
Metaviromes Reveal the Dynamics of Pseudomonas Host-Specific Phages Cultured and Uncultured by Plaque Assay

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacterial Host Strain and Organic Waste Sample Preparation
2.2. Plaque Assay and Isolation of Single Phages
2.3. Virome Spike and Extraction
2.4. DNA Extraction, Purification, and Library Prep
2.5. Quality Control and Assembly
2.6. Viral Identification and Clustering
2.7. Abundance Calculations
2.8. Host Prediction
2.9. Diversity Analysis and Statistics
3. Results
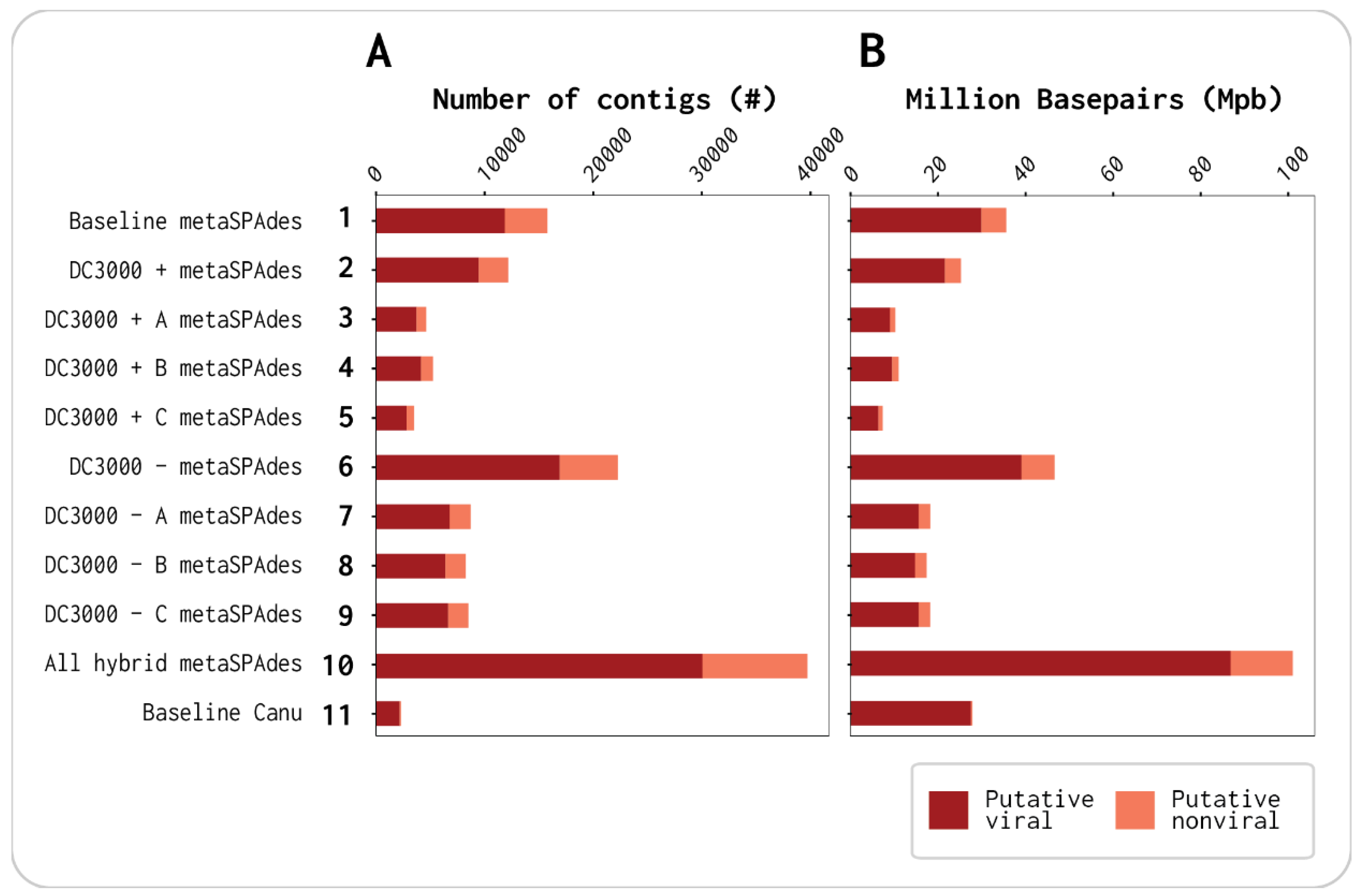
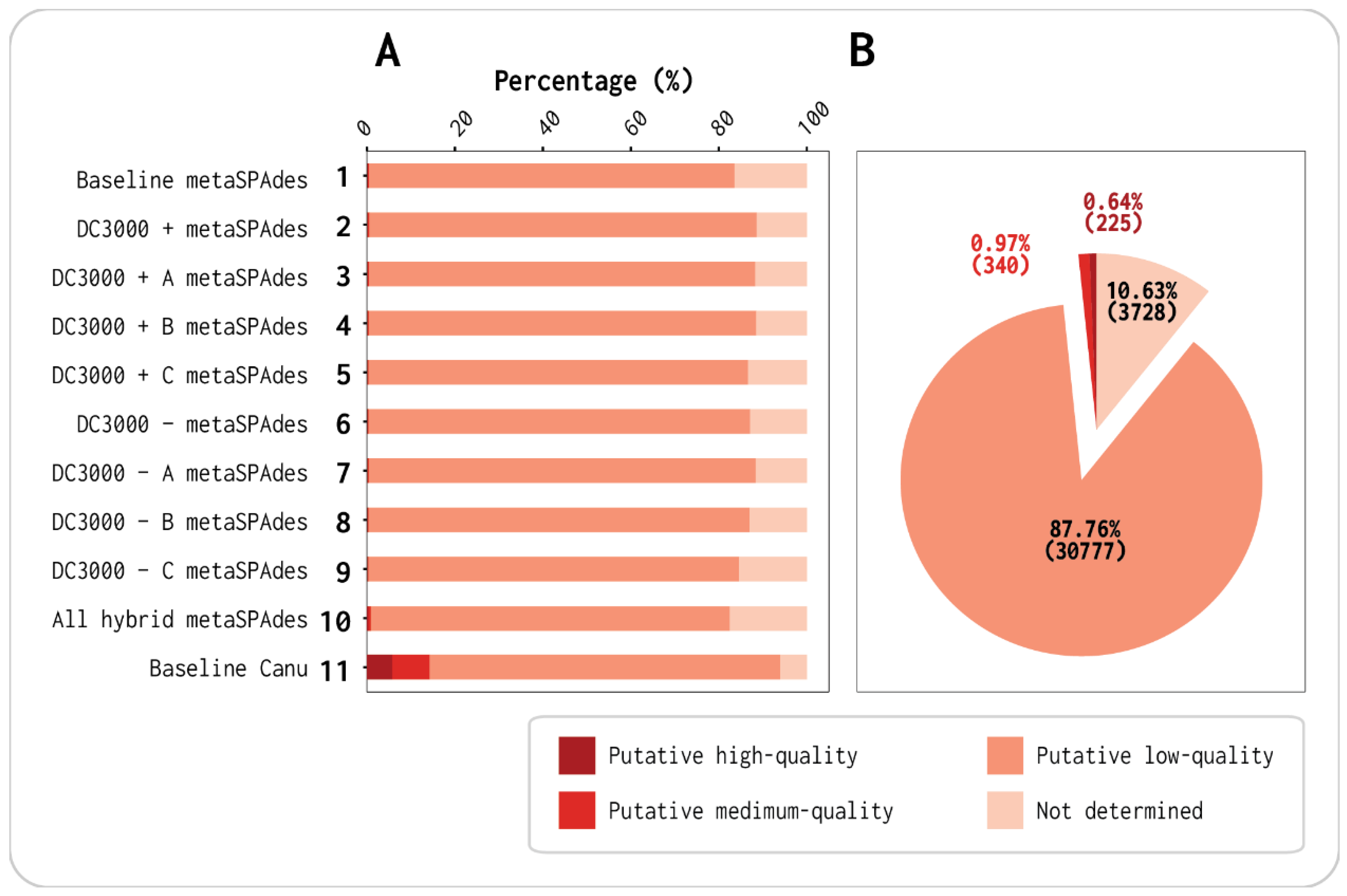
3.1. Viral Contigs Constitute the Majority of Each Assembly
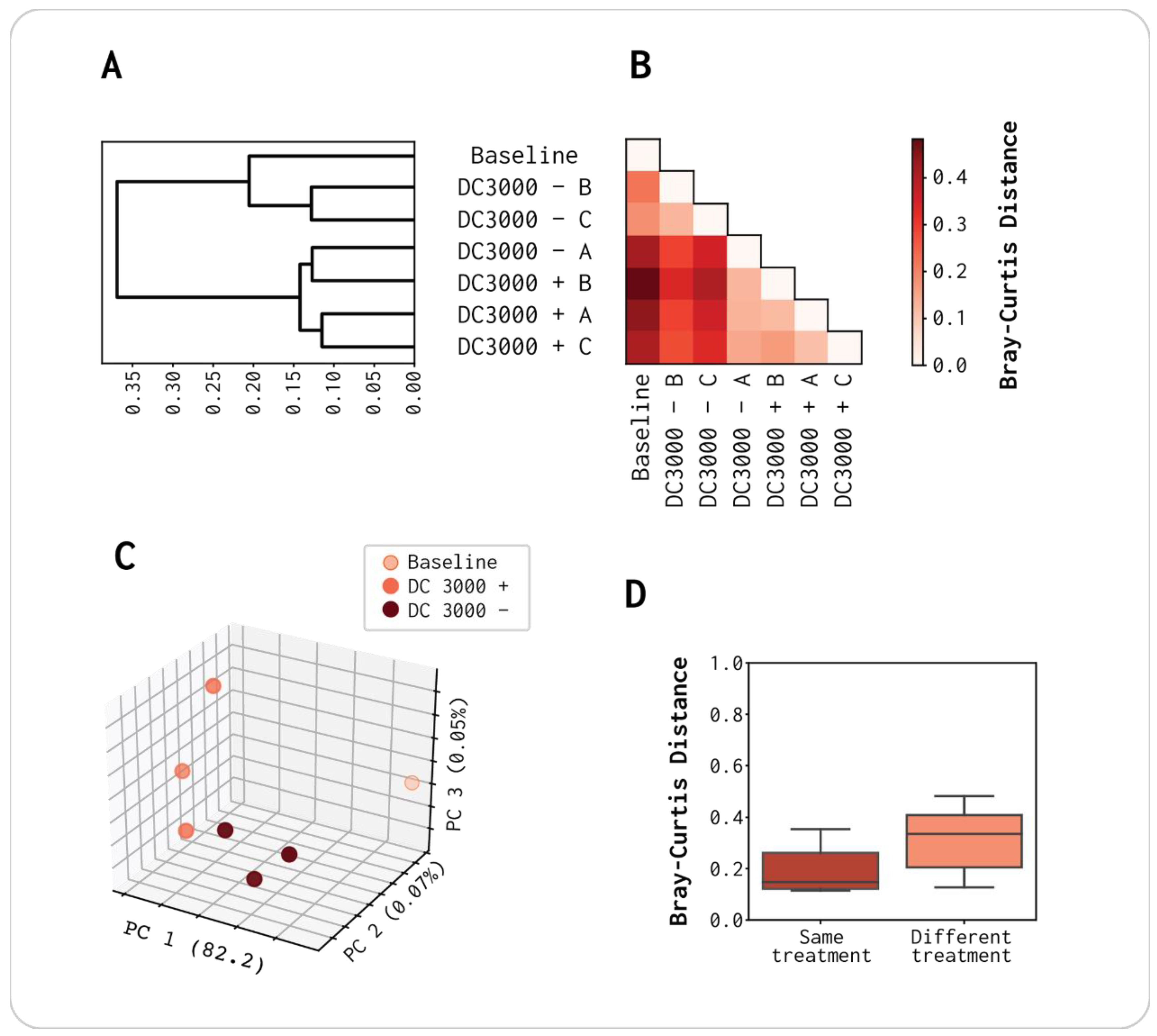
3.2. Differences in Viral Composition Correlates with the Presence/Absence of vOTUs Rather Than High/Low Abundance
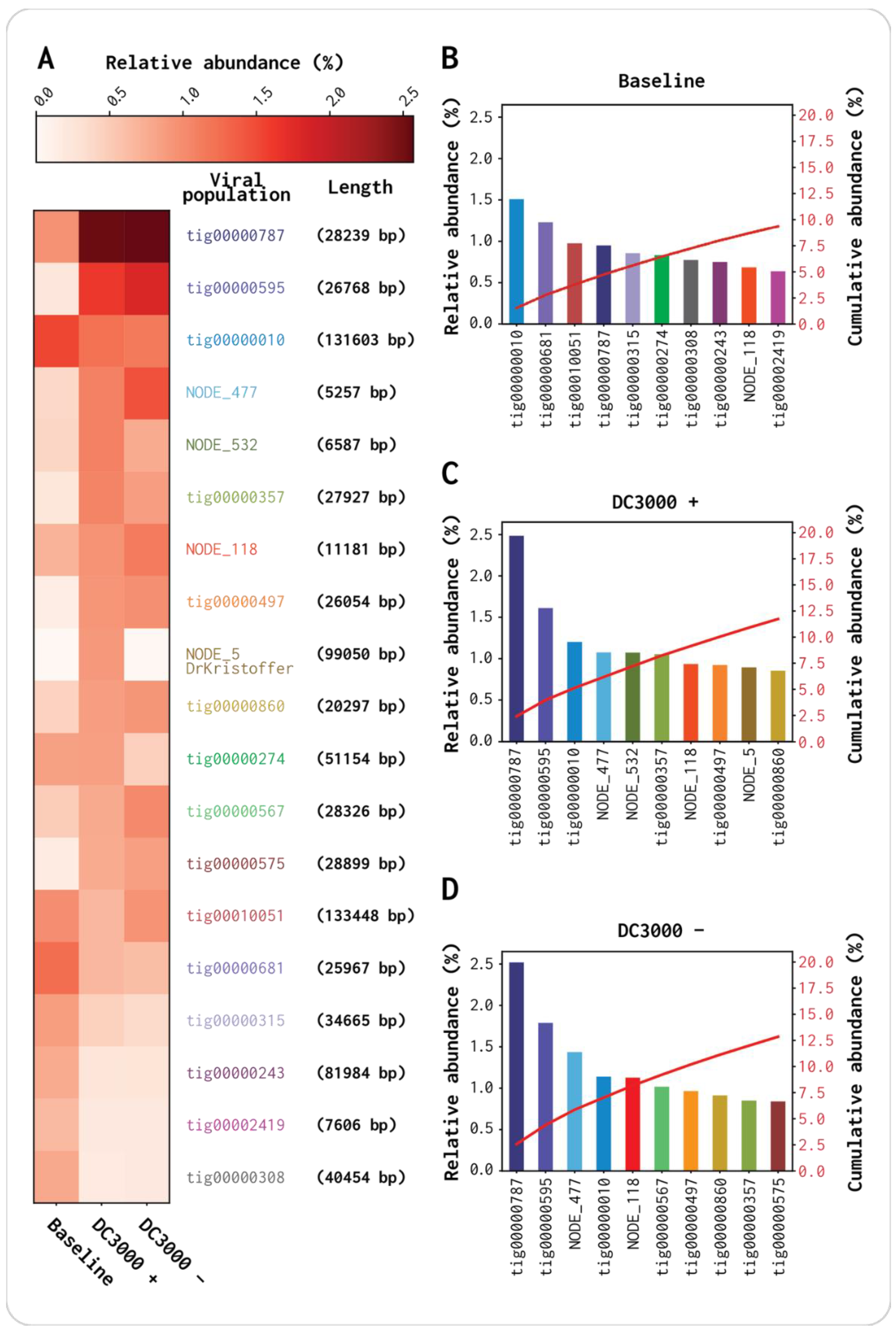
3.3. Most Abundant vOTUs Indicate a Population Change
3.4. DrKristoffer, Hovsa, and OtownIsak Cluster Together with Two Contigs from the OW Viromes
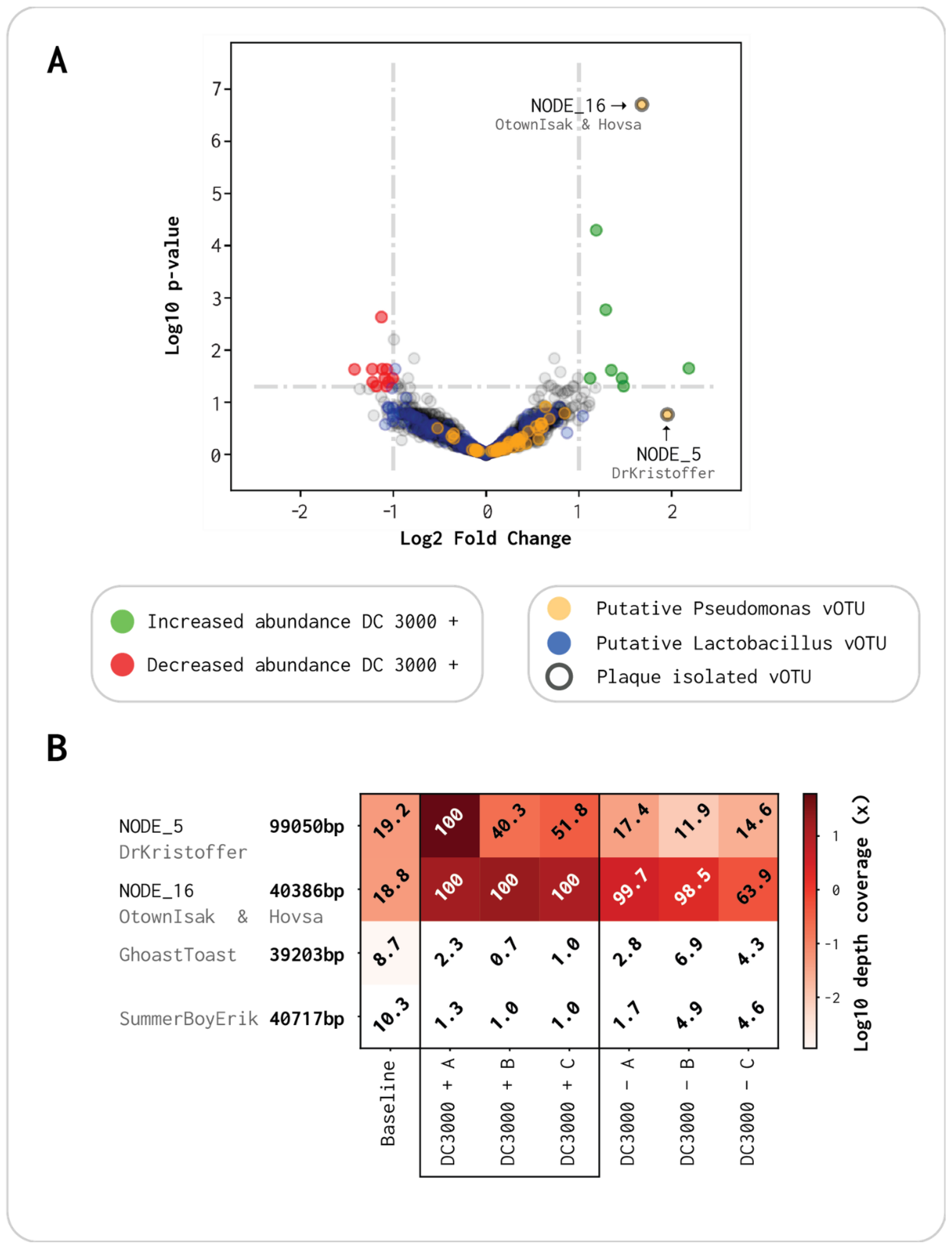
3.5. Pseudomonas Phages Are Significantly Enriched When Exposed to DC3000
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding

Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chibani-chennoufi, S.; Bruttin, A.; Brüssow, H.; Dillmann, M.; Bru, H. Phage-Host Interaction: An Ecological Perspective MINIREVIEW Phage-Host Interaction: An Ecological Perspective. J. Bacteriol. 2004, 186, 3677–3686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mann, N.H. The Third Age of Phage. PLoS Biol. 2005, 3, e182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breitbart, M.; Thompson, L.; Suttle, C.; Sullivan, M. Exploring the Vast Diversity of Marine Viruses. Oceanography 2007, 20, 135–139. [Google Scholar] [CrossRef]
- Batinovic, S.; Wassef, F.; Knowler, S.A.; Rice, D.T.; Stanton, C.R.; Rose, J.; Tucci, J.; Nittami, T.; Vinh, A.; Drummond, G.R.; et al. Bacteriophages in Natural and Artificial Environments. Pathogens 2019, 8, 100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinbauer, M.G.; Rassoulzadegan, F. Are viruses driving microbial diversification and diversity? Environ. Microbiol. 