
The Bacteriophage pEp_SNUABM_08 Is a Novel Singleton Siphovirus with High Host Specificity for Erwinia pyrifoliae

 ,
,  , ,
, , 
Abstract
:1. Introduction
2. Material and Methods
2.1. Phage Isolation
2.2. Phage Propagation and Purification
2.3. Transmission Electron Microscopy
2.4. Host Range Analysis
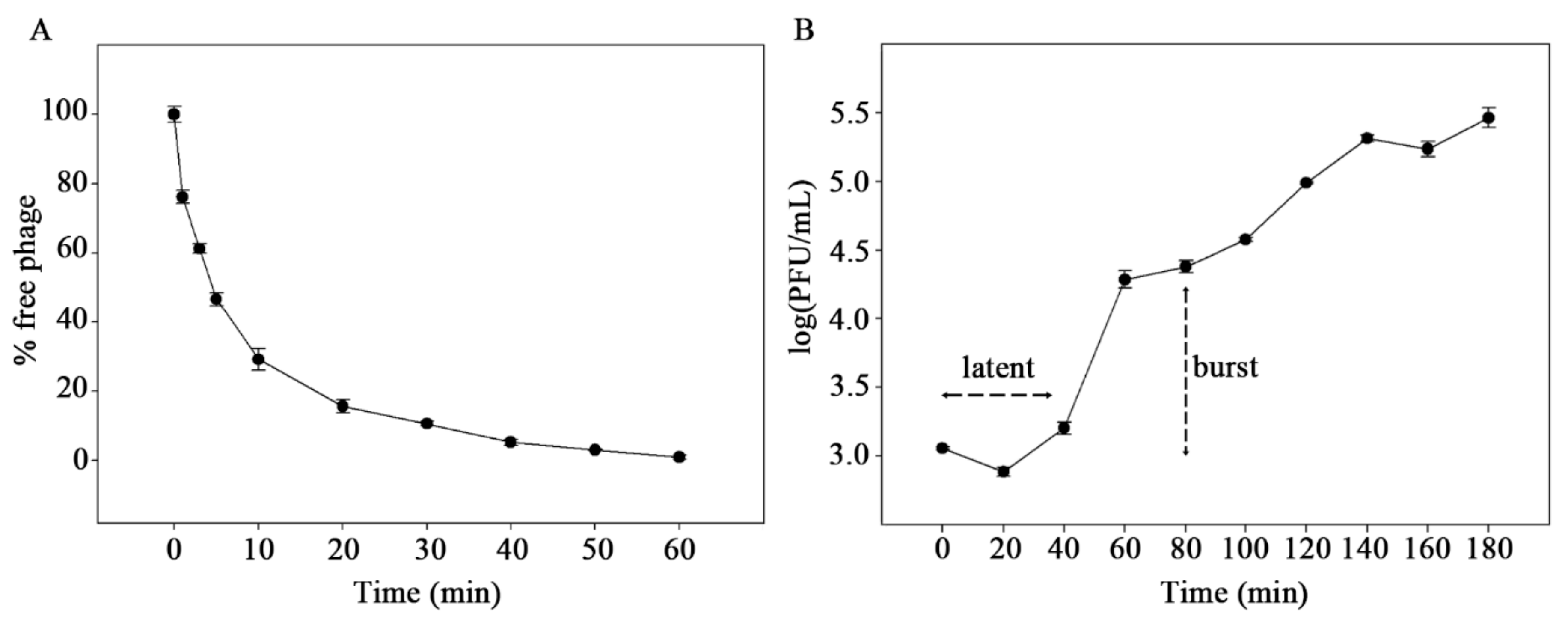
2.5. Adsorption and One-Step Growth Curve
2.6. DNA Extraction and Sequencing
2.7. Genome Analysis
2.8. Proteome Analysis
3. Results
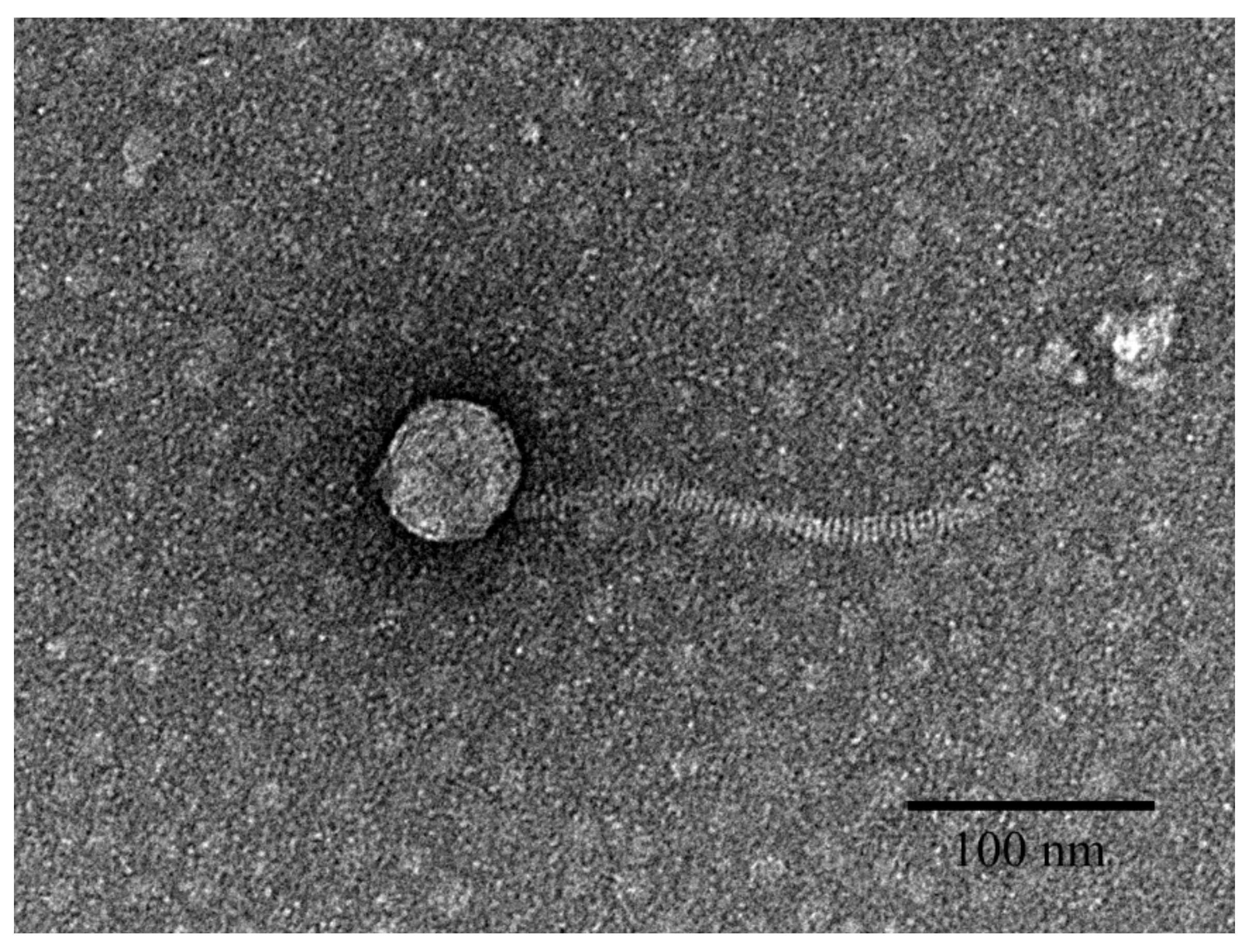
3.1. Biological Analysis of pEp_SNUABM_08
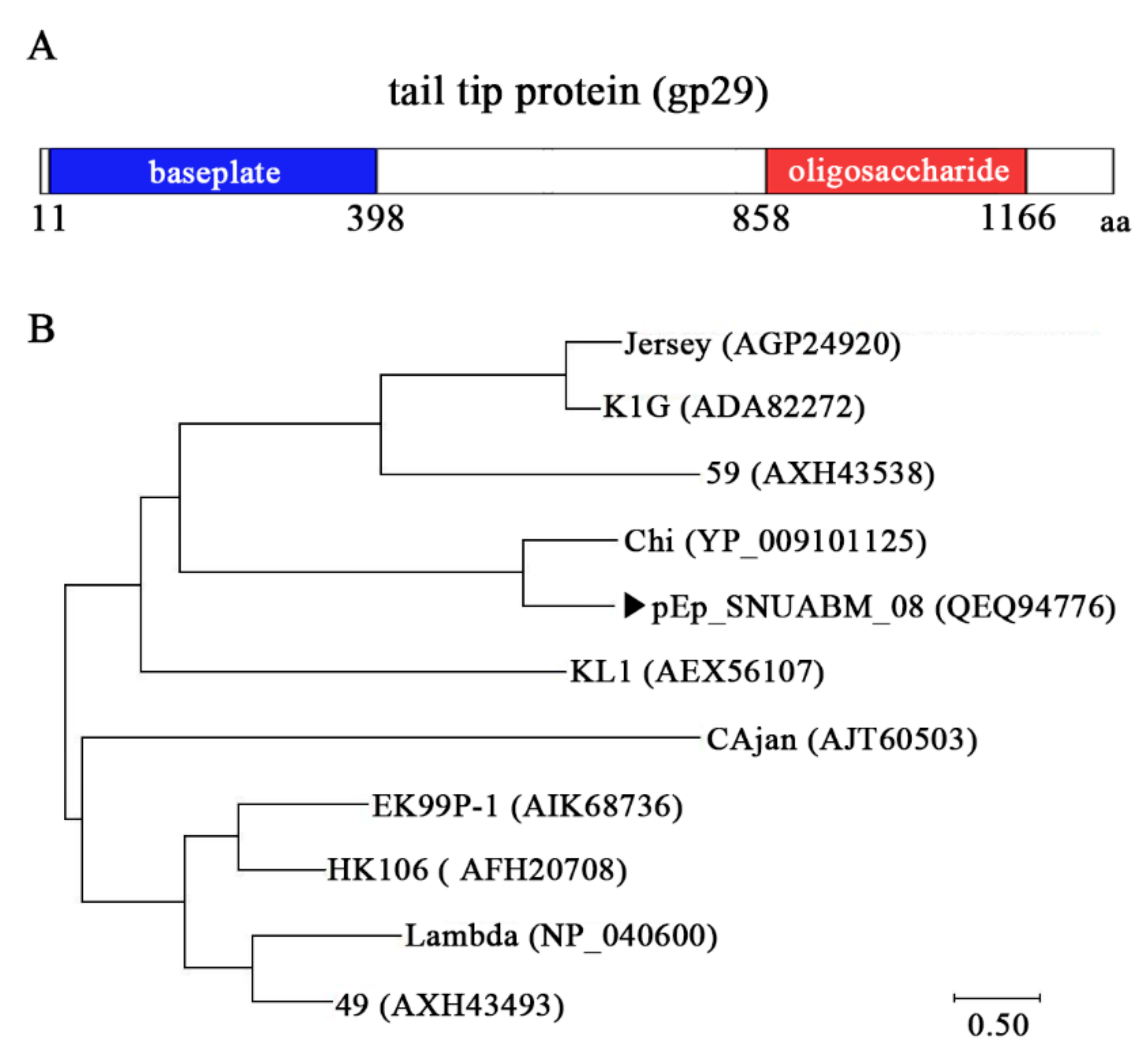
3.2. Protein Analysis of pEp_SNUABM_08
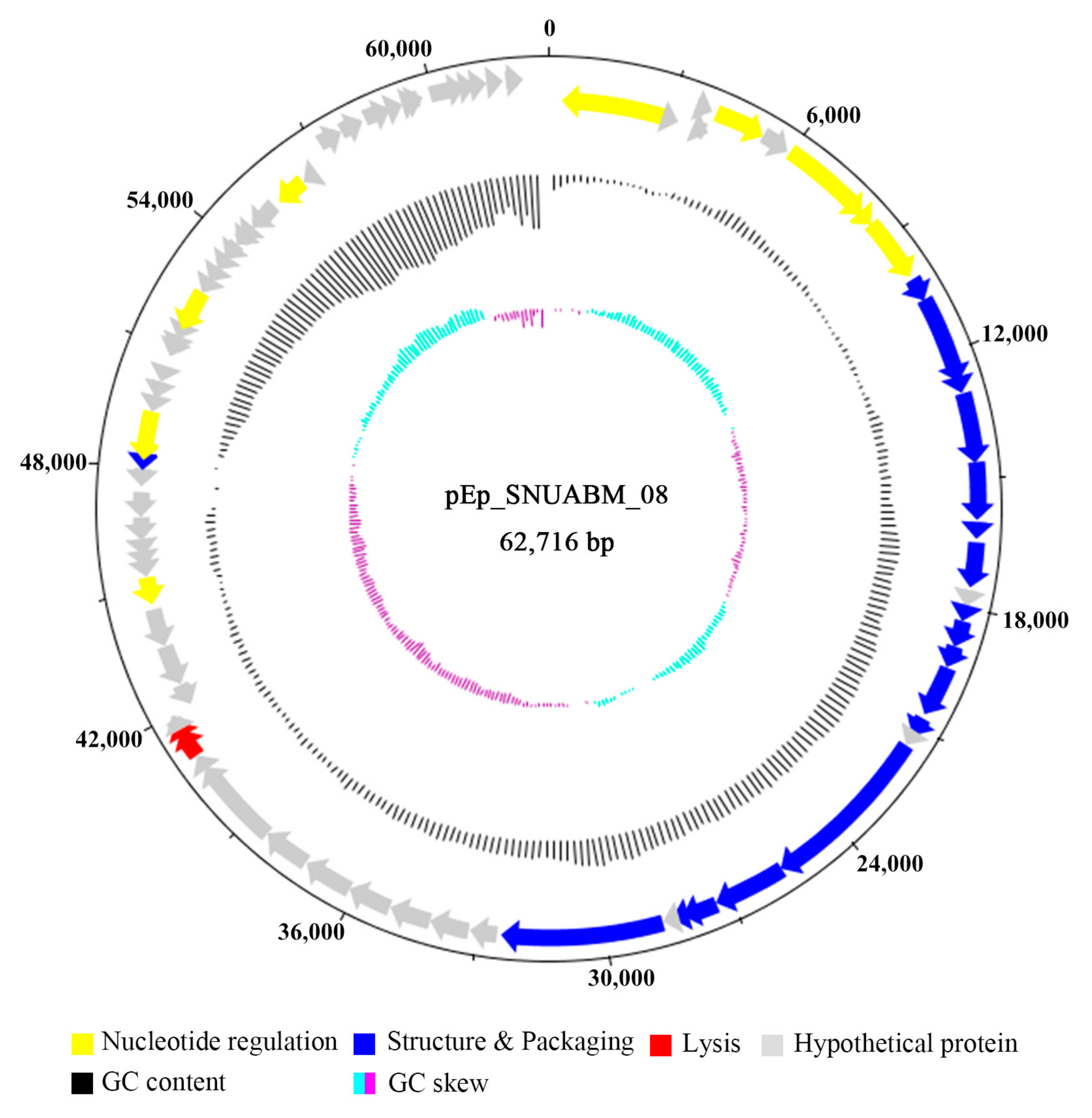
3.3. Genomic Analysis of pEp_SNUABM_08
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kim, W.S.; Gardan, L.; Rhim, S.L.; Geider, K. Erwinia pyrifoliae sp. nov., a novel pathogen that affects Asian pear trees (Pyrus pyrifolia Nakai). Int. J. Syst. Evol. Microbiol. 1999, 49, 899–906. [Google Scholar] [CrossRef]
- Shrestha, R.; Koo, J.H.; Park, D.H.; Hwang, I.; Hur, J.H.; Lim, C.K. Erwinia pyrifoliae, a causal endemic pathogen of shoot blight of Asian pear. Plant Pathol. J. 2003, 9, 294–300. [Google Scholar] [CrossRef]
- Kim, W.S.; Jock, S.; Paulin, J.P.; Rhim, S.L.; Geider, K. Molecular detection and differentiation of Erwinia pyrifoliae and host range analysis of the Asian pear pathogen. Plant Dis. 2001, 85, 1183–1188. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Qi, M. Comparative genomics of Erwinia amylovora and related Erwinia species—What do we learn? Genes 2011, 2, 627–639. [Google Scholar] [CrossRef] [PubMed]
- Gehring, I.; Geider, K. Differentiation of Erwinia amylovora and Erwinia pyrifoliae strains with single nucleotide polymorphisms and by synthesis of dihydrophenylalanine. Curr. Microbiol. 2012, 65, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.M.; Ko, S.; Kim, D.; Oh, C.S. Comparative genomic analysis reveals an evolutionary trace in the genome of Erwinia pyrifoliae, a black shoot blight pathogen. In Proceedings of the 2019 KSBB Fall Meeting and International Symposium, Daegu, Korea, 9–11 October 2019; p. 400. [Google Scholar]
