, L.; Avgousti, D.C.; Kim, E.T.; Bricker, D.K.; Spruce, L.A.; Koniski, S.A.; Seeholzer, S.H.; et al. Identifying Host Factors Associated with DNA Replicated During Virus Infection. Mol. Cell. Proteom. 2017, 16, 2079–2097. [Google Scholar] [CrossRef] [Green Version]
- Bao, X.; Indukuri, H.; Liu, T.; Liao, S.L.; Tian, B.; Brasier, A.R.; Garofalo, R.P.; Casola, A. IKKepsilon modulates RSV-induced NF-kappaB-dependent gene transcription. Virology 2010, 408, 224–231. [Google Scholar] [CrossRef] [Green Version]
- Wild, C.T.; Zhu, Y.; Na, Y.; Mei, F.; Ynalvez, M.A.; Chen, H.; Cheng, X.; Zhou, J. Functionalized N,N-Diphenylamines as Potent and Selective EPAC2 Inhibitors. ACS Med. Chem. Lett. 2016, 7, 460–464. [Google Scholar] [CrossRef] [Green Version]
- Ren, J.; Wang, Q.; Kolli, D.; Prusak, D.J.; Tseng, C.-T.K.; Chen, Z.; Li, K.; Wood, T.G.; Bao, X. Human Metapneumovirus M2-2 Protein Inhibits Innate Cellular Signaling by Targeting MAVS. J. Virol. 2012, 86, 13049–13061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Ma, Y.; Escaffre, O.; Ivanciuc, T.; Komaravelli, N.; Kelley, J.P.; Coletta, C.; Szabo, C.; Rockx, B.; Garofalo, R.P.; et al. Role of Hydrogen Sulfide in Paramyxovirus Infections. J. Virol. 2015, 89, 5557–5568. [Google Scholar] [CrossRef] [Green Version]
- Guerrero-Plata, A.; Casola, A.; Suarez, G.; Yu, X.; Spetch, L.; Peeples, M.E.; Garofalo, R.P. Differential Response of Dendritic Cells to Human Metapneumovirus and Respiratory Syncytial Virus. Am. J. Respir. Cell Mol. Biol. 2006, 34, 320–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guerrero-Plata, A.; Casola, A.; Garofalo, R.P. Human Metapneumovirus Induces a Profile of Lung Cytokines Distinct from That of Respiratory Syncytial Virus. J. Virol. 2005, 79, 14992–14997. [Google Scholar] [CrossRef] [Green Version]
- Huck, B.; Neumann-Haefelin, D.; Schmitt-Graeff, A.; Weckmann, M.; Mattes, J.; Ehl, S.; Falcone, V. Human metapneumovirus induces more severe disease and stronger innate immune response in BALB/c mice as compared with respiratory syncytial virus. Respir. Res. 2007, 8, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malmo, J.; Moe, N.; Krokstad, S.; Ryan, L.; Loevenich, S.; Johnsen, I.B.; Espevik, T.; Nordbø, S.A.; Døllner, H.; Anthonsen, M.W. Cytokine Profiles in Human Metapneumovirus Infected Children: Identification of Genes Involved in the Antiviral Response and Pathogenesis. PLoS ONE 2016, 11, e0155484. [Google Scholar] [CrossRef] [PubMed]
- Pancham, K.; Perez, G.F.; Huseni, S.; Jain, A.; Kurdi, B.; Rodriguez-Martinez, C.E.; Preciado, D.; Rose, M.C.; Nino, G. Prem-ature infants have impaired airway antiviral IFNgamma responses to human metapneumovirus compared to respiratory syncytial virus. Pediatr. Res. 2015, 78, 389–394. [Google Scholar] [CrossRef] [Green Version]
- Melchjorsen, J.; Siren, J.; Julkunen, I.; Paludan, S.R.; Matikainen, S. Induction of cytokine expression by herpes simplex virus in human monocyte-derived macrophages and dendritic cells is dependent on virus replication and is counteracted by ICP27 targeting NF-kappaB and IRF-3. J. Gen. Virol. 2006, 87, 1099–1108. [Google Scholar] [CrossRef]
