
Retrospective Characterization of the 2006–2007 Swine Vesicular Disease Epidemic in Northern Italy by Whole Genome Sequence Analysis

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
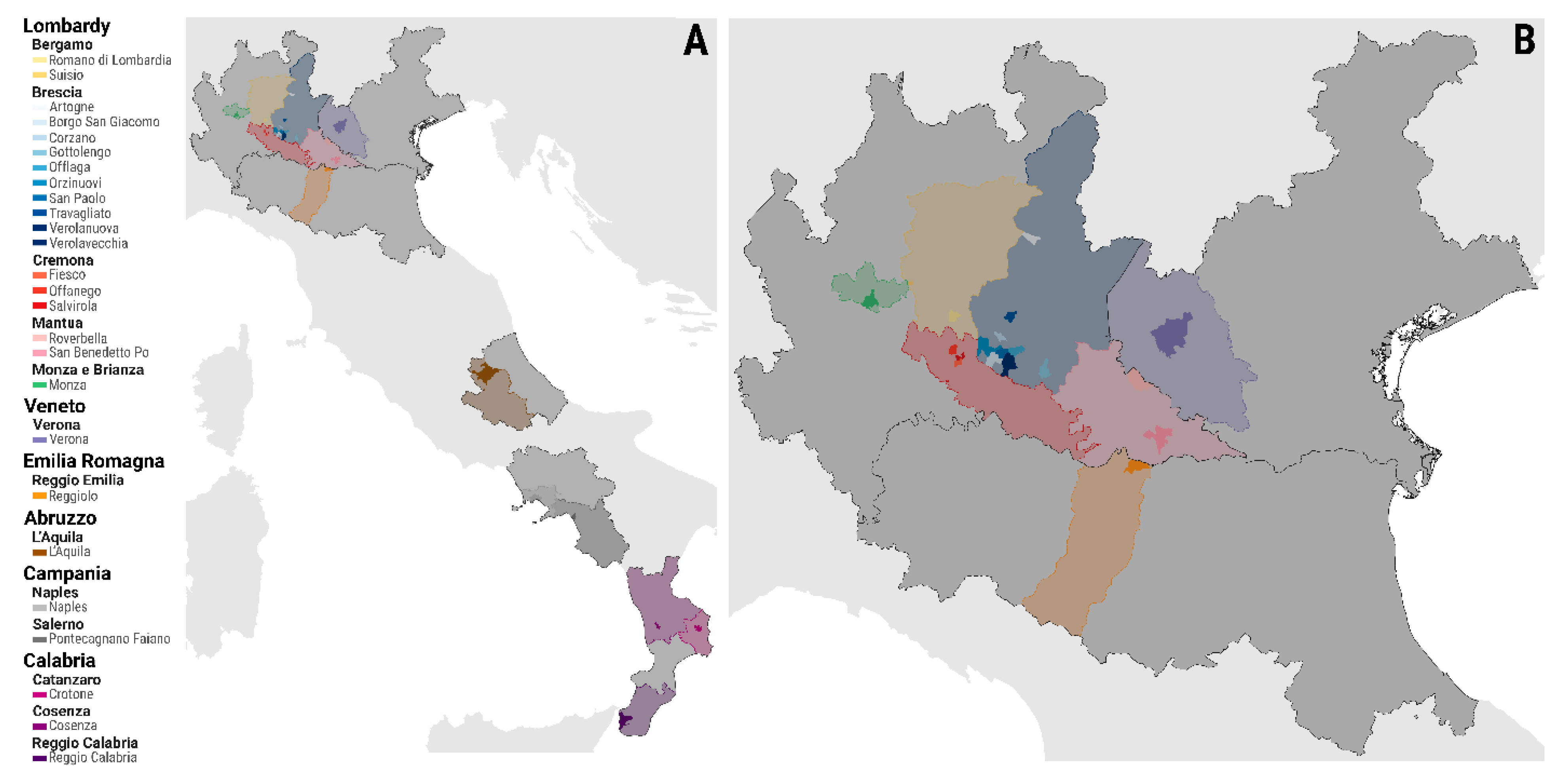
2.1. Epidemiological Field Investigation and Data Collection
2.2. Clinical Samples
2.3. RNA Isolation, PCR Amplification and Sequencing
2.4. Phylogenetic Inference and Phylogeography Reconstruction
2.5. Transmission Tree Reconstruction
2.6. Testing for Selection in the SVDV Genome
3. Results
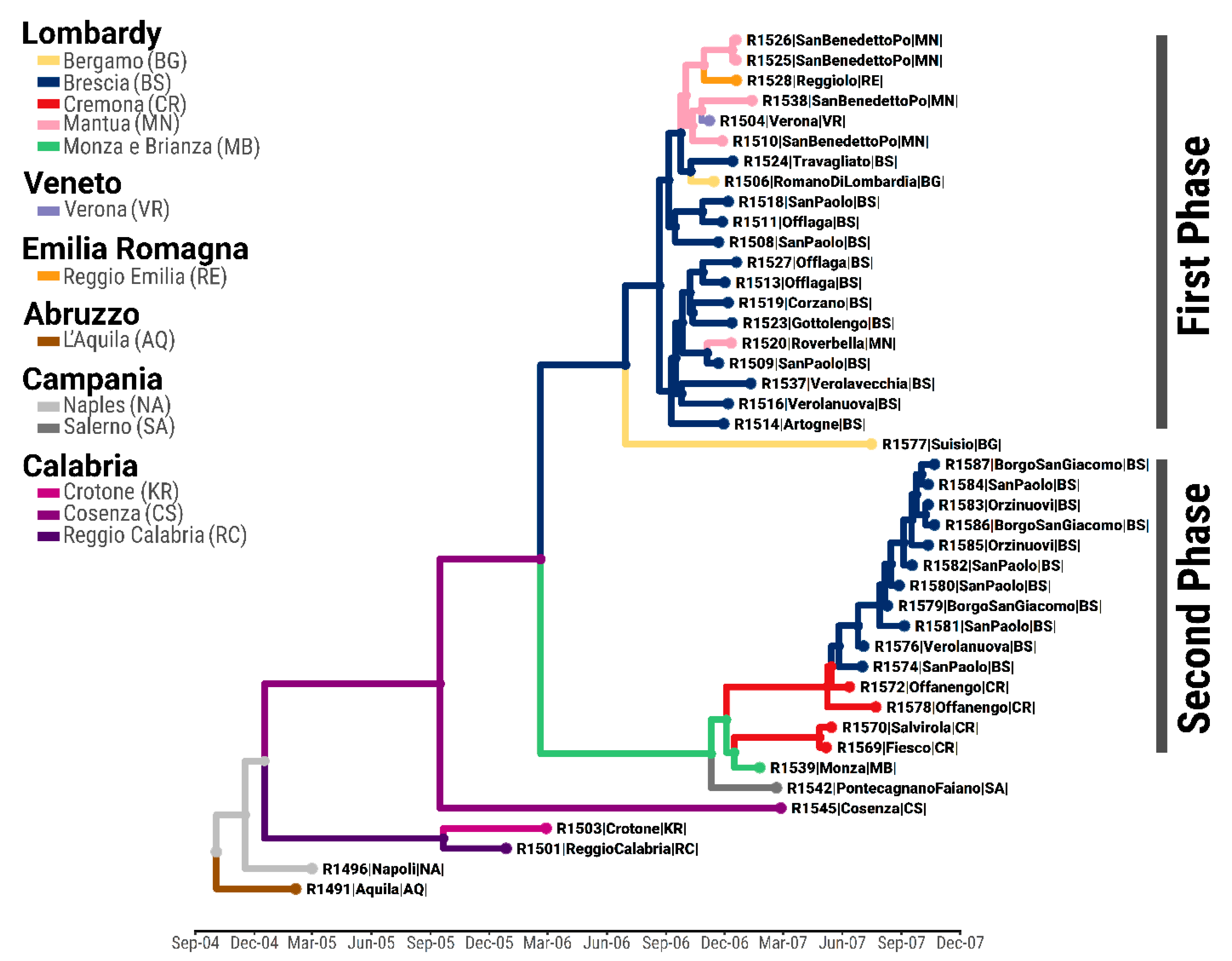
3.1. Phylogenetic and Phylogeography Analyses
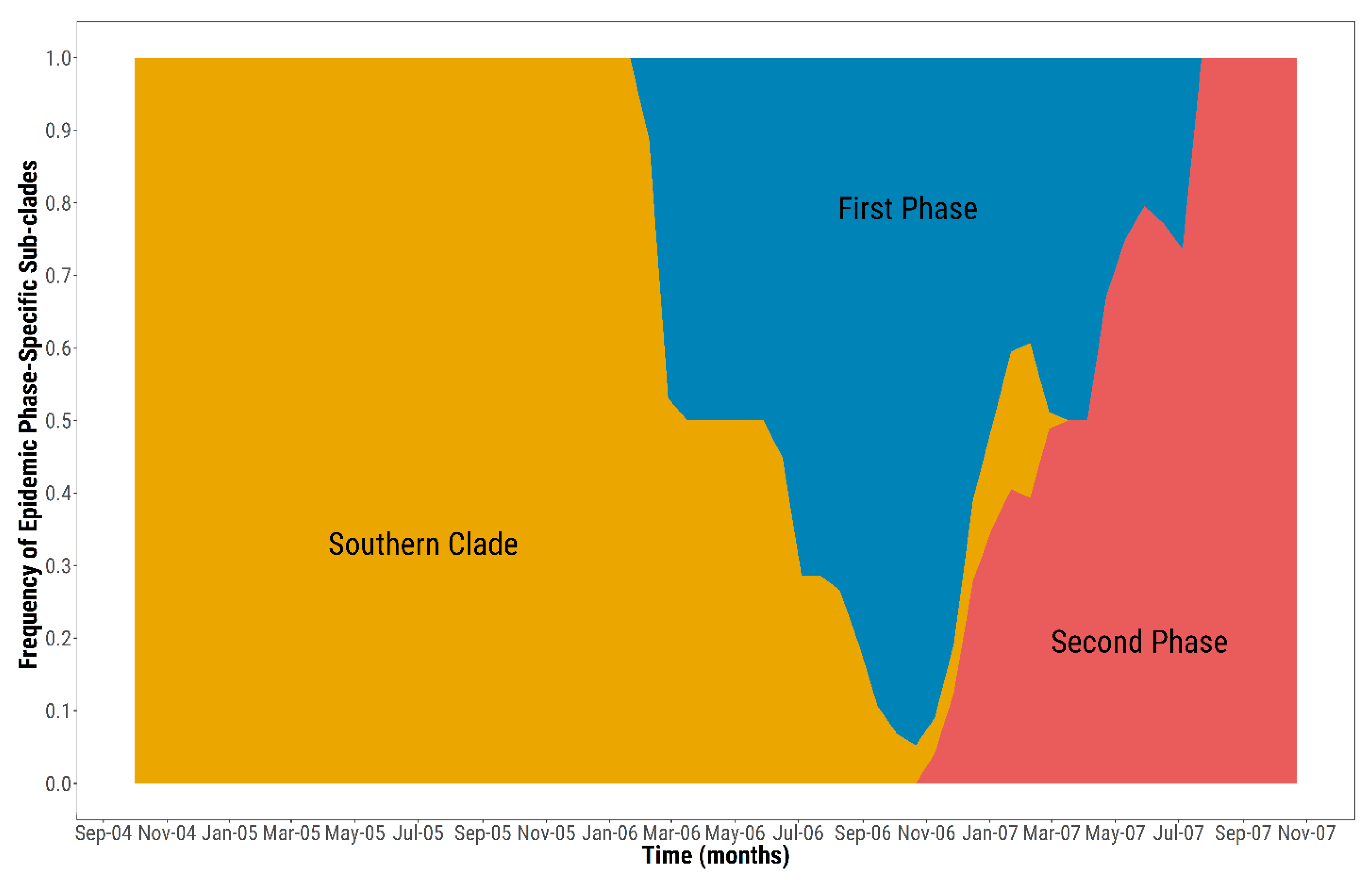
3.2. Ancestry of the First Epidemic Phase
3.3. Ancestry of the Second Epidemic Phase
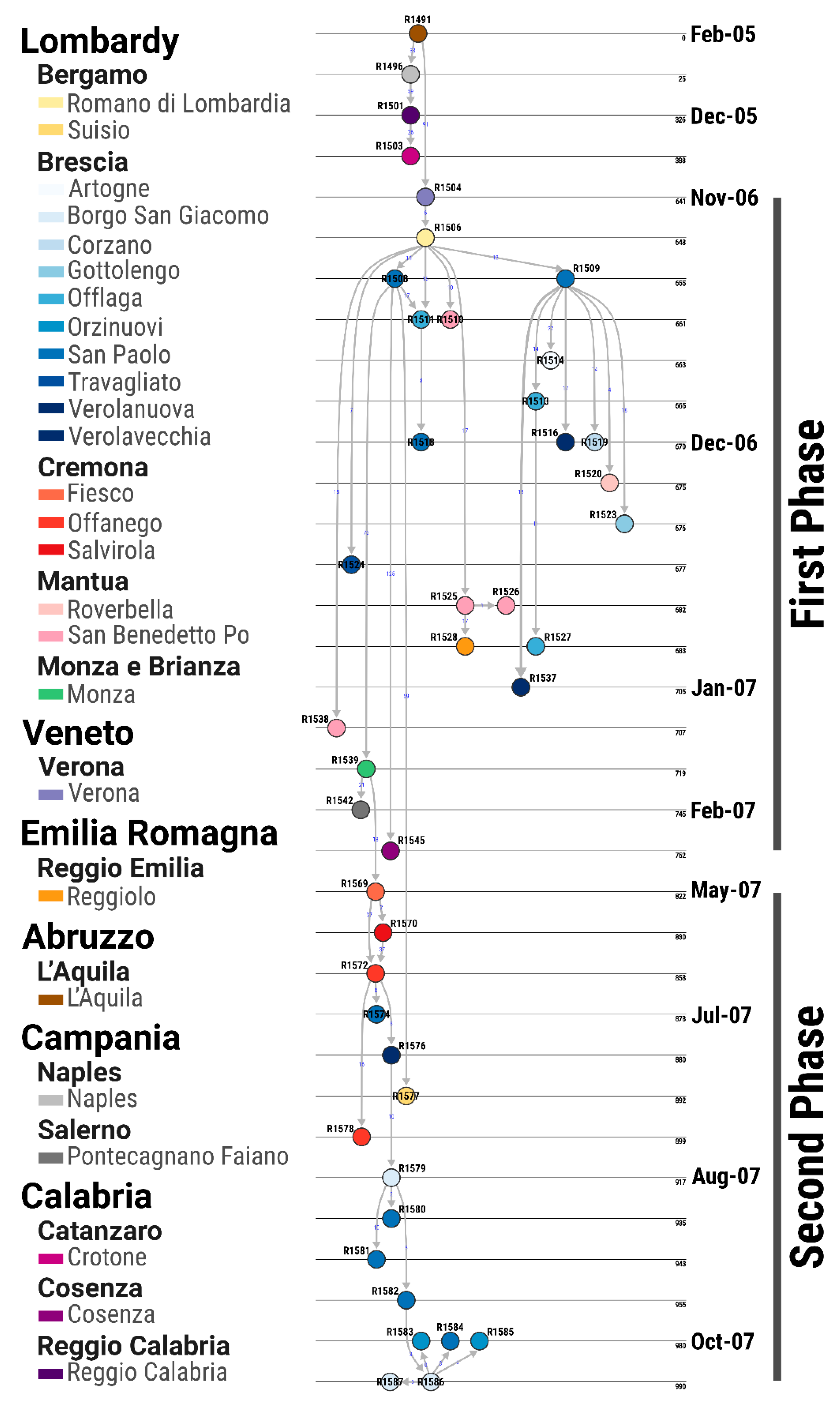
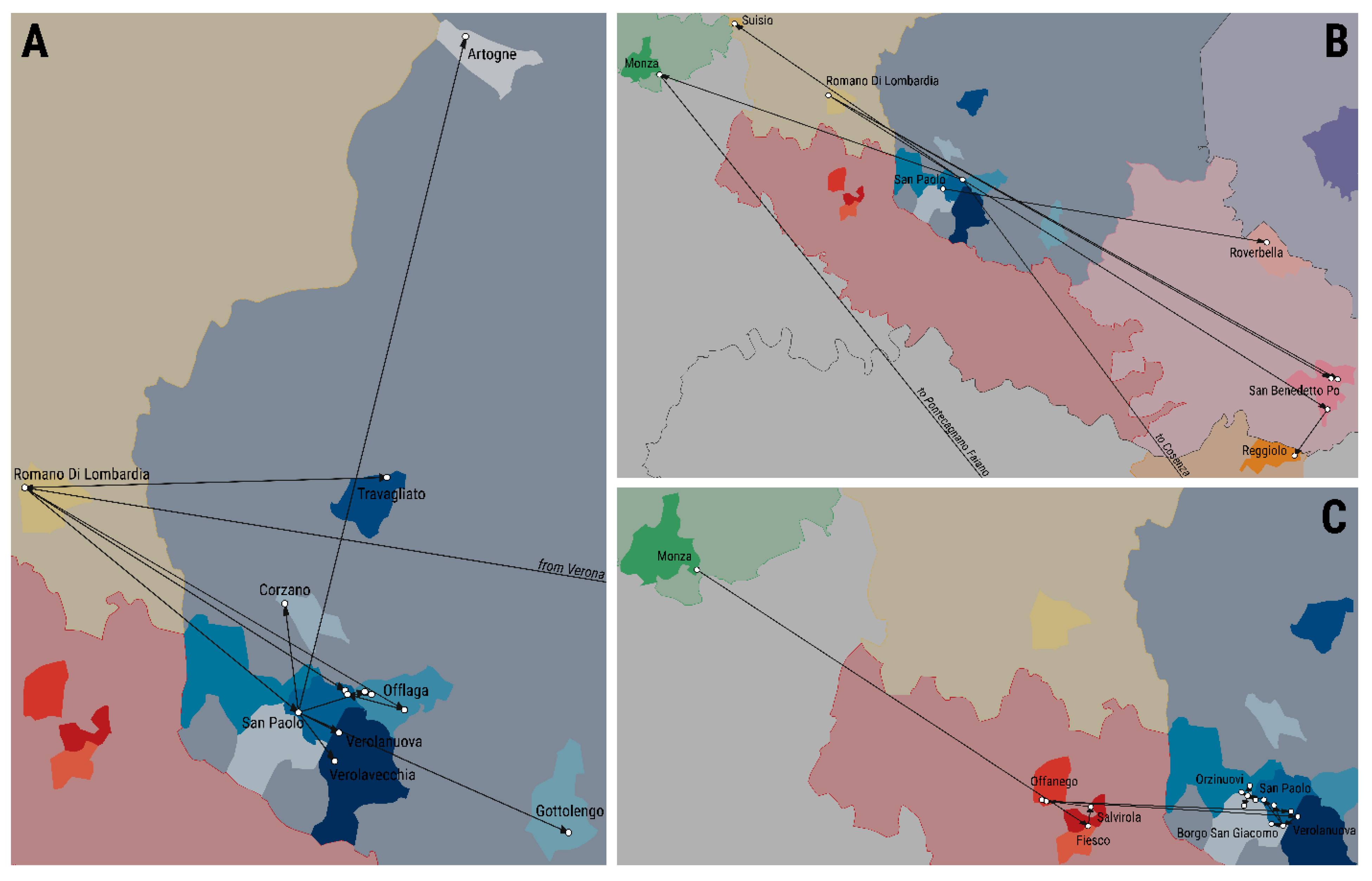
3.4. Transmission Tree Structure and Epidemiological Features of the Epidemic
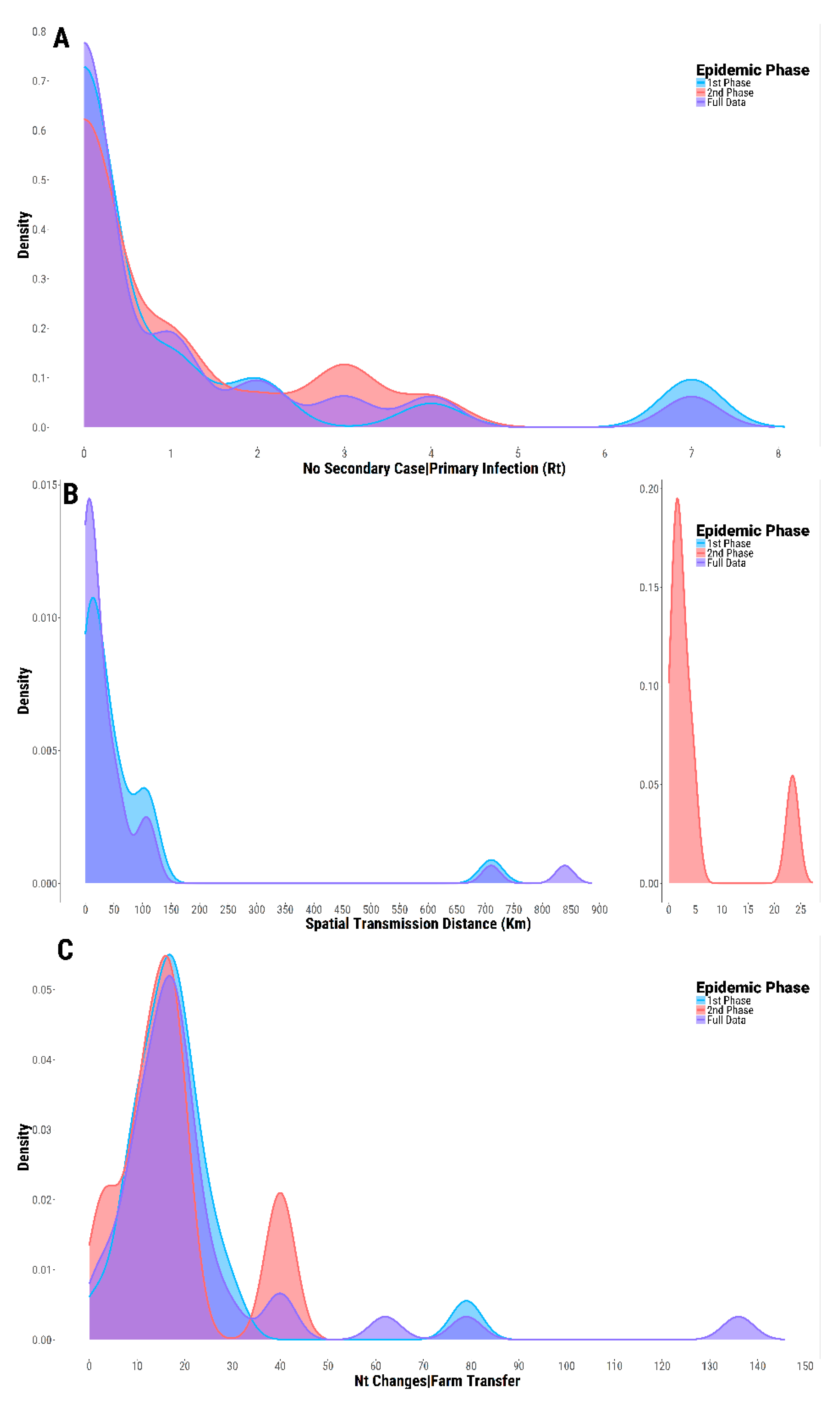
3.5. Case-Reproduction Ratio and Spatial Connectivity
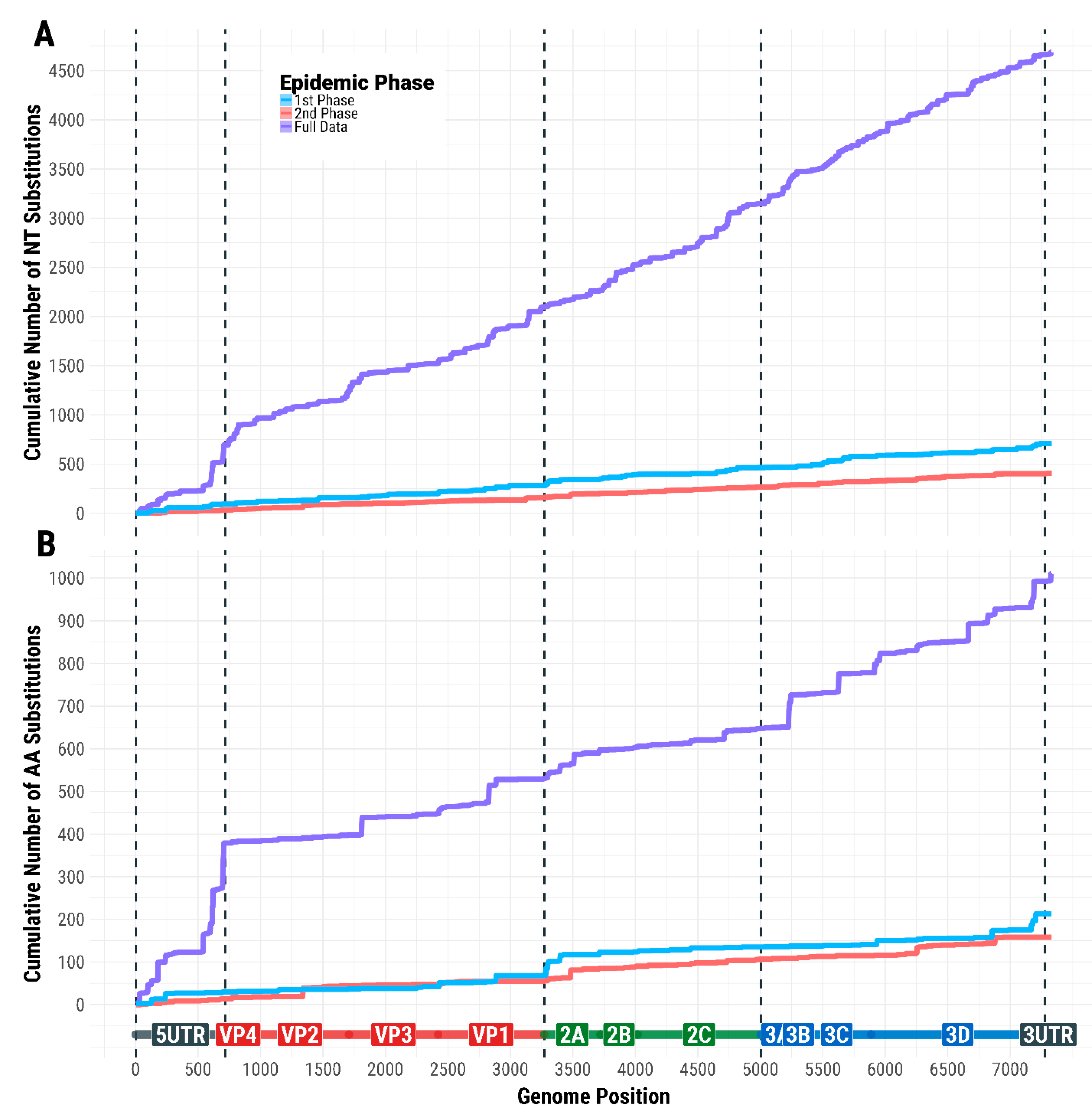
3.6. Evolutionary Profile of SVDV Transmissions
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nardelli, L.; Lodetti, E.; Galandi, G.L.; Burrows, R.; Goodridge, D.; Brown, F.; Cartwright, B. A Foot and Mouth Disease Syndrome in Pigs caused by an Enterovirus. Nat. Cell Biol. 1968, 219, 1275–1276. [Google Scholar] [CrossRef] [PubMed]
- Dekker, A.; Moonen, P.; De Boer-Luijtze, E.; Terpstra, C. Pathogenesis of swine vesicular disease after exposure of pigs to an infected environment. Vet. Microbiol. 1995, 45, 243–250. [Google Scholar] [CrossRef]
- Bellini, S.; Alborali, L.; Zanardi, G.; Bonazza, V.; Brocchi, E. Swine vesicular disease in northern Italy: Diffusion through densely populated pig areas. Rev. Sci. Tech. 2010, 29, 639–648. [Google Scholar] [CrossRef] [PubMed]
