
Genetic and Chemical Capsid Modifications of Adenovirus Vectors to Modulate Vector–Host Interactions

 and
and
Abstract
:1. Introduction
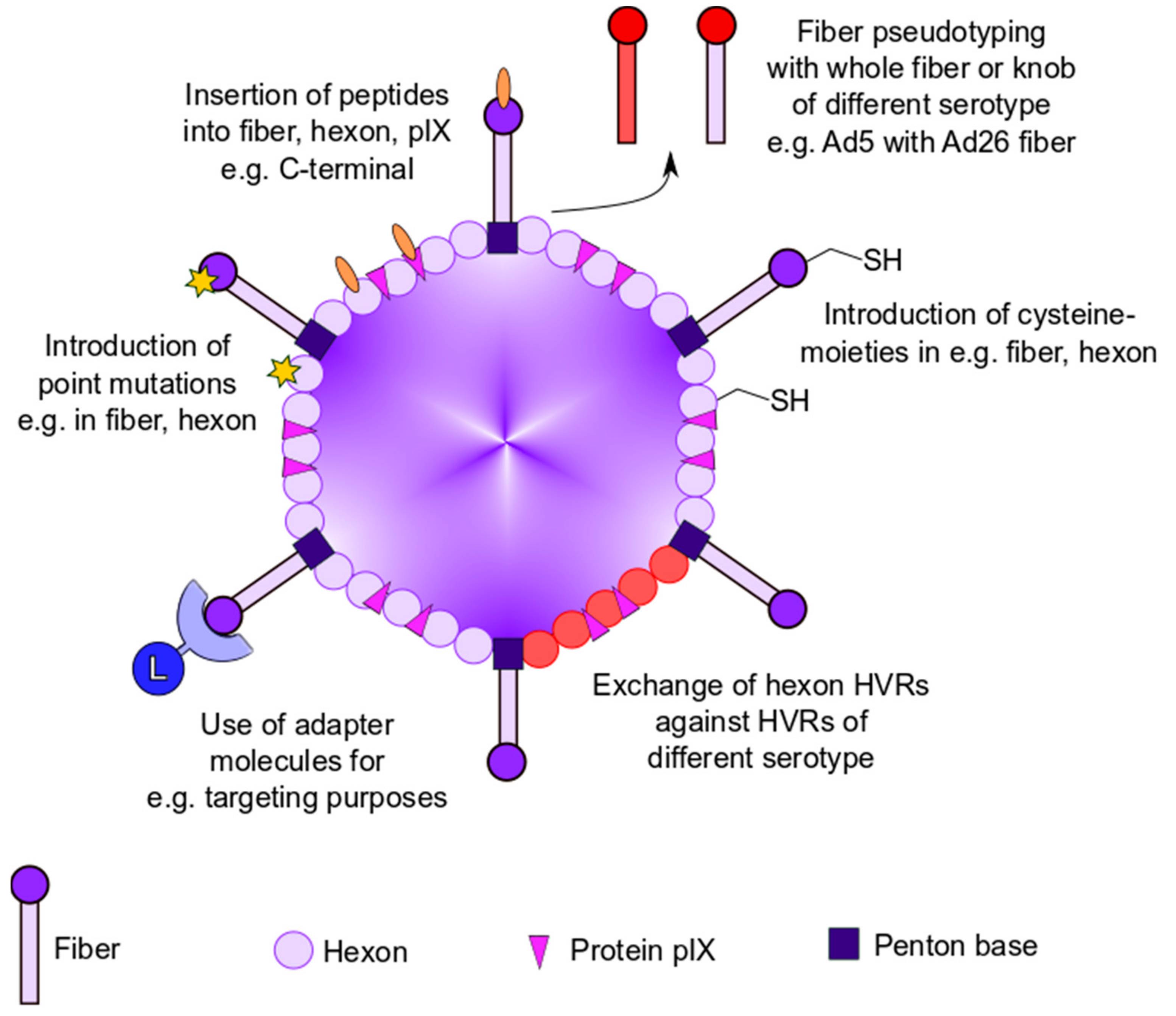
2. Genetic Capsid Modifications
2.1. Genetic Modifications to Ablate Vector Tropism
2.1.1. Generation of Chimeric Ad Vectors by Fiber Pseudotyping
2.1.2. Insertion of Targeting Peptides into Ad Capsid Proteins
2.1.3. Adapter-Based Strategies to Ablate Ad Tropism
2.2. Genetic Modifications to Circumvent Anti-Ad Immune Responses
2.2.1. Removal of Viral Genes from the Vector Genome
2.2.2. Geneti-Chemical Modification Strategies
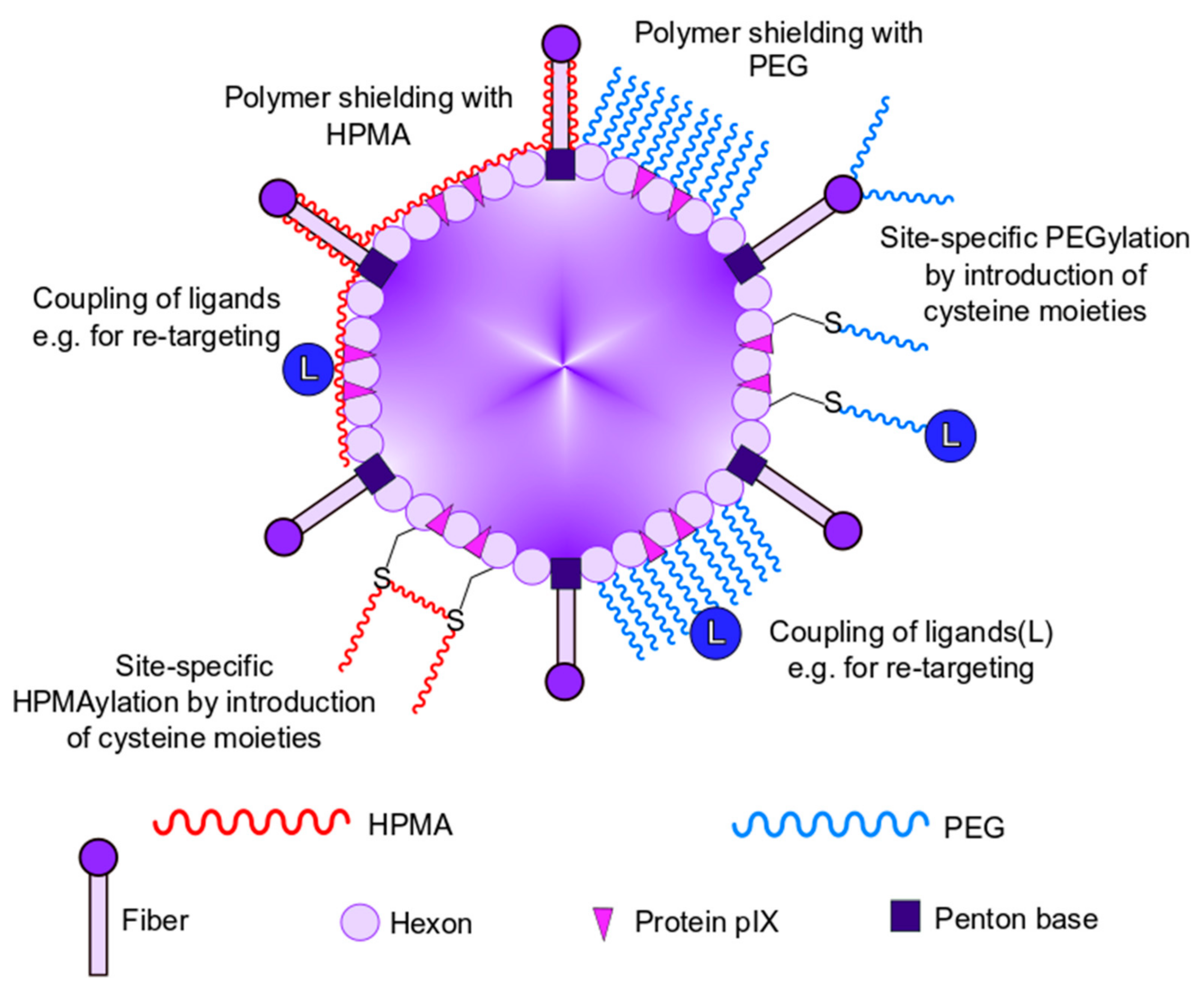
3. Chemical Shielding of Ad Vectors
4. Shielding with HPMA
4.1. HPMA-Coating of Ad
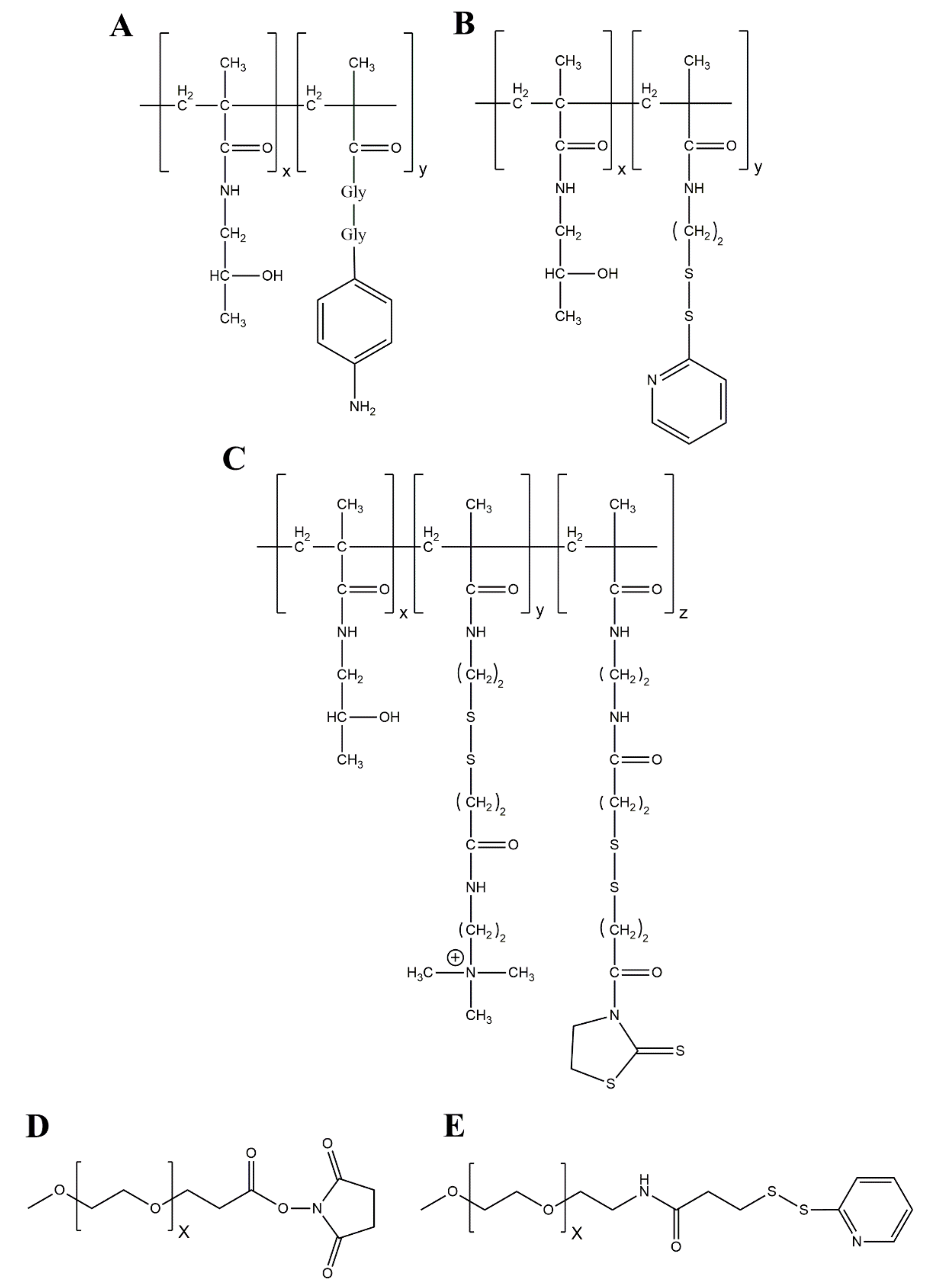
4.2. Chemistry of HPMA Coupling
4.2.1. Amine-Directed HPMA Coupling
4.2.2. Thiol-Directed HPMA Coupling
4.2.3. Next Generation HPMA Polymers—Bioresponsive HPMA Coupling
4.3. Detargeting and Immune Evasion of HPMAylated Ad Vectors
4.4. Retargeting of HPMAylated Ad Vectors
5. Shielding with Polyethylene Glycol (PEG)
5.1. Chemistry of PEGylation
Bioresponsive PEGylation Approaches
5.2. Detargeting and Retargeting with PEGylation
5.3. Immune Evasion with PEGylation and PEGylated Ad Vectors as Vaccines
5.4. Preventing Ad-Mediated Toxicity with PEGylation
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shaw, A.R.; Suzuki, M. Immunology of Adenoviral Vectors in Cancer Therapy. Mol. Ther. Methods Clin. Dev. 2019, 15, 418–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bulcha, J.T.; Wang, Y.; Ma, H.; Tai, P.W.L.; Gao, G. Viral vector platforms within the gene therapy landscape. Signal Transduct. Target. Ther. 2021, 6, 1–24. [Google Scholar] [CrossRef]
- Folegatti, P.M.; Ewer, K.J.; Aley, P.K.; Angus, B.; Becker, S.; Belij-Rammerstorfer, S.; Bellamy, D.; Bibi, S.; Bittaye, M.; Clutterbuck, E.A.; et al. Safety and immunogenicity of the ChAdOx1 nCoV-19 vaccine against SARS-CoV-2: A preliminary report of a phase 1/2, single-blind, randomised controlled trial. Lancet 2020, 396, 467–478. [Google Scholar] [CrossRef]
- Ewer, K.J.; The Oxford COVID Vaccine Trial Group; Barrett, J.R.; Belij-Rammerstorfer, S.; Sharpe, H.; Makinson, R.; Morter, R.; Flaxman, A.; Wright, D.; Bellamy, D.; et al. T cell and antibody responses induced by a single dose of ChAdOx1 nCoV-19 (AZD1222) vaccine in a phase 1/2 clinical trial. Nat. Med. 2021, 27, 270–278. [Google Scholar] [CrossRef]
- Logunov, D.Y.; Dolzhikova, I.V.; Zubkova, O.V.; Tukhvatullin, A.I.; Shcheblyakov, D.V.; Dzharullaeva, A.S.; Grousova, D.M.; Erokhova, A.S.; Kovyrshina, A.V.; Botikov, A.G.; et al. Safety and immunogenicity of an rAd26 and rAd5 vector-based heterologous prime-boost COVID-19 vaccine in two formulations: Two open, non-randomised phase 1/2 studies from Russia. Lancet 2020, 396, 887–897. [Google Scholar] [CrossRef]
- Logunov, D.Y.; Dolzhikova, I.V.; Shcheblyakov, D.V.; Tukhvatulin, A.I.; Zubkova, O.V.; Dzharullaeva, A.S.; Kovyrshina, A.V.; Lubenets, N.L.; Grousova, D.M.; Erokhova, A.S.; et al. Safety and efficacy of an rAd26 and rAd5 vector-based heterologous prime-boost COVID-19 vaccine: An interim analysis of a randomised controlled phase 3 trial in Russia. Lancet 2021, 397, 671–681. [Google Scholar] [CrossRef]
- Van Oostrum, J.; Burnett, R.M. Molecular composition of the adenovirus type 2 virion. J. Virol. 1985, 56, 439–448. [Google Scholar] [CrossRef] [Green Version]
- Stewart, P.; Fuller, S.; Burnett, R. Difference imaging of adenovirus: Bridging the resolution gap between X-ray crystallography and electron microscopy. EMBO J. 1993, 12, 2589–2599. [Google Scholar] [CrossRef]
- Stewart, P.L.; Burnett, R.M.; Cyrklaff, M.; Fuller, S.D. Image reconstruction reveals the complex molecular organization of adenovirus. Cell 1991, 67, 145–154. [Google Scholar] [CrossRef]