- Park, D.H.; Lee, Y.G.; Kim, J.S.; Cha, J.S.; Oh, C.S. Current status of fire blight caused by Erwinia amylovora and action for its management in Korea. J. Plant Pathol. 2017, 99, 59–63. [Google Scholar]
- Lee, M.H.; Ji, S.; Ham, H.H.; Kong, G.H.; Park, D.S.; Lee, Y.H. First report of fire blight of Apricot (Prunus armeniaca) caused by Erwinia amylovora in Korea. Plant Dis. 2020. [Google Scholar] [CrossRef]
- Bertozzi Silva, J.; Storms, Z.; Sauvageau, D. Host receptors for bacteriophage adsorption. FEMS Microbiol. Lett. 2016. [Google Scholar] [CrossRef]
- Santos, S.B.; Azeredo, J. Bacteriophage-based biotechnological applications. Viruses 2019, 11, 737. [Google Scholar] [CrossRef] [PubMed]
- Melo, L.D.; Oliveira, H.; Pires, D.P.; Dabrowska, K.; Azeredo, J. Phage therapy efficacy: A review of the last 10 years of preclinical studies. Crit. Rev. Microbiol. 2020, 46, 78–99. [Google Scholar] [CrossRef]
- Kakasis, A.; Panitsa, G. Bacteriophage therapy as an alternative treatment for human infections. A comprehensive review. Int. J. Antimicrob. Agents 2019, 53, 16–21. [Google Scholar] [CrossRef] [PubMed]
- Meile, S.; Kilcher, S.; Loessner, M.J.; Dunne, M. Reporter phage-based detection of bacterial pathogens: Design guidelines and recent developments. Viruses 2020, 12, 944. [Google Scholar] [CrossRef]
- Nagy, J.; Király, L.; Schwarczinger, I. Phage therapy for plant disease control with a focus on fire blight. Open Life Sci. 2012, 7, 1–12. [Google Scholar] [CrossRef]
- Boulé, J.; Sholberg, P.L.; Lehman, S.M.; O’gorman, D.T.; Svircev, A.M. Isolation and characterization of eight bacteriophages infecting Erwinia amylovora and their potential as biological control agents in British Columbia, Canada. Can. J. Plant Pathol. 2011, 33, 308–317. [Google Scholar] [CrossRef]
- Schwarczinger, I.; Nagy, J.K.; Künstler, A.; Szabó, L.; Geider, K.; Király, L.; Pogány, M. Characterization of Myoviridae and Podoviridae family bacteriophages of Erwinia amylovora from Hungary-potential of application in biological control of fire blight. Eur. J. Plant Pathol. 2017, 149, 639–652. [Google Scholar] [CrossRef]