- Bellini, S.; Santucci, U.; Zanardi, G.; Brocchi, E.; Marabelli, R. Swine vesicular disease surveillance and eradication activities in Italy. Rev. Sci. Tech. OIE 2007, 26, 585–593. [Google Scholar] [CrossRef]
- Brocchi, E.; Zhang, G.; Knowles, N.J.; Wilsden, G.; McCauley, J.W.; Marquardt, O.; Ohlinger, V.F.; De Simone, F. Molecular epidemiology of recent outbreaks of swine vesicular disease: Two genetically and antigenically distinct variants in Europe, 1987–94. Epidemiol. Infect. 1997, 118, 51–61. [Google Scholar] [CrossRef]
- Mowat, G.N.; Darbyshire, J.H.; Huntley, J.F. Differentiation of a vesicular disease of pigs in Hong Kong from foot-and-mouth disease. Vet. Rec. 1972, 90, 618–621. [Google Scholar] [CrossRef]
- Bruhn, C.A.W.; Nielsen, S.C.A.; Samaniego, J.A.; Wadsworth, J.; Knowles, N.J.; Gilbert, M. Viral meningitis epidemics and a single, recent, recombinant and anthroponotic origin of swine vesicular disease virus. Evol. Med. Public Health 2015, 2015, 289–303. [Google Scholar] [CrossRef] [Green Version]
- Knowles, N.J.; Wilsden, G.; Reid, S.M.; Ferris, N.P.; King, D.P.; Paton, D.J.; Fevereiro, M.; Brocchi, E. Reappearance of swine vesicular disease virus in Portugal. Vet. Rec. 2007, 161, 71. [Google Scholar] [CrossRef]
- Pezzoni, G.; Benedetti, D.; Bregoli, A.; Barbieri, I.; Foglia, E.A.; Grazioli, S.; Brocchi, E. Diagnostic Performances of Different Genome Amplification Assays for the Detection of Swine Vesicular Disease Virus in Relation to Genomic Lineages That Circulated in Italy. Viruses 2020, 12, 1336. [Google Scholar] [CrossRef]