2004, 6, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Paez-Espino, D.; Eloe-Fadrosh, E.A.; Pavlopoulos, G.A.; Thomas, A.D.; Huntemann, M.; Mikhailova, N.; Rubin, E.; Ivanova, N.N.; Kyrpides, N.C. Uncovering Earth’s virome. Nat. Cell Biol. 2016, 536, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Shkoporov, A.N.; Clooney, A.G.; Sutton, T.D.; Ryan, F.J.; Daly, K.M.; Nolan, J.A.; McDonnell, S.A.; Khokhlova, E.V.; Draper, L.A.; Forde, A.; et al. The Human Gut Virome Is Highly Diverse, Stable, and Individual Specific. Cell Host Microbe 2019, 26, 527–541.e5. [Google Scholar] [CrossRef] [PubMed]
- Suttle, C.A. Marine viruses—Major players in the global ecosystem. Nat. Rev. Genet. 2007, 5, 801–812. [Google Scholar] [CrossRef] [PubMed]
- Olsen, N.S.; Nielsen, K.J.; Plöger, M.; Kot, W.; Hansen, L.H. Enterococcus phage Nonaheksakonda infecting clinical isolates of Enterococcus faecalis represents a new lineage in the family Siphoviridae. Arch. Virol. 2021, 166, 593–599. [Google Scholar] [CrossRef] [PubMed]
- Bétermier, M. Large-scale genome remodelling by the developmentally programmed elimination of germ line sequences in the ciliate Paramecium. Res. Microbiol. 2004, 155, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Carstens, A.B.; Djurhuus, A.M.; Kot, W.; Hansen, L.H. A novel six-phage cocktail reduces Pectobacterium atrosepticum soft rot infection in potato tubers under simulated storage conditions. FEMS Microbiol. Lett. 2019, 366, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Dykhuizen, D. Species Numbers in Bacteria. Proc. Calif. Acad. Sci. 2005, 56, 62–71. [Google Scholar] [PubMed]
- Roux, S.; Adriaenssens, E.M.; E Dutilh, B.; Koonin, E.V.; Kropinski, A.M.; Krupovic, M.; Kuhn, J.H.; Lavigne, R.; Brister, J.R.; Varsani, A.; et al. Minimum Information about an Uncultivated Virus Genome (MIUViG). Nat. Biotechnol. 2019, 37, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Roux, S.; Páez-Espino, D.; A Chen, I.-M.; Palaniappan, K.; Ratner, A.; Chu, K.; Reddy, T.B.K.; Nayfach, S.; Schulz, F.; Call, L.; et al. IMG/VR v3: An integrated ecological and evolutionary framework for interrogating genomes of uncultivated viruses. Nucleic Acids Res. 2021, 49, D764–D775. [Google Scholar] [CrossRef] [PubMed]
- Simmonds, P.; Adams, M.J.; Benkő, M.; Breitbart, M.; Brister, J.R.; Carstens, E.B.; Davison, A.J.; Delwart, E.; Gorbalenya, A.E.; Harrach, B.; et al. Virus taxonomy in the age of metagenomics. Nat. Rev. Genet. 2017, 15, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Summers, W.C. History of virology: Bacteriophages. In Encyclopedia of Virology; Bamford, D.H., Zuckerman, H., Eds.; Elsevier Science Publishing Co Inc.: Amsterdam, The Netherlands, 2021; Volume 4, pp. 3–9. ISBN 9780123744104. [Google Scholar]