- Shirley, J.L.; de Jong, Y.P.; Terhorst, C.; Herzog, R.W. Immune Responses to Viral Gene Therapy Vectors. Mol. Ther. 2020, 28, 709–722. [Google Scholar] [CrossRef]
- Atasheva, S.; Yao, J.; Shayakhmetov, D.M. Innate immunity to adenovirus: Lessons from mice. FEBS Lett. 2019, 593, 3461–3483. [Google Scholar] [CrossRef]
- Shayakhmetov, D.M.; Gaggar, A.; Ni, S.; Li, Z.-Y.; Lieber, A. Adenovirus Binding to Blood Factors Results in Liver Cell Infection and Hepatotoxicity. J. Virol. 2005, 79, 7478–7491. [Google Scholar] [CrossRef] [Green Version]
- Doronin, K.; Flatt, J.; Di Paolo, N.C.; Khare, R.; Kalyuzhniy, O.; Acchione, M.; Sumida, J.P.; Ohto, U.; Shimizu, T.; Akashi-Takamura, S.; et al. Coagulation Factor X Activates Innate Immunity to Human Species C Adenovirus. Science 2012, 338, 795–798. [Google Scholar] [CrossRef] [Green Version]
- Schnell, M.A.; Zhang, Y.; Tazelaar, J.; Gao, G.-P.; Yu, Q.; Qian, R.; Chen, S.-J.; Varnavski, A.N.; LeClair, C.; Raper, S.E.; et al. Activation of Innate Immunity in Nonhuman Primates Following Intraportal Administration of Adenoviral Vectors. Mol. Ther. 2001, 3, 708–722. [Google Scholar] [CrossRef]
- Muruve, D.A.; Cotter, M.J.; Zaiss, A.K.; White, L.R.; Liu, Q.; Chan, T.; Clark, S.A.; Ross, P.J.; Meulenbroek, R.A.; Maelandsmo, G.M.; et al. Helper-Dependent Adenovirus Vectors Elicit Intact Innate but Attenuated Adaptive Host Immune Responses In Vivo. J. Virol. 2004, 78, 5966–5972. [Google Scholar] [CrossRef] [Green Version]
- Allen, R.J.; Byrnes, A.P. Interaction of adenovirus with antibodies, complement, and coagulation factors. FEBS Lett. 2019, 593, 3449–3460. [Google Scholar] [CrossRef]
- Alemany, R.; Suzuki, K.; Curiel, D.T. Blood clearance rates of adenovirus type 5 in mice. J. Gen. Virol. 2000, 81, 2605–2609. [Google Scholar] [CrossRef]
- Xu, Z.; Tian, J.; Smith, J.S.; Byrnes, A.P. Clearance of Adenovirus by Kupffer Cells Is Mediated by Scavenger Receptors, Natural Antibodies, and Complement. J. Virol. 2008, 82, 11705–11713. [Google Scholar] [CrossRef] [Green Version]
- Moskalenko, M.; Chen, L.; van Roey, M.; Donahue, B.A.; Snyder, R.O.; McArthur, J.G.; Patel, S.D. Epitope Mapping of Human Anti-Adeno-Associated Virus Type 2 Neutralizing Antibodies: Implications for Gene Therapy and Virus Structure. J. Virol. 2000, 74, 1761–1766. [Google Scholar] [CrossRef] [Green Version]
- Zhi, Y.; Figueredo, J.; Kobinger, G.P.; Hagan, H.; Calcedo, R.; Miller, J.R.; Gao, G.; Wilson, J.M. Efficacy of Severe Acute Respiratory Syndrome Vaccine Based on a Nonhuman Primate Adenovirus in the Presence of Immunity Against Human Adenovirus. Hum. Gene Ther. 2006, 17, 500–506. [Google Scholar] [CrossRef]
- Perreau, M.; Pantaleo, G.; Kremer, E.J. Activation of a dendritic cell–T cell axis by Ad5 immune complexes creates an improved environment for replication of HIV in T cells. J. Exp. Med. 2008, 205, 2717–2725. [Google Scholar] [CrossRef]
- Cotter, M.J.; Zaiss, A.K.; Muruve, D.A. Neutrophils Interact with Adenovirus Vectors via Fc Receptors and Complement Receptor. J. Virol. 2005, 79, 14622–14631. [Google Scholar] [CrossRef] [Green Version]
- Zaiss, A.K.; Vilaysane, A.; Cotter, M.J.; Clark, S.A.; Meijndert, H.C.; Colarusso, P.; Yates, R.M.; Petrilli, V.; Tschopp, J.; Muruve, D.A. Antiviral Antibodies Target Adenovirus to Phagolysosomes and Amplify the Innate Immune Response. J. Immunol. 2009, 182, 7058–7068. [Google Scholar] [CrossRef] [Green Version]
- McEwan, W.A.; Tam, J.; Watkinson, R.E.; Bidgood, S.; Mallery, D.L.; James, L.C. Intracellular antibody-bound pathogens stimulate immune signaling via the Fc receptor TRIM21. Nat. Immunol. 2013, 14, 327–336. [Google Scholar] [CrossRef] [Green Version]