- Saba, T.G.; Chung, Y.; Hong, J.Y.; Sajjan, U.S.; Bentley, J.K.; Hershenson, M.B. Rhinovirus-Induced Macrophage Cytokine Expression Does Not Require Endocytosis or Replication. Am. J. Respir. Cell Mol. Biol. 2014, 50, 974–984. [Google Scholar] [CrossRef]
- Forbester, J.L.; Clement, M.; Wellington, D.; Yeung, A.; Dimonte, S.; Marsden, M.; Chapman, L.; Coomber, E.L.; Tolley, C.; Lees, E.; et al. IRF5 Promotes Influenza Virus-Induced Inflam-matory Responses in Human Induced Pluripotent Stem Cell-Derived Myeloid Cells and Murine Models. J. Virol. 2020, 94. [Google Scholar] [CrossRef] [Green Version]
- Hayney, M.S.; Henriquez, K.M.; Barnet, J.H.; Ewers, T.; Champion, H.M.; Flannery, S.; Barrett, B. Serum IFN-gamma-induced protein 10 (IP-10) as a biomarker for severity of acute respiratory infection in healthy adults. J. Clin. Virol. 2017, 90, 32–37. [Google Scholar] [CrossRef]
- Korpi-Steiner, N.L.; Bates, M.E.; Lee, W.-M.; Hall, D.J.; Bertics, P.J. Human rhinovirus induces robust IP-10 release by monocytic cells, which is independent of viral replication but linked to type I interferon receptor ligation and STAT1 activation. J. Leukoc. Biol. 2006, 80, 1364–1374. [Google Scholar] [CrossRef]
- Hamamdzic, D.; Phillips-Dorsett, T.; Altman-Hamamdzic, S.; London, S.D.; London, L. Reovirus triggers cell type-specific proinflammatory responses dependent on the autocrine action of IFN-beta. Am. J. Physiol. Lung Cell. Mol. Physiol. 2001, 280, L18–L29. [Google Scholar] [CrossRef] [PubMed]
- Kruijer, W.; van Schaik, F.M.; Speijer, J.G.; Sussenbach, J.S. Structure and function of adenovirus DNA binding protein: Comparison of the amino acid sequences of the Ad5 and Ad12 proteins derived from the nucleotide sequence of the corre-sponding genes. Virology 1983, 128, 140–153. [Google Scholar] [CrossRef]
- Hornsleth, A.; Loland, L.; Larsen, L.B. Cytokines and chemokines in respiratory secretion and severity of disease in infants with respiratory syncytial virus (RSV) infection. J. Clin. Virol. 2001, 21, 163–170. [Google Scholar] [CrossRef]
- Teigler, J.E.; Iampietro, M.J.; Barouch, D.H. Vaccination with Adenovirus Serotypes 35, 26, and 48 Elicits Higher Levels of Innate Cytokine Responses than Adenovirus Serotype 5 in Rhesus Monkeys. J. Virol. 2012, 86, 9590–9598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Booth, J.L.; Metcalf, J.P. Type-Specific Induction of Interleukin-8 by Adenovirus. Am. J. Respir. Cell Mol. Biol. 1999, 21, 521–527. [Google Scholar] [CrossRef]
- Tamanini, A.; Nicolis, E.; Bonizzato, A.; Bezzerri, V.; Melotti, P.; Assael, B.M.; Cabrini, G. Interaction of Adenovirus Type 5 Fiber with the Coxsackievirus and Adenovirus Receptor Activates Inflammatory Response in Human Respiratory Cells. J. Virol. 2006, 80, 11241–11254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oeckinghaus, A.; Ghosh, S. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb. Perspect. Biol. 2009, 1, a000034. [Google Scholar] [CrossRef] [PubMed]
- Song, K.; Li, S. The Role of Ubiquitination in NF-kappaB Signaling during Virus Infection. Viruses 2021, 13, 145. [Google Scholar] [CrossRef]
- Bao, X.; Liu, T.; Spetch, L.; Kolli, D.; Garofalo, R.; Casola, A. Airway epithelial cell response to human metapneumovirus infection. J. Virol. 2007, 368, 91–101. [Google Scholar] [CrossRef] [Green Version]