- Olsen, N.S.; Hendriksen, N.B.; Hansen, L.H.; Kot, W. A New High-Throughput Screening Method for Phages: Enabling Crude Isolation and Fast Identification of Diverse Phages with Therapeutic Potential. Phage 2020, 1, 137–148. [Google Scholar] [CrossRef]
- Ghugare, G.S.; Nair, A.; Nimkande, V.; Sarode, P.; Rangari, P.; Khairnar, K. Membrane filtration immobilization technique-a simple and novel method for primary isolation and enrichment of bacteriophages. J. Appl. Microbiol. 2016, 122, 531–539. [Google Scholar] [CrossRef]
- Buchholz, H.H.; Michelsen, M.L.; Bolaños, L.M.; Browne, E.; Allen, M.J.; Temperton, B. Efficient dilution-to-extinction isolation of novel virus–host model systems for fastidious heterotrophic bacteria. ISME J. 2021, 1–14. [Google Scholar] [CrossRef]
- Gratia, A. Des relations numériques entre bacteries lysogenes et particules de bactériophage. The numerical relation between lysogenic bacteria and the phage particles which they carry. Ann. Inst. Pasteur 1936, 57, 652–676. [Google Scholar]
- van Twest, R.; Kropinski, A.M. Bacteriophage enrichment from water and soil. In Bacteriophages. Methods in Molecular Biology; Clokie, M.R., Kropinski, A.M., Eds.; Humane Press: Totowa, NJ, USA, 2009; Volume 501, pp. 15–21. ISBN 9781603271646. [Google Scholar]
- Serwer, P.; Hayes, J.S.; Thomas, A.J.; Demeler, B.; Hardies, C.S. Isolation of novel large and aggregating bacteriophages. In Bacteriophages. Methods in Molecular Biology; Clokie, M.R.J., Kropinski, M.A., Eds.; Humane Press: Totowa, NJ, USA, 2009; pp. 55–66. ISBN 978-1-58829-682-5. [Google Scholar]
- Castromejia, J.L.; Muhammed, M.K.; Kot, W.; Neve, H.; Franz, C.M.A.P.; Hansen, L.H.; Vogensen, F.K.; Nielsen, D.S. Optimizing protocols for extraction of bacteriophages prior to metagenomic analyses of phage communities in the human gut. Microbiome 2015, 3, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Hyman, P. Phages for Phage Therapy: Isolation, Characterization, and Host Range Breadth. Pharmaceuticals 2019, 12, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rinke, C.; Low, S.; Woodcroft, B.J.; Raina, J.-B.; Skarshewski, A.; Le, X.H.; Butler, M.K.; Stocker, R.; Seymour, J.; Tyson, G.W.; et al. Validation of picogram- and femtogram-input DNA libraries for microscale metagenomics. PeerJ 2016, 4, e2486. [Google Scholar] [CrossRef] [Green Version]