- Khare, R.; Hillestad, M.L.; Xu, Z.; Byrnes, A.P.; Barry, M.A. Circulating Antibodies and Macrophages as Modulators of Adenovirus Pharmacology. J. Virol. 2013, 87, 3678–3686. [Google Scholar] [CrossRef] [Green Version]
- Bottermann, M.; Foss, S.; van Tienen, L.M.; Vaysburd, M.; Cruickshank, J.; O’Connell, K.; Clark, J.; Mayes, K.; Higginson, K.; Hirst, J.C.; et al. TRIM21 mediates antibody inhibition of adenovirus-based gene delivery and vaccination. Proc. Natl. Acad. Sci. USA 2018, 115, 10440–10445. [Google Scholar] [CrossRef] [Green Version]
- Groß, R.; Zanoni, M.; Seidel, A.; Conzelmann, C.; Gilg, A.; Krnavek, D.; Erdemci-Evin, S.; Mayer, B.; Hoffmann, M.; Pöhlmann, S.; et al. Heterologous ChAdOx1 NCoV-19 and BNT162b2 Prime-Boost Vaccination Elicits Potent Neutralizing Antibody Responses and T Cell Reactivity. medRxiv 2021. [Google Scholar] [CrossRef]
- Coyne, C.B.; Bergelson, J.M. CAR: A virus receptor within the tight junction. Adv. Drug Deliv. Rev. 2005, 57, 869–882. [Google Scholar] [CrossRef]
- Wickham, T.J.; Mathias, P.; Cheresh, D.A.; Nemerow, G.R. Integrins αvβ3 and αvβ5 promote adenovirus internalization but not virus attachment. Cell 1993, 73, 309–319. [Google Scholar] [CrossRef]
- Wu, E.; Nemerow, G.R. Virus yoga: The role of flexibility in virus host cell recognition. Trends Microbiol. 2004, 12, 162–169. [Google Scholar] [CrossRef]
- Meier, O.; Boucke, K.; Hammer, S.V.; Keller, S.; Stidwill, R.P.; Hemmi, S.; Greber, U.F. Adenovirus triggers macropinocytosis and endosomal leakage together with its clathrin-mediated uptake. J. Cell Biol. 2002, 158, 1119–1131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiethoff, C.M.; Wodrich, H.; Gerace, L.; Nemerow, G.R. Adenovirus Protein VI Mediates Membrane Disruption following Capsid Disassembly. J. Virol. 2005, 79, 1992–2000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greber, U.F.; Fornerod, M. Nuclear Import in Viral Infections. Curr. Top. Microbiol. Immunol. 2004, 285, 109–138. [Google Scholar] [CrossRef]
- Carlisle, R.C.; Di, Y.; Cerny, A.M.; Sonnen, A.F.-P.; Sim, R.; Green, N.K.; Subr, V.; Ulbrich, K.; Gilbert, R.; Fisher, K.D.; et al. Human erythrocytes bind and inactivate type 5 adenovirus by presenting Coxsackie virus-adenovirus receptor and complement receptor. Blood 2009, 113, 1909–1918. [Google Scholar] [CrossRef]
- Rojas, L.A.; Moreno, R.; Calderón, H.; Alemany, R. Adenovirus coxsackie adenovirus receptor-mediated binding to human erythrocytes does not preclude systemic transduction. Cancer Gene Ther. 2016, 23, 411–414. [Google Scholar] [CrossRef]
- Kirby, I.; Davison, E.; Beavil, A.J.; Soh, C.P.C.; Wickham, T.J.; Roelvink, P.W.; Kovesdi, I.; Sutton, B.J.; Santis, G. Identification of Contact Residues and Definition of the CAR-Binding Site of Adenovirus Type 5 Fiber Protein. J. Virol. 2000, 74, 2804–2813. [Google Scholar] [CrossRef] [Green Version]
- Krutzke, L.; Prill, J.; Engler, T.; Schmidt, C.; Xu, Z.; Byrnes, A.; Simmet, T.; Kreppel, F. Substitution of blood coagulation factor X-binding to Ad5 by position-specific PEGylation: Preventing vector clearance and preserving infectivity. J. Control. Release 2016, 235, 379–392. [Google Scholar] [CrossRef] [Green Version]
- Alemany, R.; Curiel, D.T. CAR-binding ablation does not change biodistribution and toxicity of adenoviral vectors. Gene Ther. 2001, 8, 1347–1353. [Google Scholar] [CrossRef] [Green Version]
- Waddington, S.N.; McVey, J.; Bhella, D.; Parker, A.L.; Barker, K.; Atoda, H.; Pink, R.; Buckley, S.M.; Greig, J.A.; Denby, L.; et al. Adenovirus Serotype 5 Hexon Mediates Liver Gene Transfer. Cell 2008, 132, 397–409. [Google Scholar] [CrossRef] [Green Version]
- Kalyuzhniy, O.; Di Paolo, N.C.; Silvestry, M.; Hofherr, S.E.; Barry, M.A.; Stewart, P.L.; Shayakhmetov, D.M. Adenovirus serotype 5 hexon is critical for virus infection of hepatocytes in vivo. Proc. Natl. Acad. Sci. USA 2008, 105, 5483–5488. [Google Scholar] [CrossRef] [Green Version]