- Ministero delle Politiche Agricole Alimentari e forestali; MIPAAF Piano Nazionale di Eradicazione e Sorveglianza della Malattia Vescicolare e Sorveglianza della Peste Suina Classica. Available online: https://www.politicheagricole.it/flex/cm/pages/ServeBLOB.php/L/IT/IDPagina/2720 (accessed on 16 June 2021).
- Taylor, L. Swine Vesicular Disease. In Manual of Diagnostic Tests and Vaccines for Terrestrial Animals, 8th ed.; OIE Press: Paris, France, 2018; pp. 1608–1617. [Google Scholar]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Tavaré, S. Some probabilistic and statistical problems in the analysis of DNA sequences. Lect. Math. Life Sci. 1986, 17, 57–86. [Google Scholar]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. j Model Test 2: More models, new heuristics and parallel computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guindon, S.; Gascuel, O. A Simple, Fast, and Accurate Algorithm to Estimate Large Phylogenies by Maximum Likelihood. Syst. Biol. 2003, 52, 696–704. [Google Scholar] [CrossRef] [Green Version]
- Drummond, A.J.; Ho, S.Y.W.; Phillips, M.J.; Rambaut, A. Relaxed Phylogenetics and Dating with Confidence. PLoS Biol. 2006, 4, e88. [Google Scholar] [CrossRef] [PubMed]
- Drummond, A.J.; Rambaut, A.; Shapiro, B.; Pybus, O.G. Bayesian Coalescent Inference of Past Population Dynamics from Molecular Sequences. Mol. Biol. Evol. 2005, 22, 1185–1192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- A Suchard, M.; Lemey, P.; Baele, G.; Ayres, D.L.; Drummond, A.J.; Rambaut, A. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 2018, 4, vey016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemey, P.; Suchard, M.; Rambaut, A. Reconstructing the initial global spread of a human influenza pandemicA Bayesian spatial-temporal model for the global spread of H1N1pdm. PLoS Curr. 2009, 1, RRN1031. [Google Scholar] [CrossRef]