- Rico, A.; Preston, G.M. Pseudomonas syringae pv. tomato DC3000 Uses Constitutive and Apoplast-Induced Nutrient Assimilation Pathways to Catabolize Nutrients that Are Abundant in the Tomato Apoplast. Mol. Plant-Microbe Interact. 2008, 21, 269–282. [Google Scholar] [CrossRef] [Green Version]
- Xin, X.-F.; He, S.Y. Pseudomonas syringae pv. tomato DC3000: A Model Pathogen for Probing Disease Susceptibility and Hormone Signaling in Plants. Annu. Rev. Phytopathol. 2013, 51, 473–498. [Google Scholar] [CrossRef] [PubMed]
- Bertani, G. Lysogeny at Mid-Twentieth Century: P1, P2, and Other Experimental Systems. J. Bacteriol. 2004, 186, 595–600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jørgensen, J.B.; Djurhuus, A.M.; Carstens, A.B.; Kot, W.; Neve, H.; Morris, C.E.; Hansen, L.H. Presentation of Three Novel Tailed Phages Targeting Multiple Strains of Pseudomonas syringae. Phage 2020, 1, 245–250. [Google Scholar] [CrossRef]
- Kropinski, A.M.; Mazzocco, A.; Waddell, T.E.; Lingohr, E.; Johnson, R.P. Enumeration of bacteriophages by double agar overlay plaque assay. In Bacteriophages: Methods and Protocols, Volume 1: Isolation, Characterization, and Interactions; Martha, R.J., Clokie, A.M.K., Eds.; Humane Press: Totowa, NJ, USA, 2009; Volume 501, pp. 69–76. ISBN 9781603271646. [Google Scholar]
- Green, R.M.; Sambrook, J. Molecular Cloning—A Laboratory Manual, 4th ed.; Cold Spring Harbor Laboratory Press: New York, NY, USA, 2012; Volume 1, ISBN 9781936113415. [Google Scholar]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Bushnell, B.; Rood, J.; Singer, E. BBMerge—Accurate paired shotgun read merging via overlap. PLoS ONE 2017, 12, e0185056. [Google Scholar] [CrossRef]
- de Coster, W.; D’Hert, S.; Schultz, D.T.; Cruts, M.; van Broeckhoven, C. NanoPack: Visualizing and processing long-read sequencing data. Bioinformatics 2018, 34, 2666–2669. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 Genome Project Data Processing Subgroup the Sequence Alignment/Map Format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Nurk, S.; Meleshko, D.; Korobeynikov, A.; Pevzner, P.A. metaSPAdes: A new versatile metagenomic assembler. Genome Res. 2017, 27, 824–834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Bergman, N.H.; Phillippy, A.M. Canu: Scalable and accurate long-read assembly via adaptivek-mer weighting and repeat separation. Genome Res. 2017, 27, 722–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaser, R.; Sović, I.; Nagarajan, N.; Šikić, M. Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res. 2017, 27, 737–746. [Google Scholar] [CrossRef] [Green Version]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K.; et al. Pilon: An Integrated Tool for Comprehensive Microbial Variant Detection and Genome Assembly Improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef] [PubMed]
- Roux, S.; Enault, F.; Hurwitz, B.L.; Sullivan, M.B. VirSorter: Mining viral signal from microbial genomic data. PeerJ 2015, 3, e985. [Google Scholar] [CrossRef]
- Nayfach, S.; Camargo, A.P.; Schulz, F.; Eloe-Fadrosh, E.; Roux, S.; Kyrpides, N.C. CheckV assesses the quality and completeness of metagenome-assembled viral genomes. Nat. Biotechnol. 2021, 39, 578–585. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, R.; Mirdita, M.; Karin, E.L.; Norroy, C.; Galiez, C.; Söding, J. SpacePHARER: Sensitive identification of phages from CRISPR spacers in prokaryotic hosts. Bioinformatics 2021. [Google Scholar] [CrossRef] [PubMed]
- Shmakov, S.A.; Sitnik, V.; Makarova, K.S.; Wolf, Y.I.; Severinov, K.V.; Koonin, E.V. The CRISPR Spacer Space Is Dominated by Sequences from Species-Specific Mobilomes. mBio 2017, 8, e01397-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Russel, J.; Thorsen, J.; Brejnrod, A.D.; Bisgaard, H.; Sørensen, S.J.; Burmølle, M. DAtest: A framework for choosing differential abundance or expression method. bioRxiv 2018, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Paulson, J.N.; Stine, O.C.; Bravo, H.C.; Pop, M. Differential abundance analysis for microbial marker-gene surveys. Nat. Methods 2013, 10, 1200–1202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jost, L.; Chao, A.; Chazdon, L.R. Compositional similarity and β (beta) diversity. In Biological Diversity: Frontiers in Measurement and Assessment; Magurran, E.A., McGrill, J.B., Eds.; Oxford University Press: Oxford, UK, 2011; pp. 66–84. ISBN 978-0-19-958066-8. [Google Scholar]
- Pagliarini, S.; Korobeinikov, A. A mathematical model of marine bacteriophage evolution. R. Soc. Open Sci. 2018, 5, 171661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fabreti, L.G.; Castro, D.; Gorzoni, B.; Janini, L.M.R.; Antoneli, F. Stochastic Modeling and Simulation of Viral Evolution. Bull. Math. Biol. 2018, 81, 1031–1069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marić, J. Long Read RNA-seq Mapper. Ph.D. Thesis, Fakultet Elektrotehnike i Računarstva, Sveučilište u Zagrebu, FakultetSveučilište u Zagrebu, Zagreb, Croatia, 2015. [Google Scholar]