- Schofield, D.A.; Bull, C.T.; Rubio, I.; Wechter, W.P.; Westwater, C.; Molineux, I.J. Development of an engineered bioluminescent reporter phage for detection of bacterial blight of crucifers. Appl. Environ. Microbiol. 2012, 78, 3592–3598. [Google Scholar] [CrossRef] [PubMed]
- Van der Merwe, R.G.; Van Helden, P.D.; Warren, R.M.; Sampson, S.L.; Van Pittius, N.G. Phage-based detection of bacterial pathogens. Analyst 2014, 139, 2617–2626. [Google Scholar] [CrossRef]
- Vu, N.T.; Oh, C.S. Bacteriophage usage for bacterial disease management and diagnosis in plants. Plant Pathol. J. 2020, 36, 204. [Google Scholar] [CrossRef]
- Born, Y.; Fieseler, L.; Thöny, V.; Leimer, N.; Duffy, B.; Loessner, M.J. Engineering of bacteriophages Y2::dpoL1-C and Y2::luxAB for efficient control and rapid detection of the fire blight pathogen, Erwinia amylovora. Appl. Environ. Microbiol. 2017, 83, e00341-17. [Google Scholar] [CrossRef]
- Parmar, K.M.; Gaikwad, S.L.; Dhakephalkar, P.K.; Kothari, R.; Singh, R.P. Intriguing interaction of bacteriophage-host association: An understanding in the era of omics. Front. Microbiol. 2017, 8, 559. [Google Scholar] [CrossRef]
- Li, M.; Wang, Y.; Li, F.; Zhao, Y.; Liu, M.; Zhang, S.; Bin, Y.; Smith, A.I.; Webb, G.; Li, J.; et al. A deep learning-based method for identification of bacteriophage-host interaction. IEEE/ACM Trans. Comput. Biol. Bioinform. 2020. [Google Scholar] [CrossRef]
- Kim, S.G.; Lee, S.B.; Giri, S.S.; Kim, H.J.; Kim, S.W.; Kwon, J.; Park, J.; Roh, E.; Park, S.C. Characterization of novel Erwinia amylovora jumbo bacteriophages from Eneladusvirus genus. Viruses 2020, 12, 1373. [Google Scholar] [CrossRef] [PubMed]
- Besarab, N.V.; Akhremchuk, A.E.; Zlatohurska, M.A.; Romaniuk, L.V.; Valentovich, L.N.; Tovkach, F.I.; Lagonenko, A.L.; Evtushenkov, A.N. Isolation and characterization of Hena1–a novel Erwinia amylovora bacteriophage. FEMS Microbiol. Lett. 2020, 367, fnaa070. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.; Pielstick, B.A.; Bell, K.A.; Nieman, T.B.; Stubbs, O.A.; Yeates, E.L.; Baltrus, D.A.; Grose, J.H. A novel, highly related jumbo family of bacteriophages that were isolated against Erwinia. Front. Microbiol. 2019, 10, 1533. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.; Berg, J.A.; Beatty, N.J.; Choi, M.C.; Cowger, A.E.; Cozzens, B.J.R.; Duncan, S.G.; Fajardo, C.P.; Ferguson, H.P.; Galbraith, T.; et al. Genome sequences of nine Erwinia amylovora bacteriophages. Microbiol. Resour. Announc. 2019, 7, e00944-18. [Google Scholar]
- Arens, D.K.; Brady, T.S.; Carter, J.L.; Pape, J.A.; Robinson, D.M.; Russell, K.A.; Staley, L.A.; Stettler, J.M.; Tateoka, O.B.; Townsend, M.H.; et al. Characterization of two related Erwinia myoviruses that are distant relatives of the PhiKZ-like jumbo phages. PLoS ONE 2018, 13, e0200202. [Google Scholar] [CrossRef] [PubMed]
- Müller, I.; Lurz, R.; Kube, M.; Quedenau, C.; Jelkmann, W.; Geider, K. Molecular and physiological properties of bacteriophages from North America and Germany affecting the fire blight pathogen Erwinia amylovora. Microb. Biotechnol. 2011, 4, 735–745. [Google Scholar] [CrossRef]
- Lehman, S.M.; Kropinski, A.M.; Castle, A.J.; Svircev, A.M. Complete genome of the broad-host-range Erwinia amylovora phage ΦEa21-4 and its relationship to Salmonella phage Felix O1. Appl. Environ. Microbiol. 2009, 75, 2139–2147. [Google Scholar] [CrossRef]
- Thompson, D.W.; Casjens, S.R.; Sharma, R.; Grose, J.H. Genomic comparison of 60 completely sequenced bacteriophages that infect Erwinia and/or Pantoea bacteria. Virology 2019, 535, 59–73. [Google Scholar] [CrossRef]
- Lu, Z.; Breidt, F., Jr.; Fleming, H.P.; Altermann, E.; Klaenhammer, T.R. Isolation and characterization of a Lactobacillus plantarum bacteriophage, ΦJL-1, from a cucumber fermentation. Int. J. Food Microbiol. 2003, 84, 225–235. [Google Scholar] [CrossRef]
- Kim, S.G.; Jun, J.W.; Giri, S.S.; Yun, S.; Kim, H.J.; Kim, S.W.; Kang, J.W.; Han, S.J.; Jeong, D.; Park, S.C. Isolation and characterisation of pVa-21, a giant bacteriophage with anti-biofilm potential against Vibrio alginolyticus. Sci. Rep. 2019, 9, 1–10. [Google Scholar] [CrossRef]