- Vigant, F.; Descamps, D.; Jullienne, B.; Esselin, S.; Connault, E.; Opolon, P.; Tordjmann, T.; Vigne, E.; Perricaudet, M.; Benihoud, K. Substitution of Hexon Hypervariable Region 5 of Adenovirus Serotype 5 Abrogates Blood Factor Binding and Limits Gene Transfer to Liver. Mol. Ther. 2008, 16, 1474–1480. [Google Scholar] [CrossRef]
- Stasiak, A.C.; Stehle, T. Human adenovirus binding to host cell receptors: A structural view. Med. Microbiol. Immunol. 2019, 209, 325–333. [Google Scholar] [CrossRef] [Green Version]
- Krasnykh, V.N.; Mikheeva, G.V.; Douglas, J.T.; Curiel, D.T. Generation of recombinant adenovirus vectors with modified fibers for altering viral tropism. J. Virol. 1996, 70, 6839–6846. [Google Scholar] [CrossRef] [Green Version]
- Shayakhmetov, D.M.; Papayannopoulou, T.; Stamatoyannopoulos, G.; Lieber, A. Efficient Gene Transfer into Human CD34(+) Cells by a Retargeted Adenovirus Vector. J. Virol. 2000, 74, 2567–2583. [Google Scholar] [CrossRef] [Green Version]
- Kanerva, A.; Mikheeva, G.V.; Krasnykh, V.; Coolidge, C.J.; Lam, J.T.; Mahasreshti, P.J.; Barker, S.D.; Straughn, M.; Barnes, M.N.; Alvarez, R.D.; et al. Targeting adenovirus to the serotype 3 receptor increases gene transfer efficiency to ovarian cancer cells. Clin. Cancer Res. 2002, 8, 275–280. [Google Scholar]
- Kanerva, A.; Zinn, K.R.; Chaudhuri, T.R.; Lam, J.T.; Suzuki, K.; Uil, T.G.; Hakkarainen, T.; Bauerschmitz, G.J.; Wang, M.; Liu, B.; et al. Enhanced therapeutic efficacy for ovarian cancer with a serotype 3 receptor-targeted oncolytic adenovirus. Mol. Ther. 2003, 8, 449–458. [Google Scholar] [CrossRef]
- Raddi, N.; Vigant, F.; Wagner-Ballon, O.; Giraudier, S.; Custers, J.; Hemmi, S.; Benihoud, K. Pseudotyping Serotype 5 Adenovirus with the Fiber from Other Serotypes Uncovers a Key Role of the Fiber Protein in Adenovirus 5-Induced Thrombocytopenia. Hum. Gene Ther. 2016, 27, 193–201. [Google Scholar] [CrossRef] [Green Version]
- Krasnykh, V.; Dmitriev, I.; Mikheeva, G.; Miller, C.R.; Belousova, N.; Curiel, D.T. Characterization of an Adenovirus Vector Containing a Heterologous Peptide Epitope in the HI Loop of the Fiber Knob. J. Virol. 1998, 72, 1844–1852. [Google Scholar] [CrossRef] [Green Version]
- Belousova, N.; Krendelchtchikova, V.; Curiel, D.T.; Krasnykh, V. Modulation of Adenovirus Vector Tropism via Incorporation of Polypeptide Ligands into the Fiber Protein. J. Virol. 2002, 76, 8621–8631. [Google Scholar] [CrossRef] [Green Version]
- Parrott, M.; Adams, K.E.; Mercier, G.T.; Mok, H.; Campos, S.K.; Barry, M.A. Metabolically biotinylated adenovirus for cell targeting, ligand screening, and vector purification. Mol. Ther. 2003, 8, 688–700. [Google Scholar] [CrossRef]
- Dmitriev, I.; Krasnykh, V.; Miller, C.R.; Wang, M.; Kashentseva, E.; Mikheeva, G.; Belousova, N.; Curiel, D.T. An Adenovirus Vector with Genetically Modified Fibers Demonstrates Expanded Tropism via Utilization of a Coxsackievirus and Adenovirus Receptor-Independent Cell Entry Mechanism. J. Virol. 1998, 72, 9706–9713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, H.; Seki, T.; Dmitriev, I.; Uil, T.; Kashentseva, E.; Han, T.; Curiel, D.T. Double Modification of Adenovirus Fiber with RGD and Polylysine Motifs Improves Coxsackievirus–Adenovirus Receptor-Independent Gene Transfer Efficiency. Hum. Gene Ther. 2002, 13, 1647–1653. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Han, T.; Lam, J.T.; A Leath, C.; Dmitriev, I.; Kashentseva, E.; Barnes, M.N.; Alvarez, R.D.; Curiel, D.T. Preclinical evaluation of a class of infectivity-enhanced adenoviral vectors in ovarian cancer gene therapy. Gene Ther. 2004, 11, 874–878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimball, K.J.; Preuss, M.A.; Barnes, M.N.; Wang, M.; Siegal, G.P.; Wan, W.; Kuo, H.; Saddekni, S.; Stockard, C.R.; Grizzle, W.E.; et al. A Phase I Study of a Tropism-Modified Conditionally Replicative Adenovirus for Recurrent Malignant Gynecologic Diseases. Clin. Cancer Res. 2010, 16, 5277–5287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, K.H.; Dmitriev, I.; O’Malley, J.P.; Wang, M.; Saddekni, S.; You, Z.; Preuss, M.A.; Harris, R.D.; Aurigemma, R.; Siegal, G.P.; et al. A Phase I Clinical Trial of Ad5.SSTR/TK.RGD, a Novel Infectivity-Enhanced Bicistronic Adenovirus, in Patients with Recurrent Gynecologic Cancer. Clin. Cancer Res. 2012, 18, 3440–3451. [Google Scholar] [CrossRef] [Green Version]
- MacLeod, S.H.; Elgadi, M.M.; Bossi, G.; Sankar, U.; Pisio, A.; Agopsowicz, K.; Sharon, D.; Graham, F.L.; Hitt, M.M. HER3 targeting of adenovirus by fiber modification increases infection of breast cancer cells in vitro, but not following intratumoral injection in mice. Cancer Gene Ther. 2012, 19, 888–898. [Google Scholar] [CrossRef] [Green Version]
- Behr, M.; Kaufmann, J.K.; Ketzer, P.; Engelhardt, S.; Muck-Hausl, M.; Okun, P.M.; Petersen, G.; Neipel, F.; Hassel, J.C.; Ehrhardt, A.; et al. Adenoviruses Using the Cancer Marker EphA2 as a Receptor In Vitro and In Vivo by Genetic Ligand Insertion into Different Capsid Scaffolds. PLoS ONE 2014, 9, e95723. [Google Scholar] [CrossRef]
- Dmitriev, I.P.; Kashentseva, E.A.; Curiel, D.T. Engineering of Adenovirus Vectors Containing Heterologous Peptide Sequences in the C Terminus of Capsid Protein IX. J. Virol. 2002, 76, 6893–6899. [Google Scholar] [CrossRef] [Green Version]
- Le, L.P.; Everts, M.; Dmitriev, I.P.; Davydova, J.G.; Yamamoto, M.; Curiel, D.T. Fluorescently Labeled Adenovirus with PIX-EGFP for Vector Detection. Mol. Imaging 2004, 3, 105–116. [Google Scholar] [CrossRef]
- Wu, H.; Han, T.; Belousova, N.; Krasnykh, V.; Kashentseva, E.; Dmitriev, I.; Kataram, M.; Mahasreshti, P.J.; Curiel, D.T. Identification of Sites in Adenovirus Hexon for Foreign Peptide Incorporation. J. Virol. 2005, 79, 3382–3390. [Google Scholar] [CrossRef] [Green Version]
- Vigne, E.; Mahfouz, I.; Dedieu, J.-F.; Brie, A.; Perricaudet, M.; Yeh, P. RGD Inclusion in the Hexon Monomer Provides Adenovirus Type 5-Based Vectors with a Fiber Knob-Independent Pathway for Infection. J. Virol. 1999, 73, 5156–5161. [Google Scholar] [CrossRef] [Green Version]
- Xu, Z.; Qiu, Q.; Tian, J.; Smith, J.S.; Conenello, G.M.; Morita, T.; Byrnes, A.P. Coagulation factor X shields adenovirus type 5 from attack by natural antibodies and complement. Nat. Med. 2013, 19, 452–457. [Google Scholar] [CrossRef]
- Watkins, S.; Mesyanzhinov, V.; Kurochkina, L.; Hawkins, R. The ‘Adenobody’ Approach to Viral Targeting: Specific and Enhanced Adenoviral Gene Delivery. Gene Ther. 1997, 4, 1004–1012. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.Y.; May, S.M.; Barry, M.A. Targeting Adenoviruses with Factor X–Single-Chain Antibody Fusion Proteins. Hum. Gene Ther. 2010, 21, 739–749. [Google Scholar] [CrossRef] [Green Version]
- Campos, S.K.; Parrott, M.B.; Barry, M.A. Avidin-based targeting and purification of a protein IX-modified, metabolically biotinylated adenoviral vector. Mol. Ther. 2004, 9, 942–954. [Google Scholar] [CrossRef]
- Dmitriev, I.; Kashentseva, E.; Rogers, B.E.; Krasnykh, V.; Curiel, D.T. Ectodomain of Coxsackievirus and Adenovirus Receptor Genetically Fused to Epidermal Growth Factor Mediates Adenovirus Targeting to Epidermal Growth Factor Receptor-Positive Cells. J. Virol. 2000, 74, 6875–6884. [Google Scholar] [CrossRef] [Green Version]
- Roberts, D.M.; Nanda, A.; Havenga, M.J.E.; Abbink, P.; Lynch, D.M.; Ewald, B.A.; Liu, J.; Thorner, A.R.; Swanson, P.E.; Gorgone, D.A.; et al. Hexon-chimaeric adenovirus serotype 5 vectors circumvent pre-existing anti-vector immunity. Nature 2006, 441, 239–243. [Google Scholar] [CrossRef]