- Minin, V.N.; Suchard, M.A. Counting labeled transitions in continuous-time Markov models of evolution. J. Math. Biol. 2007, 56, 391–412. [Google Scholar] [CrossRef] [Green Version]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; A Suchard, M. Posterior Summarization in Bayesian Phylogenetics Using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing, version 4.0.4; R Foundation for Statistical Computing: Vienna, Austria, 2021. [Google Scholar]
- Yu, G.; Smith, D.K.; Zhu, H.; Guan, Y.; Lam, T.T. ggtree: An r package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol. Evol. 2016, 8, 28–36. [Google Scholar] [CrossRef]
- Campbell, F.; Didelot, X.; Fitzjohn, R.; Ferguson, N.; Cori, A.; Jombart, T. outbreaker2: A modular platform for outbreak reconstruction. BMC Bioinform. 2018, 19, 363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bøtner, A.; Broom, D.; Doherr, M.G.; Domingo, M.; Hartung, J.; Keeling, L.; Koenen, F.; More, S.; Morton, D.; Oltenacu, P.; et al. EFSA Panel Animal Health Welfare, Scientific Opinion on Swine Vesicular Disease and Vesicular Stomatitis. EFSA J. 2012, 10, 2631–2728. [Google Scholar]
- Brandes, U.; Wagner, D. Visone-Analysis and visualization of social network. In Graph Drawing Software; Junger, M., Mutzel, P., Eds.; Springer: Heidelberg/Berlin, Germany, 2004; pp. 321–340. [Google Scholar]
- Pond, S.L.K.; Poon, A.F.Y.; Velazquez, R.; Weaver, S.; Hepler, N.L.; Murrell, B.; Shank, S.D.; Magalis, B.R.; Bouvier, D.; Nekrutenko, A.; et al. HyPhy 2.5—A Customizable Platform for Evolutionary Hypothesis Testing Using Phylogenies. Mol. Biol. Evol. 2020, 37, 295–299. [Google Scholar] [CrossRef]