- Besemer, J.; Lomsadze, A.; Borodovsky, M. GeneMarkS: A self-training method for prediction of gene starts in microbial genomes. Implications for finding sequence motifs in regulatory regions. Nucleic Acids Res. 2001, 29, 2607–2618. [Google Scholar]
- Aziz, R.K.; Bartels, D.; Best, A.A.; De Jong, M.; Dis, T.; Edward, R.A.; Formsma, K.; Gerdes, S.; Glass, E.M.; Kubal, M.; et al. The RAST Server: Rapid annotations using subsystems technology. BMC Genom. 2008, 9, 75. [Google Scholar] [CrossRef]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef] [PubMed]
- Zankari, E.; Hasman, H.; Cosentino, S.; Vestergaard, M.; Rasmussen, S.; Lund, O.; Aarestrup, F.M.; Larsen, M.V. Identification of acquired antimicrobial resistance genes. J. Antimicrob. Chemother. 2012, 67, 2640–2644. [Google Scholar] [CrossRef] [PubMed]
- Joensen, K.G.; Scheutz, F.; Lund, O.; Hasman, H.; Kaas, R.S.; Nielsen, E.M.; Aarestrup, F.M. Real-time whole-genome sequencing for routine typing, surveillance, and outbreak detection of verotoxigenic Escherichia coli. J. Clin. Microbiol. 2014, 52, 1501–1510. [Google Scholar] [CrossRef]
- Turner, D.; Reynolds, D.; Seto, D.; Mahadevan, P. CoreGenes3.5: A webserver for the determination of core genes from sets of viral and small bacterial genomes. BMC Res. Notes 2013, 6, 140. [Google Scholar] [CrossRef]
- Krumsiek, J.; Arnold, R.; Rattei, T. Gepard: A rapid and sensitive tool for creating dotplots on genome scale. Bioinformatics 2007, 23, 1026–1028. [Google Scholar] [CrossRef]
- Sullivan, M.J.; Petty, N.K.; Beatson, S.A. Easyfig: A genome comparison visualizer. Bioinformatics 2011, 27, 1009–1010. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Meier-Kolthoff, J.P.; Göker, M. VICTOR: Genome-based phylogeny and classification of prokaryotic viruses. Bioinformatics 2017, 33, 3396–3404. [Google Scholar] [CrossRef]
- Lim, S.R.; Kim, I.K. New Bacteriophage φEp14. KR20130078092A, 10 July 2013. [Google Scholar]