- Yoboua, F.; Martel, A.; Duval, A.; Mukawera, E.; Grandvaux, N. Respiratory syncytial virus-mediated NF-kappa B p65 phosphorylation at serine 536 is dependent on RIG-I, TRAF6, and IKK beta. J. Virol. 2010, 84, 7267–7277. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, M.; Dekker, F.; Maarsingh, H. Exchange Protein Directly Activated by cAMP (epac): A Multidomain cAMP Mediator in the Regulation of Diverse Biological Functions. Pharmacol. Rev. 2013, 65, 670–709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, P.; Liu, Z.; Chen, H.; Ye, N.; Cheng, X.; Zhou, J. Exchange proteins directly activated by cAMP (EPACs): Emerging therapeutic targets. Bioorganic Med. Chem. Lett. 2017, 27, 1633–1639. [Google Scholar] [CrossRef] [PubMed]
- Courilleau, D.; Bisserier, M.; Jullian, J.C.; Lucas, A.; Bouyssou, P.; Fischmeister, R.; Blondeau, J.P.; Lezoualc’’h, F. Identification of a tetrahydroquinoline analog as a pharmacological inhibitor of the cAMP-binding protein Epac. J. Biol. Chem. 2012, 287, 44192–44202. [Google Scholar] [CrossRef] [Green Version]
- Laudette, M.; Coluccia, A.; Sainte-Marie, Y.; Solari, A.; Fazal, L.; Sicard, P.; Silvestri, R.; Mialet-Perez, J.; Pons, S.; Ghaleh, B.; et al. Identification of a pharmacological inhibitor of Epac1 that protects the heart against acute and chronic models of cardiac stress. Cardiovasc. Res. 2019, 115, 1766–1777. [Google Scholar] [CrossRef]
- Waner, J.L. Mixed viral infections: Detection and management. Clin. Microbiol. Rev. 1994, 7, 143–151. [Google Scholar] [CrossRef]
- Calvo, C.; García-García, M.L.; Blanco, C.; Vázquez, M.C.; Frías, M.E.; Pérez-Breña, P.; Casas, I. Multiple simultaneous viral infections in infants with acute respiratory tract infections in Spain. J. Clin. Virol. 2008, 42, 268–272. [Google Scholar] [CrossRef]
- Brand, H.K.; de Groot, R.; Galama, J.M.; Brouwer, M.L.; Teuwen, K.; Hermans, P.W.; Melchers, W.J.; Warris, A. Infection with multiple viruses is not associated with increased disease severity in children with bronchiolitis. Pediatr. Pulmonol. 2012, 47, 393–400. [Google Scholar] [CrossRef]
- Martin, E.T.; Fairchok, M.P.; Stednick, Z.J.; Kuypers, J.; Englund, J.A. Epidemiology of Multiple Respiratory Viruses in Childcare Attendees. J. Infect. Dis. 2013, 207, 982–989. [Google Scholar] [CrossRef] [Green Version]
- Martin, E.T.; Kuypers, J.; Wald, A.; Englund, J.A. Multiple versus single virus respiratory infections: Viral load and clinical disease severity in hospitalized children. Influenza Other Respir. Viruses 2012, 6, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Pinky, L.; Dobrovolny, H.M. Coinfections of the Respiratory Tract: Viral Competition for Resources. PLoS ONE 2016, 11, e0155589. [Google Scholar] [CrossRef]