- Bradley, R.R.; Maxfield, L.F.; Lynch, D.M.; Iampietro, M.J.; Borducchi, E.N.; Barouch, D.H. Adenovirus Serotype 5-Specific Neutralizing Antibodies Target Multiple Hexon Hypervariable Regions. J. Virol. 2011, 86, 1267–1272. [Google Scholar] [CrossRef] [Green Version]
- Rojas, L.A.; Condezo, G.N.; Moreno, R.; Fajardo, C.A.; Arias-Badia, M.; Martín, C.S.; Alemany, R. Albumin-binding adenoviruses circumvent pre-existing neutralizing antibodies upon systemic delivery. J. Control. Release 2016, 237, 78–88. [Google Scholar] [CrossRef]
- Atasheva, S.; Emerson, C.C.; Yao, J.; Young, C.; Stewart, P.L.; Shayakhmetov, D.M. Systemic cancer therapy with engineered adenovirus that evades innate immunity. Sci. Transl. Med. 2020, 12, eabc6659. [Google Scholar] [CrossRef]
- Alba, R.; Bosch, A.; Chillon, M. Gutless adenovirus: Last-generation adenovirus for gene therapy. Gene Ther. 2005, 12, S18–S27. [Google Scholar] [CrossRef] [Green Version]
- Rosewell, A.; Vetrini, F.; Ng, P. Helper-Dependent Adenoviral Vectors. J. Genet. Syndr. Gene Ther. 2011, (Suppl. 5), 1. [Google Scholar] [CrossRef] [Green Version]
- Parks, R.J.; Graham, F.L. A helper-dependent system for adenovirus vector production helps define a lower limit for efficient DNA packaging. J. Virol. 1997, 71, 3293–3298. [Google Scholar] [CrossRef] [Green Version]
- Parks, R.J.; Bramson, J.L.; Wan, Y.; Addison, C.L.; Graham, F.L. Effects of Stuffer DNA on Transgene Expression from Helper-Dependent Adenovirus Vectors. J. Virol. 1999, 73, 8027–8034. [Google Scholar] [CrossRef] [Green Version]
- Cerullo, V.; Seiler, M.P.; Mane, V.; Brunetti-Pierri, N.; Clarke, C.; Bertin, T.K.; Rodgers, J.R.; Lee, B. Toll-like Receptor 9 Triggers an Innate Immune Response to Helper-dependent Adenoviral Vectors. Mol. Ther. 2007, 15, 378–385. [Google Scholar] [CrossRef]
- Liu, J. Helper virus-free gutless adenovirus (HF-GLAd): A new platform for gene therapy. BMB Rep. 2020, 53, 565–575. [Google Scholar] [CrossRef]
- Brunetti-Pierri, N.; Ng, T.; Iannitti, D.; Cioffi, W.; Stapleton, G.; Law, M.; Breinholt, J.; Palmer, D.; Grove, N.; Rice, K.; et al. Transgene Expression up to 7 Years in Nonhuman Primates Following Hepatic Transduction with Helper-Dependent Adenoviral Vectors. Hum. Gene Ther. 2013, 24, 761–765. [Google Scholar] [CrossRef] [Green Version]
- Rastall, D.P.W.; Seregin, S.S.; Aldhamen, Y.A.; Kaiser, L.; Mullins, C.; Liou, A.; Ing, F.; Pereria-Hicks, C.; Godbehere-Roosa, S.; Palmer, D.; et al. Long-term, high-level hepatic secretion of acid α-glucosidase for Pompe disease achieved in non-human primates using helper-dependent adenovirus. Gene Ther. 2016, 23, 743–752. [Google Scholar] [CrossRef]
- Kreppel, F.; Gackowski, J.; Schmidt, E.; Kochanek, S. Combined Genetic and Chemical Capsid Modifications Enable Flexible and Efficient De- and Retargeting of Adenovirus Vectors. Mol. Ther. 2005, 12, 107–117. [Google Scholar] [CrossRef]
- Prill, J.-M.; Espenlaub, S.; Samen, U.; Engler, T.; Schmidt, E.; Vetrini, F.; Rosewell, A.; Grove, N.; Palmer, D.; Ng, P.; et al. Modifications of Adenovirus Hexon Allow for Either Hepatocyte Detargeting or Targeting With Potential Evasion From Kupffer Cells. Mol. Ther. 2011, 19, 83–92. [Google Scholar] [CrossRef] [Green Version]
- Corjon, S.; Wortmann, A.; Engler, T.; Van Rooijen, N.; Kochanek, S.; Kreppel, F. Targeting of Adenovirus Vectors to the LRP Receptor Family with the High-affinity Ligand RAP via Combined Genetic and Chemical Modification of the pIX Capsomere. Mol. Ther. 2008, 16, 1813–1824. [Google Scholar] [CrossRef] [PubMed]