- Pond, S.L.K.; Wisotsky, S.R.; Escalante, A.; Magalis, B.R.; Weaver, S. Contrast-FEL—A Test for Differences in Selective Pressures at Individual Sites among Clades and Sets of Branches. Mol. Biol. Evol. 2021, 38, 1184–1198. [Google Scholar] [CrossRef]
- Nassuato, C.; Boender, G.J.; Eblé, P.L.; Alborali, L.; Bellini, S.; Hagenaars, T.J. Spatial Transmission of Swine Vesicular Disease Virus in the 2006–2007 Epidemic in Lombardy. PLOS ONE 2013, 8, e62878. [Google Scholar] [CrossRef] [Green Version]
- Zwart, M.P.; Elena, S.F. Matters of Size: Genetic Bottlenecks in Virus Infection and Their Potential Impact on Evolution. Annu. Rev. Virol. 2015, 2, 161–179. [Google Scholar] [CrossRef] [PubMed]
- McCrone, J.T.; Lauring, A.S. Genetic bottlenecks in intraspecies virus transmission. Curr. Opin. Virol. 2018, 28, 20–25. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sequence ID | Municipality (Province) | Region | Sampling Date | Source ID | Material Samples | GenBank Accession No | SRA Accession No |
|---|---|---|---|---|---|---|---|
| R1491 | Villa Sant’Angelo (AQ) | Abruzzo | 2 April 2005 | Faeces | MW984391 | SAMN19667915 | |
| R1496 | Torre del Greco (NA) | Campania | 1 March 2005 | Faeces | MW984392 | SAMN19667916 | |
| R1501 | Locri (RC) | Calabria | 27 December 2005 | Faeces | MW984393 | SAMN19667917 | |
| R1503 | Casabona (KR) | Calabria | 27 February 2006 | Faeces | MW984394 | SAMN19667918 | |
| R1504 | Verona (VR) | Veneto | 7 November 2006 | * | Faeces | MW984370 | SAMN19667894 |
| R1506 | Romano Di Lombardia (BG) | Lombardy | 14 November 2006 | R1504 | Faeces | MW984371 | SAMN19667895 |
| R1508 | San Paolo (BS) | Lombardy | 21 November 2006 | R1504 | Faeces | MW984372 | SAMN19667896 |