- Park, J.; Lee, G.M.; Kim, D.; Park, D.H.; Oh, C.S. Characterization of the lytic bacteriophage phiEaP-8 effective against both Erwinia amylovora and Erwinia pyrifoliae causing severe diseases in apple and pear. Plant Pathol. J. 2018, 34, 445. [Google Scholar] [CrossRef] [PubMed]
- Davidson, A.R.; Cardarelli, L.; Pell, L.G.; Radford, D.R.; Maxwell, K.L. Long noncontractile tail machines of bacteriophages. In Viral Molecular Machines; Rossmann, M.G., Rao, V.B., Eds.; Springer: Boston, MA, USA, 2012; pp. 115–142. [Google Scholar]
- Islam, M.Z.; Fokine, A.; Mahalingam, M.; Zhang, Z.; Garcia-Doval, C.; Van Raaij, M.J.; Rossmann, M.G.; Rao, V.B. Molecular anatomy of the receptor binding module of a bacteriophage long tail fiber. PLoS Pathog. 2019, 15, e1008193. [Google Scholar] [CrossRef]
- Goulet, A.; Spinelli, S.; Mahony, J.; Cambillau, C. Conserved and diverse traits of adhesion devices from Siphoviridae recognizing proteinaceous or saccharidic receptors. Viruses 2020, 12, 512. [Google Scholar] [CrossRef] [PubMed]
- Pell, L.G.; Gasmi-Seabrook, G.M.; Morais, M.; Neudecker, P.; Kanelis, V.; Bona, D.; Donaldson, L.W.; Edwards, A.M.; Howell, P.L.; Davidson, A.R.; et al. The solution structure of the C-terminal Ig-like domain of the bacteriophage λ tail tube protein. J. Mol. Biol. 2010, 403, 468–479. [Google Scholar] [CrossRef] [PubMed]
- Rajagopala, S.V.; Casjens, S.; Uetz, P. The protein interaction map of bacteriophage lambda. BMC Microbiol. 2011, 11, 213. [Google Scholar] [CrossRef]



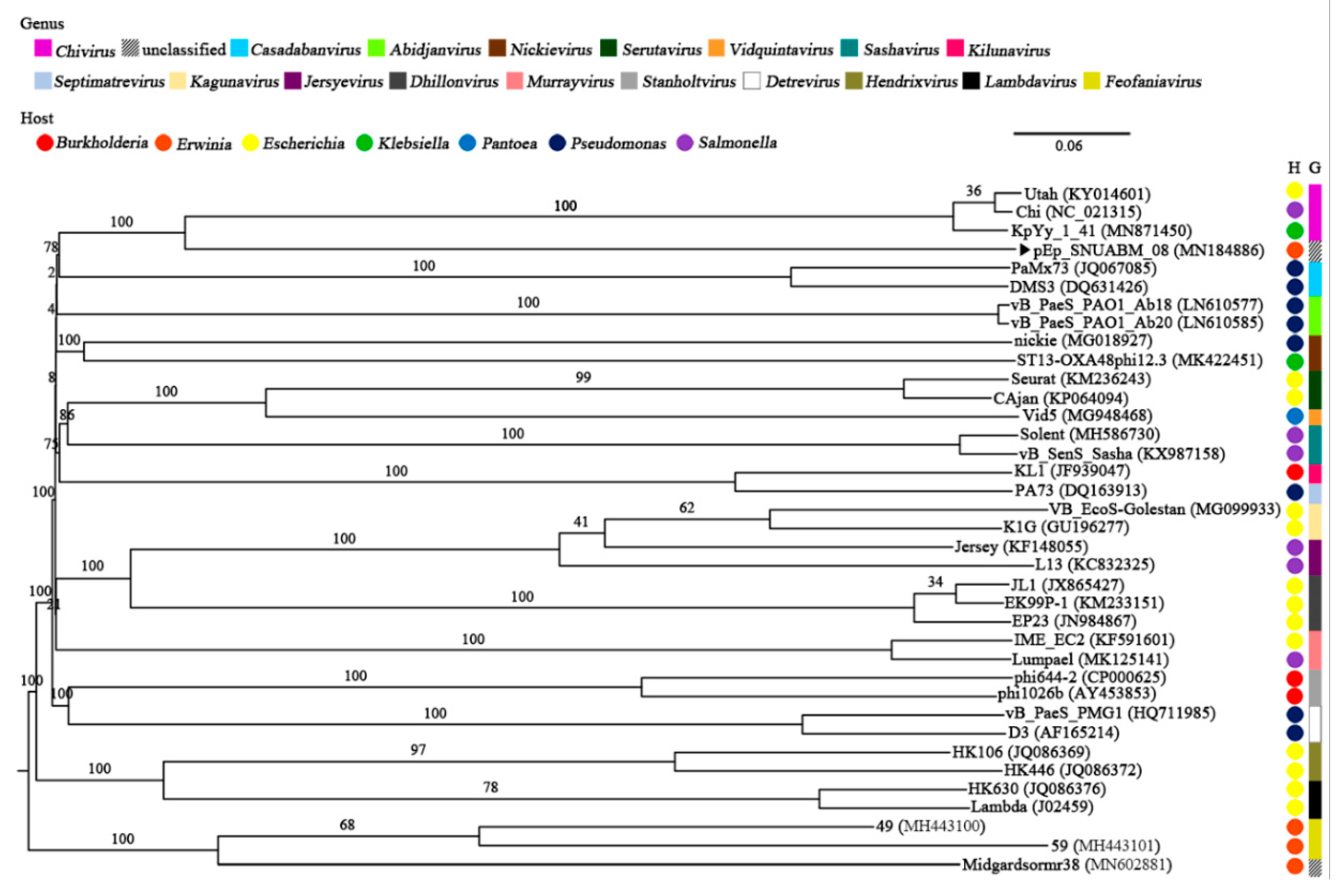
 ). The genera (Chivirus, magenta; unclassified, mosaic; Casadabanvirus, cyan; Abidjanvirus, light green; Nickievirus, brown; Serutavirus, dark green; Vidquintavirus, orange; Sashavirus, turquoise; Kilunavirus, crimson; Septimavirus, sky blue; Kagunavirus, bisque; Jersyevirus, violet; Dhillonvirus, kakhi; Murrayvirus, petal; Stanboltvirus, gray; Detrevirus, white; Hendrixvirus, chartreuse; Lambdavirus, black; Feofaniavirus, chrome yellow) and host (Burkholderia, red; Erwinia, orange; Escherichia, yellow; Klebsiella, green; Pantoea, blue; Pseudomonas, dark blue; Salmonella, purple) are indicated using respective colors.