- Deng, L.; Silins, R.; Castro-Mejía, J.L.; Kot, W.; Jessen, L.; Thorsen, J.; Shah, S.; Stokholm, J.; Bisgaard, H.; Moineau, S.; et al. A Protocol for Extraction of Infective Viromes Suitable for Metagenomics Sequencing from Low Volume Fecal Samples. Viruses 2019, 11, 667. [Google Scholar] [CrossRef] [Green Version]
- Henson, M.W.; Pitre, D.M.; Weckhorst, J.L.; Lanclos, V.C.; Webber, A.T.; Thrash, J.C. Second Correction for Henson et al., Artificial Seawater Media Facilitate Cultivating Members of the Microbial Majority from the Gulf of Mexico. mSphere 2018, 3, e00415-18. [Google Scholar] [CrossRef] [PubMed]
- Nabergoj, D.; Modic, P.; Podgornik, A. Effect of bacterial growth rate on bacteriophage population growth rate. Microbiologyopen 2018, 7, e00558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abedon, S.T.; Herschler, T.D.; Stopar, D. Bacteriophage Latent-Period Evolution as a Response to Resource Availability. Appl. Environ. Microbiol. 2001, 67, 4233–4241. [Google Scholar] [CrossRef] [Green Version]
- Hasegawa, H.; Naganuma, K.; Nakagawa, Y.; Matsuyama, T. Membrane filter (pore size, 0.22–0.45 μm; Thickness, 150 μm) passing-through activity of Pseudomonas aeruginosa and other bacterial species with indigenous infiltration ability. FEMS Microbiol. Lett. 2003, 223, 41–46. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Li, B.; Wang, Y.; Zhang, G.; Jiang, X.; Li, X. Passage and community changes of filterable bacteria during microfiltration of a surface water supply. Environ. Int. 2019, 131, 104998. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Hammes, F.; Boon, A.N.; Egli, T. Quantification of the Filterability of Freshwater Bacteria through 0.45, 0.22, and 0.1 μm Pore Size Filters and Shape-Dependent Enrichment of Filterable Bacterial Communities. Environ. Sci. Technol. 2007, 41, 7080–7086. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.-C.; Chen, C.-R.; Lin, J.-W.; Shen, G.-H.; Chang, K.-M.; Tseng, Y.-H.; Weng, S.-F. Isolation and Characterization of Novel Giant Stenotrophomonas maltophilia Phage φSMA5. Appl. Environ. Microbiol. 2005, 71, 1387–1393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Shayeb, B.; Sachdeva, R.; Chen, L.