| R1509 | San Paolo (BS) | Lombardy | 21 November 2006 | R1504 | Faeces | MW984373 | SAMN19667897 |
| R1510 | San Benedetto Po (MN) | Lombardy | 27 November 2006 | R1504 | Epithelium | MW984374 | SAMN19667898 |
| R1511 | Offlaga (BS) | Lombardy | 27 November 2006 | R1508 | Epithelium | MW984375 | SAMN19667899 |
| R1513 | Offlaga (BS) | Lombardy | 1 December 2006 | R1508 | Epithelium | MW984376 | SAMN19667900 |
| R1514 | Artogne (BS) | Lombardy | 29 November 2006 | R1508 | Faeces | MW984377 | SAMN19667901 |
| R1516 | Verolanuova (BS) | Lombardy | 6 December 2006 | R1513 | Epithelium | MW984378 | SAMN19667902 |
| R1518 | San Paolo (BS) | Lombardy | 6 December 2006 | R1508/R1509 | Faeces | MW984379 | SAMN19667903 |
| R1519 | Corzano (BS) | Lombardy | 6 December 2006 | R1513 | Faeces | MW984380 | SAMN19667904 |
| R1520 | Roverbella (MN) | Lombardy | 11 December 2006 | Faeces | MW984381 | SAMN19667905 | |
| R1523 | Gottolengo (BS) | Lombardy | 12 December 2006 | R1513 | Faeces | MW984382 | SAMN19667906 |
| R1524 | Travagliato (BS) | Lombardy | 13 December 2006 | * | Faeces | MW984383 | SAMN19667907 |
| R1525 | San Benedetto Po (MN) | Lombardy | 18 December 2006 | * | Epithelium | MW984384 | SAMN19667908 |
| R1526 | San Benedetto Po (MN) | Lombardy | 18 December 2006 | * | Epithelium | MW984385 | SAMN19667909 |
| R1527 | Offlaga (BS) | Lombardy | 19 December 2006 | R1513 | Epithelium | MW984386 | SAMN19667910 |
| R1528 | Reggiolo (RE) | Emilia Romagna | 19 December 2006 | * | Faeces | MW984387 | SAMN19667911 |
| R1537 | Verolavecchia (BS) | Lombardy | 10 January 2007 | R1518 | Epithelium | MW984388 | SAMN19667912 |
| R1538 | San Benedetto Po (MN) | Lombardy | 12 January 2007 | R1510 | Epithelium | MW984389 | SAMN19667913 |