). The genera (Chivirus, magenta; unclassified, mosaic; Casadabanvirus, cyan; Abidjanvirus, light green; Nickievirus, brown; Serutavirus, dark green; Vidquintavirus, orange; Sashavirus, turquoise; Kilunavirus, crimson; Septimavirus, sky blue; Kagunavirus, bisque; Jersyevirus, violet; Dhillonvirus, kakhi; Murrayvirus, petal; Stanboltvirus, gray; Detrevirus, white; Hendrixvirus, chartreuse; Lambdavirus, black; Feofaniavirus, chrome yellow) and host (Burkholderia, red; Erwinia, orange; Escherichia, yellow; Klebsiella, green; Pantoea, blue; Pseudomonas, dark blue; Salmonella, purple) are indicated using respective colors.
). The genera (Chivirus, magenta; unclassified, mosaic; Casadabanvirus, cyan; Abidjanvirus, light green; Nickievirus, brown; Serutavirus, dark green; Vidquintavirus, orange; Sashavirus, turquoise; Kilunavirus, crimson; Septimavirus, sky blue; Kagunavirus, bisque; Jersyevirus, violet; Dhillonvirus, kakhi; Murrayvirus, petal; Stanboltvirus, gray; Detrevirus, white; Hendrixvirus, chartreuse; Lambdavirus, black; Feofaniavirus, chrome yellow) and host (Burkholderia, red; Erwinia, orange; Escherichia, yellow; Klebsiella, green; Pantoea, blue; Pseudomonas, dark blue; Salmonella, purple) are indicated using respective colors.
). The genera (Chivirus, magenta; unclassified, mosaic; Casadabanvirus, cyan; Abidjanvirus, light green; Nickievirus, brown; Serutavirus, dark green; Vidquintavirus, orange; Sashavirus, turquoise; Kilunavirus, crimson; Septimavirus, sky blue; Kagunavirus, bisque; Jersyevirus, violet; Dhillonvirus, kakhi; Murrayvirus, petal; Stanboltvirus, gray; Detrevirus, white; Hendrixvirus, chartreuse; Lambdavirus, black; Feofaniavirus, chrome yellow) and host (Burkholderia, red; Erwinia, orange; Escherichia, yellow; Klebsiella, green; Pantoea, blue; Pseudomonas, dark blue; Salmonella, purple) are indicated using respective colors. ). The arrangement of sequences was paralleled with the whole-genome sequence phylogeny (Figure 4).
). The arrangement of sequences was paralleled with the whole-genome sequence phylogeny (Figure 4).
). The arrangement of sequences was paralleled with the whole-genome sequence phylogeny (Figure 4).
). The arrangement of sequences was paralleled with the whole-genome sequence phylogeny (Figure 4). ). The numbers above the branches represent Genome-BLAST Distance Phylogeny (GBDP) pseudo-bootstrap support values based on the conduction of 100 replications.
). The numbers above the branches represent Genome-BLAST Distance Phylogeny (GBDP) pseudo-bootstrap support values based on the conduction of 100 replications.
). The numbers above the branches represent Genome-BLAST Distance Phylogeny (GBDP) pseudo-bootstrap support values based on the conduction of 100 replications.
). The numbers above the branches represent Genome-BLAST Distance Phylogeny (GBDP) pseudo-bootstrap support values based on the conduction of 100 replications. ) and related phages (B).
) and related phages (B).
) and related phages (B).