-X.; Ward, F.; Munk, P.; Devoto, A.; Castelle, C.J.; Olm, M.R.; Bouma-Gregson, K.; Amano, Y.; et al. Clades of huge phages from across Earth’s ecosystems. Nat. Cell Biol. 2020, 578, 425–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tannock, G.W. A Special Fondness for Lactobacilli. Appl. Environ. Microbiol. 2004, 70, 3189–3194. [Google Scholar] [CrossRef] [Green Version]
- Daeschel, M.; Andersson, R.; Fleming, H. Microbial ecology of fermenting plant materials. FEMS Microbiol. Lett. 1987, 46, 357–367. [Google Scholar] [CrossRef]
- Feyereisen, M.; Mahony, J.; Lugli, G.A.; Ventura, M.; Neve, H.; Franz, C.M.A.P.; Noben, J.-P.; O’Sullivan, T.; van Sinderen, D. Isolation and Characterization of Lactobacillus brevis Phages. Viruses 2019, 11, 393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carstens, A.B.; Kot, W.; Hansen, L.H. Complete Genome Sequences of Four Novel Escherichia coli Bacteriophages Belonging to New Phage Groups. Genome Announc. 2015, 3, e00741-15. [Google Scholar] [CrossRef] [Green Version]
- Parras-Moltó, M.; Rodríguez-Galet, A.; Suárez-Rodríguez, P.; López-Bueno, A. Evaluation of bias induced by viral enrichment and random amplification protocols in metagenomic surveys of saliva DNA viruses. Microbiome 2018, 6, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Warwick-Dugdale, J.; Solonenko, N.; Moore, K.; Chittick, L.; Gregory, A.C.; Allen, M.J.; Sullivan, M.B.; Temperton, B. Long-read viral metagenomics captures abundant and microdiverse viral populations and their niche-defining genomic islands. PeerJ 2019, 7, e6800. [Google Scholar] [CrossRef] [Green Version]
- Duhaime, M.B.; Deng, L.; Poulos, B.T.; Sullivan, M.B. Towards quantitative metagenomics of wild viruses and other ultra-low concentration DNA samples: A rigorous assessment and optimization of the linker amplification method. Environ. Microbiol. 2012, 14, 2526–2537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gregory, A.C.; Zayed, A.A.; Conceição-Neto, N.; Temperton, B.; Bolduc, B.; Alberti, A.; Ardyna, M.; Arkhipova, K.; Carmichael, M.; Cruaud, C.; et al. Marine DNA Viral Macro- and Microdiversity from Pole to Pole. Cell 2019, 177, 1109–1123.e14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ross, A.; Ward, S.; Hyman, P. More Is Better: Selecting for Broad Host Range Bacteriophages. Front. Microbiol. 