- Barry, M.A.; Rubin, J.D.; Lu, S. Retargeting adenoviruses for therapeutic applications and vaccines. FEBS Lett. 2020, 594, 1918–1946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kreppel, F.; Kochanek, S. Modification of Adenovirus Gene Transfer Vectors With Synthetic Polymers: A Scientific Review and Technical Guide. Mol. Ther. 2008, 16, 16–29. [Google Scholar] [CrossRef] [PubMed]
- Prill, J.-M.; Šubr, V.; Pasquarelli, N.; Engler, T.; Hoffmeister, A.; Kochanek, S.; Ulbrich, K.; Kreppel, F. Traceless Bioresponsive Shielding of Adenovirus Hexon with HPMA Copolymers Maintains Transduction Capacity In Vitro and In Vivo. PLoS ONE 2014, 9, e82716. [Google Scholar] [CrossRef] [Green Version]
- Fella, C.; Walker, G.F.; Ogris, M.; Wagner, E. Amine-reactive pyridylhydrazone-based PEG reagents for pH-reversible PEI polyplex shielding. Eur. J. Pharm. Sci. 2008, 34, 309–320. [Google Scholar] [CrossRef]
- Hofherr, S.E.; Shashkova, E.V.; Weaver, E.A.; Khare, R.; Barry, M.A. Modification of Adenoviral Vectors with Polyethylene Glycol Modulates In Vivo Tissue Tropism and Gene Expression. Mol. Ther. 2008, 16, 1276–1282. [Google Scholar] [CrossRef]
- Říhová, B.; Kovar, M. Immunogenicity and immunomodulatory properties of HPMA-based polymers. Adv. Drug Deliv. Rev. 2010, 62, 184–191. [Google Scholar] [CrossRef]
- Volfova, I.; Říhová, B.; VetviČKa, V.; Rossmann, P.; Ulbrich, K. Biocompatibility of Biopolymers. J. Bioact. Compat. Polym. 1992, 7, 175–190. [Google Scholar] [CrossRef]
- Říhová, B.; Ulbrich, K.; Kopeček, J.; Mančal, P. Immunogenicity of N-(2-hydroxypropyl)-methacrylamide copolymers—Potential hapten or drug carriers. Folia Microbiol. 1983, 28, 217–227. [Google Scholar] [CrossRef]
- Fisher, K.; Stallwood, Y.; Green, N.K.; Ulbrich, K.; Mautner, V.; Seymour, L.W. Polymer-coated adenovirus permits efficient retargeting and evades neutralising antibodies. Gene Ther. 2001, 8, 341–348. [Google Scholar] [CrossRef] [Green Version]
- Subr, V.; Kostka, L.; Selby-Milic, T.; Fisher, K.; Ulbrich, K.; Seymour, L.W.; Carlisle, R.C. Coating of Adenovirus Type 5 with Polymers Containing Quaternary Amines Prevents Binding to Blood Components. J. Control. Release 2009, 135, 152–158. [Google Scholar] [CrossRef]
- Fisher, K.D.; Green, N.K.; Hale, A.; Subr, V.; Ulbrich, K.; Seymour, L.W. Passive tumour targeting of polymer-coated adenovirus for cancer gene therapy. J. Drug Target. 2007, 15, 546–551. [Google Scholar] [CrossRef]
- Šubr, V.; Ulbrich, K. Synthesis and properties of new N-(2-hydroxypropyl)methacrylamide copolymers containing thiazolidine-2-thione reactive groups. React. Funct. Polym. 2006, 66, 1525–1538. [Google Scholar] [CrossRef]
- Ulbrich, K. Polymeric Drugs Based on Conjugates of Synthetic and Natural Macromolecules I. Synthesis and Physico-Chemical Characterisation. J. Control. Release 2000, 64, 63–79. [Google Scholar] [CrossRef]
- Espenlaub, S.; Wortmann, A.; Engler, T.; Corjon, S.; Kochanek, S.; Kreppel, F. Reductive amination as a strategy to reduce adenovirus vector promiscuity by chemical capsid modification with large polysaccharides. J. Gene Med. 2008, 10, 1303–1314. [Google Scholar] [CrossRef]
- Green, N.K.; Hale, A.; Cawood, R.; Illingworth, S.; Herbert, C.; Hermiston, T.; Subr, V.; Ulbrich, K.; Van Rooijen, N.; Seymour, L.W.; et al. Tropism ablation and stealthing of oncolytic adenovirus enhances systemic delivery to tumors and improves virotherapy of cancer. Nanomedicine 2012, 7, 1683–1695. [Google Scholar] [CrossRef]
- Mok, H.; Palmer, D.J.; Ng, P.; Barry, M.A. Evaluation of polyethylene glycol modification of first-generation and helper-dependent adenoviral vectors to reduce innate immune responses. Mol. Ther. 2005, 11, 66–79. [Google Scholar] [CrossRef]
- Espenlaub, S.; Corjon, S.; Engler, T.; Fella, C.; Ogris, M.; Wagner, E.; Kochanek, S.; Kreppel, F. Capsomer-Specific Fluorescent Labeling of Adenoviral Vector Particles Allows for Detailed Analysis of Intracellular Particle Trafficking and the Performance of Bioresponsive Bonds for Vector Capsid Modifications. Hum. Gene Ther. 2010, 21, 1155–1167. [Google Scholar] [CrossRef]