| R1539 | Monza (MB) | Lombardy | 24 January 2007 | * | Faeces | MW984395 | SAMN19667919 |
| R1542 | Pontecagnano Faiano (SA) | Campania | 19 February 2007 | Faeces | MW984396 | SAMN19667920 | |
| R1545 | Cosenza (CS) | Calabria | 26 February 2007 | Faeces | MW984397 | SAMN19667921 | |
| R1569 | Fiesco (CR) | Lombardy | 7 May 2007 | Epithelium | MW984398 | SAMN19667922 | |
| R1570 | Salvirola (CR) | Lombardy | 15 May 2007 | R1569 | Epithelium | MW984399 | SAMN19667923 |
| R1572 | Offanengo (CR) | Lombardy | 12 June 2007 | * | Epithelium | MW984400 | SAMN19667924 |
| R1574 | San Paolo (BS) | Lombardy | 2 July 2007 | * | Epithelium | MW984401 | SAMN19667925 |
| R1576 | Verolanuova (BS) | Lombardy | 4 July 2007 | R1574 | Epithelium | MW984402 | SAMN19667926 |
| R1577 | Suisio (BG) | Lombardy | 16 July 2007 | * | Faeces | MW984390 | SAMN19667914 |
| R1578 | Offanengo (CR) | Lombardy | 23 July 2007 | R1572 | Faeces | MW984403 | SAMN19667927 |
| R1579 | Borgo San Giacomo (BS) | Lombardy | 10 August 2007 | * | Epithelium | MW984404 | SAMN19667928 |
| R1580 | San Paolo (BS) | Lombardy | 28 August 2007 | * | Epithelium | MW984405 | SAMN19667929 |
| R1581 | San Paolo (BS) | Lombardy | 5 September 2007 | * | Epithelium | MW984406 | SAMN19667930 |
| R1582 | San Paolo (BS) | Lombardy | 17 September 2007 | * | Epithelium | MW984407 | SAMN19667931 |
| R1583 | Orzinuovi (BS) | Lombardy | 12 October 2007 | * | Epithelium | MW984408 | SAMN19667932 |
| R1584 | San Paolo (BS) | Lombardy | 12 October 2007 | * | Epithelium | MW984409 | SAMN19667933 |
| R1585 | Orzinuovi (BS) | Lombardy | 12 October 2007 | * | Epithelium | MW984410 | SAMN19667934 |
| R1586 | Borgo San Giacomo (BS) | Lombardy | 22 October 2007 | R1583 | Epithelium | MW984411 | SAMN19667935 |