2016, 7, 1352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buttimer, C.; Hendrix, H.; Lucid, A.; Neve, H.; Noben, J.-P.; Franz, C.; O’Mahony, J.; Lavigne, R.; Coffey, A. Novel N4-Like Bacteriophages of Pectobacterium atrosepticum. Pharmaceuticals 2018, 11, 45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lebeaux, D.; Merabishvili, M.; Caudron, E.; Lannoy, D.; van Simaey, L.; Duyvejonck, H.; Guillemain, R.; Thumerelle, C.; Podglajen, I.; Compain, F.; et al. A Case of Phage Therapy against Pandrug-Resistant Achromobacter xylosoxidans in a 12-Year-Old Lung-Transplanted Cystic Fibrosis Patient. Viruses 2021, 13, 60. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. of Contigs | % | No. of bp | % | ||
|---|---|---|---|---|---|
| Total | 131,073 | 100 | Total | 317,732,011 | 100 |
| Putative viral | 101,068 | 77.1 | Putative viral | 275,137,398 | 86.6 |
| Putative nonviral | 30,005 | 22.9 | Putative nonviral | 42,594,613 | 13.4 |
| Phage | Genome Size | Family/Genus | Cluster Representative (CR) | Coverage of CR | Percent Identity to CR |
|---|---|---|---|---|---|
| DrKristoffer | 98,997 | Myoviridae/Otagovirus | NODE_5 | 91% | 93.61% |
| Hovsa | 40,290 | Autographiviridae/Ghunavirus | NODE_16 | 94% | 97.41% |
| OtownIsak | 40,331 | Autographiviridae/Ghunavirus | NODE_16 | 100% | 99.99% |
| GhostToast | 39,235 | Autographiviridae/Pifdecavirus | - | - | - |
| SummerBoyErik | 40,716 | Autographiviridae/Pifdecavirus | - | - | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alanin, K.W.S.; Junco, L.M.F.; Jørgensen, J.B.; Nielsen, T.K.; Rasmussen, M.A.; Kot, W.; Hansen, L.H. Metaviromes Reveal the Dynamics of Pseudomonas Host-Specific Phages Cultured and Uncultured by Plaque Assay. Viruses 2021, 13, 959. https://0-doi-org.brum.beds.ac.uk/10.3390/v13060959
Alanin KWS, Junco LMF, Jørgensen JB, Nielsen TK, Rasmussen MA, Kot W, Hansen LH. Metaviromes Reveal the Dynamics of Pseudomonas Host-Specific Phages Cultured and Uncultured by Plaque Assay. Viruses. 2021; 13(6):959. https://0-doi-org.brum.beds.ac.uk/10.3390/v13060959
Chicago/Turabian StyleAlanin, Katrine Wacenius Skov, Laura Milena Forero Junco, Jacob Bruun Jørgensen, Tue Kjærgaard Nielsen, Morten Arendt Rasmussen, Witold Kot, and Lars Hestbjerg Hansen. 2021. "Metaviromes Reveal the Dynamics of Pseudomonas Host-Specific Phages Cultured and Uncultured by Plaque Assay" Viruses 13, no. 6: 959. https://0-doi-org.brum.beds.ac.uk/10.3390/v13060959