- Chan, K.C.; Yu, M.W.; Cheung, T.W.Y.; Lam, D.S.Y.; Leung, T.N.H.; Tsui, T.K.; Ip, K.I.; Chau, C.S.K.; Lee, S.L.; Yip, A.Y.F.; et al. Childhood bronchiolitis obliterans in Hong Kong-case series over a 20-year period. Pediatr. Pulmonol. 2021, 56, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Goka, E.A.; Vallely, P.J.; Mutton, K.J.; Klapper, P.E. Single, dual and multiple respiratory virus infections and risk of hospi-talization and mortality. Epidemiol. Infect. 2015, 143, 37–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richard, N.; Komurian-Pradel, F.; Javouhey, E.; Perret, M.; Rajoharison, A.; Bagnaud, A.; Billaud, G.; Vernet, G.; Lina, B.; Floret, D.; et al. The Impact of Dual Viral Infection in Infants Admitted to a Pediatric Intensive Care Unit Associated with Severe Bronchiolitis. Pediatr. Infect. Dis. J. 2008, 27, 213–217. [Google Scholar] [CrossRef]
- Meskill, S.D.; O’Bryant, S.C. Respiratory Virus Co-infection in Acute Respiratory Infections in Children. Curr. Infect. Dis. Rep. 2020, 22, 3. [Google Scholar] [CrossRef] [PubMed]
- Coiras, M.; Pérez-Breña, P.; García, M.; Casas, I. Simultaneous detection of influenza A, B, and C viruses, respiratory syncytial virus, and adenoviruses in clinical samples by multiplex reverse transcription nested-PCR assay. J. Med. Virol. 2003, 69, 132–144. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-kappaB signaling in inflammation. Signal Transduct. Target Ther. 2017, 2. [Google Scholar] [CrossRef] [Green Version]
- Liao, S.; Bao, X.; Liu, T.; Lai, S.; Li, K.; Garofalo, R.P.; Casola, A. Role of retinoic acid inducible gene-I in human metapneu-movirus-induced cellular signalling. J. Gen. Virol. 2008, 89 Pt 8, 1978–1986. [Google Scholar] [CrossRef]
- Alexopoulou, L.; Holt, A.C.; Medzhitov, R.; Flavell, R.A. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature 2001, 413, 732–738. [Google Scholar] [CrossRef]
- Kato, H.; Takeuchi, O.; Sato, S.; Yoneyama, M.; Yamamoto, M.; Matsui, K.; Uematsu, S.; Jung, A.; Kawai, T.; Ishii, K.J.; et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nat. Cell Biol. 2006, 441, 101–105. [Google Scholar] [CrossRef]
- Yoneyama, M.; Kikuchi, M.; Natsukawa, T.; Shinobu, N.; Imaizumi, T.; Miyagishi, M.; Taira, K.; Akira, S.; Fujita, T. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat. Immunol. 2004, 5, 730–737. [Google Scholar] [CrossRef]
- Wang, J.; Shao, Y.; Bennett, T.A.; Shankar, R.A.; Wightman, P.D.; Reddy, L.G. The Functional Effects of Physical Interactions among Toll-like Receptors 7, 8, and 9. J. Biol. Chem. 2006, 281, 37427–37434. [Google Scholar] [CrossRef] [PubMed] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choi, E.-J.; Wu, W.; Cong, X.; Zhang, K.; Luo, J.; Ye, S.; Wang, P.; Suresh, A.; Ullah, U.M.; Zhou, J.; et al. Broad Impact of Exchange Protein Directly Activated by cAMP 2 (EPAC2) on Respiratory Viral Infections. Viruses 2021, 13, 1179. https://0-doi-org.brum.beds.ac.uk/10.3390/v13061179
Choi E-J, Wu W, Cong X, Zhang K, Luo J, Ye S, Wang P, Suresh A, Ullah UM, Zhou J, et al. Broad Impact of Exchange Protein Directly Activated by cAMP 2 (EPAC2) on Respiratory Viral Infections. Viruses. 2021; 13(6):1179. https://0-doi-org.brum.beds.ac.uk/10.3390/v13061179
Chicago/Turabian StyleChoi, Eun-Jin, Wenzhe Wu, Xiaoyan Cong, Ke Zhang, Jiaqi Luo, Sha Ye, Pingyuan Wang, Adarsh Suresh, Uneeb Mohammad Ullah, Jia Zhou, and et al. 2021. "Broad Impact of Exchange Protein Directly Activated by cAMP 2 (EPAC2) on Respiratory Viral Infections" Viruses 13, no. 6: 1179. https://0-doi-org.brum.beds.ac.uk/10.3390/v13061179