) and related phages (B).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Strain | Isolated | Infectivity a | |
|---|---|---|---|---|
| Year | Province | |||
| Erwinia amylovora | YKB 14715 | 2019 | Chungcheongbuk | − |
| YKB 14740 | 2019 | Chungcheongbuk | − | |
| YKB 14742 | 2019 | Chungcheongbuk | − | |
| YKB 14748 | 2019 | Chungcheongbuk | + | |
| YKB 14750 | 2019 | Chungcheongbuk | − | |
| RA0030 | 2020 | Gyeonggi | + | |
| RA0031 | 2020 | Gyeonggi | − | |
| RA0032 | 2020 | Gyeonggi | − | |
| RA0033 | 2020 | Gyeonggi | − | |
| RA0034 | 2020 | Gyeonggi | − | |
| RA0035 | 2020 | Gyeonggi | − | |
| RA0041 | 2019 | Chungcheongnam | − | |
| RA0042 | 2020 | Chungcheongnam | − | |
| RA0043 | 2020 | Chungcheongnam | − | |
| RA0044 | 2020 | Chungcheongnam | − | |
| RA0045 | 2020 | Chungcheongbuk | − | |
| RA0051 | 2020 | Chungcheongbuk | − | |
| RA0052 | 2020 | Chungcheongbuk | − | |
| RA0053 | 2020 | Chungcheongbuk | − | |
| RA0054 | 2020 | Chungcheongbuk | − | |
| RA0055 | 2020 | Chungcheongbuk | − | |
| RA0062 | 2020 | Chungcheongbuk | − | |
| RA0063 | 2020 | Chungcheongbuk | − | |
| RA0064 | 2020 | Chungcheongbuk | − | |
| RA0065 | 2020 | Chungcheongbuk | − | |
| RA0066 | 2020 | Chungcheongbuk | − | |
| Erwinia pyrifoliae | KACC13945 | 1999 | Gangwon | + |
| KACC13946 | 1999 | Gangwon | + | |
| KACC13948 | 1999 | Gangwon | + | |
| KACC13949 | 1999 | Gangwon | + | |
| KACC13952 | 1999 | Gangwon | + | |
| RP0100 | 2020 | Gangwon | + | |
| RP0101 | 2020 | Gangwon | + | |
| RP0102 | 2020 | Gangwon | + | |
| RP0103 | 2020 | Gangwon | − | |
| RP0104 | 2020 | Gangwon | + | |
| RP0105 | 2020 | Gangwon | + | |
| RP0108 | 2020 | Gangwon | − | |
| RP0109 | 2020 | Gangwon | − | |
| RP0110 | 2020 | Gangwon | − | |
| RP0111 | 2020 | Gyeonggi | − | |
| RP0112 | 2020 | Gyeonggi | − | |
| RP0113 | 2020 | Gyeonggi | − | |
| RP0114 | 2020 | Gyeongsangbuk | − | |
| RP0115 | 2020 | Gyeongsangbuk | + | |
| RP0116 | 2020 | Chungcheongbuk | − | |
| RP0117 | 2020 | Chungcheongbuk | − | |
| RP0118 | 2020 | Chungcheongbuk | − | |
| RP0119 | 2020 | Chungcheongbuk | − | |
| RP0120 | 2020 | Gangwon | + | |
| RP0121 | 2020 | Chungcheongbuk | − | |
| Pectobacterium carotovorum | KACC17004 | N/A | Gangwon | − |
| KACC18645 | N/A | Gangwon | − | |
| Pseudomonas aeruginosa | KCCM40395 | N/A | N/A | − |
| Escherichia coli | KCTC2571 | N/A | N/A | − |
| Protein | Putative Function | Molecular Weight (kDa) | Number of Identified Peptides | Coverage (%) |
|---|---|---|---|---|
| gp12 | Head-to-tail joining protein | 9.6 | 11 | 28 |
| gp13 | Portal protein | 61.2 | 100 | 25 |
| gp15 | Head decoration protein | 14.2 | 2 | 19 |
| gp16 | Major capsid protein | 39.9 | 90 | 36 |
| gp21 | Ig-like domain-containing protein | 41.9 | 58 | 11 |
| gp22 | Putative tail assembly chaperone | 17.3 | 127 | 40 |
| gp24 | Tape measure protein | 148.4 | 528 | 48 |
| gp25 | Distal tail protein | 64.7 | 258 | 21 |
| gp29 | Putative tail protein | 139.1 | 218 | 25 |
| gp30 | Putative tail fiber protein | 23.7 | 5 | 10 |
| gp38 | Virion protein | 25.2 | 106 | 38 |
| gp51 | Tail fiber protein | 9.2 | 10 | 47 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, S.G.; Roh, E.; Park, J.; Giri, S.S.; Kwon, J.; Kim, S.W.; Kang, J.W.; Lee, S.B.; Jung, W.J.; Lee, Y.M.; et al. The Bacteriophage pEp_SNUABM_08 Is a Novel Singleton Siphovirus with High Host Specificity for Erwinia pyrifoliae. Viruses 2021, 13, 1231. https://0-doi-org.brum.beds.ac.uk/10.3390/v13071231
Kim SG, Roh E, Park J, Giri SS, Kwon J, Kim SW, Kang JW, Lee SB, Jung WJ, Lee YM, et al. The Bacteriophage pEp_SNUABM_08 Is a Novel Singleton Siphovirus with High Host Specificity for Erwinia pyrifoliae. Viruses. 2021; 13(7):1231. https://0-doi-org.brum.beds.ac.uk/10.3390/v13071231
Chicago/Turabian StyleKim, Sang Guen, Eunjung Roh, Jungkum Park, Sib Sankar Giri, Jun Kwon, Sang Wha Kim, Jeong Woo Kang, Sung Bin Lee, Won Joon Jung, Young Min Lee, and et al. 2021. "The Bacteriophage pEp_SNUABM_08 Is a Novel Singleton Siphovirus with High Host Specificity for Erwinia pyrifoliae" Viruses 13, no. 7: 1231. https://0-doi-org.brum.beds.ac.uk/10.3390/v13071231