| R1587 | Borgo San Giacomo (BS) | Lombardy | 22 October 2007 | R1583 | Epithelium | MW984412 | SAMN19667936 |
| Region | Residue | α | β (Background) | β (First Phase) | β (Second Phase) | p | q | Pairwise Test |
|---|---|---|---|---|---|---|---|---|
| VP2 | 143 | 0 | 0 | 0 | 23.02 | 0.106 | 1 | B |
| 2A | 999 | 0.073 | 0 | 0 | 15.123 | 0.101 | 1 | BS |
| 2C | 1332 | 0 | 0 | 0 | 19.789 | 0.123 | 1 | B |
| 3D | 2054 | 0 | 0 | 57.206 | 0 | 0.013 | 1 | BF, FS |
| Region | Residue | Composition | Root | Inferred Substitution | BF | |
|---|---|---|---|---|---|---|
| 2A | 861 | F7, V73 | V | V  F7 F7 | 1378.13 | |
| 2C | 1242 | F5, I75 | I | I F5 | 360.36 | |
| 3D | 2054 | A21, V59 | V | A V2, | V A3 | 5343.70 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pezzoni, G.; Bregoli, A.; Chiapponi, C.; Grazioli, S.; Di Nardo, A.; Brocchi, E. Retrospective Characterization of the 2006–2007 Swine Vesicular Disease Epidemic in Northern Italy by Whole Genome Sequence Analysis. Viruses 2021, 13, 1186. https://0-doi-org.brum.beds.ac.uk/10.3390/v13071186
Pezzoni G, Bregoli A, Chiapponi C, Grazioli S, Di Nardo A, Brocchi E. Retrospective Characterization of the 2006–2007 Swine Vesicular Disease Epidemic in Northern Italy by Whole Genome Sequence Analysis. Viruses. 2021; 13(7):1186. https://0-doi-org.brum.beds.ac.uk/10.3390/v13071186
Chicago/Turabian StylePezzoni, Giulia, Arianna Bregoli, Chiara Chiapponi, Santina Grazioli, Antonello Di Nardo, and Emiliana Brocchi. 2021. "Retrospective Characterization of the 2006–2007 Swine Vesicular Disease Epidemic in Northern Italy by Whole Genome Sequence Analysis" Viruses 13, no. 7: 1186. https://0-doi-org.brum.beds.ac.uk/10.3390/v13071186