- Carlisle, R.C.; Etrych, T.; Briggs, S.S.; Preece, J.A.; Ulbrich, K.; Seymour, L.W. Polymer-coated polyethylenimine/DNA complexes designed for triggered activation by intracellular reduction. J. Gene Med. 2004, 6, 337–344. [Google Scholar] [CrossRef]
- Wang, C.-H.K.; Chan, L.W.; Johnson, R.N.; Chu, D.S.; Shi, J.; Schellinger, J.G.; Lieber, A.; Pun, S.H. The transduction of Coxsackie and Adenovirus Receptor-negative cells and protection against neutralizing antibodies by HPMA-co-oligolysine copolymer-coated adenovirus. Biomaterials 2011, 32, 9536–9545. [Google Scholar] [CrossRef] [Green Version]
- Morrison, J.; Briggs, S.S.; Green, N.K.; Thoma, C.; Fisher, K.; Kehoe, S.T.; Seymour, L.W. Cetuximab retargeting of adenovirus via the epidermal growth factor receptor for treatment of intraperitoneal ovarian cancer. Hum. Gene Ther. 2008, 15. [Google Scholar] [CrossRef]
- Morrison, J.; Briggs, S.S.; Green, N.; Fisher, K.; Subr, V.; Ulbrich, K.; Kehoe, S.; Seymour, L.W. Virotherapy of Ovarian Cancer with Polymer-cloaked Adenovirus Retargeted to the Epidermal Growth Factor Receptor. Mol. Ther. 2008, 16, 244–251. [Google Scholar] [CrossRef]
- Jönsson, F.; Kreppel, F. Barriers to systemic application of virus-based vectors in gene therapy: Lessons from adenovirus type 5. Virus Genes 2017, 53, 692–699. [Google Scholar] [CrossRef]
- Matsumura, Y.; Maeda, H. A new concept for macromolecular therapeutics in cancer chemotherapy: Mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986, 46, 6387–6392. [Google Scholar]
- Stevenson, M.; Boos, E.; Herbert, C.; Hale, A.; Green, N.; Lyons, M.; Chandler, L.; Ulbrich, K.; Van Rooijen, N.; Mautner, V.; et al. Chick embryo lethal orphan virus can be polymer-coated and retargeted to infect mammalian cells. Gene Ther. 2005, 13, 356–368. [Google Scholar] [CrossRef] [Green Version]
- Jonker, D.J.; O’Callaghan, C.J.; Karapetis, C.S.; Zalcberg, J.R.; Tu, D.; Au, H.-J.; Berry, S.R.; Krahn, M.; Price, T.; Simes, R.J.; et al. Cetuximab for the Treatment of Colorectal Cancer. N. Engl. J. Med. 2007, 357, 2040–2048. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Chen, J.; Xu, H.; Long, J.; Xie, X.; Zhang, Y. The Targeted Transduction of MMP-Overexpressing Tumor Cells by ACPP-HPMA Copolymer-Coated Adenovirus Conjugates. PLoS ONE 2014, 9, e100670. [Google Scholar] [CrossRef]
- Sun, Y.; Lv, X.; Ding, P.; Wang, L.; Sun, Y.; Li, S.; Zhang, H.; Gao, Z. Exploring the functions of polymers in adenovirus-mediated gene delivery: Evading immune response and redirecting tropism. Acta Biomater. 2019, 97, 93–104. [Google Scholar] [CrossRef]
- Veronese, F.M. Peptide and protein PEGylation: A review of problems and solutions. Biomaterials 2001, 22, 405–417. [Google Scholar] [CrossRef]
- Veronese, F.M.; Saccà, B.; De Laureto, P.P.; Sergi, M.; Caliceti, P.; Schiavon, O.; Orsolini, P. New PEGs for peptide and protein modification, suitable for identification of the PEGylation site. Bioconjugate Chem. 2001, 12, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Pepinsky, R.B.; Lepage, D.J.; Gill, A.; Chakraborty, A.; Vaidyanathan, S.; Green, M.; Baker, D.P.; Whalley, E.; Hochman, P.S.; Martin, P. Improved pharmacokinetic properties of a polyethylene glycol-modified form of interferon-beta-1a with preserved in vitro bioactivity. J. Pharmacol. Exp. Ther. 2001, 297, 1059–1066. [Google Scholar] [PubMed]
- Wang, Y.-S.; Youngster, S.; Bausch, J.; Zhang, R.; McNemar, C.; Wyss, D.F. Identification of the Major Positional Isomer of Pegylated Interferon Alpha-2b. Biochemistry 2000, 39, 10634–10640. [Google Scholar] [CrossRef] [PubMed]
- Booth, C.; Gaspar, H.B. Pegademase bovine (PEG-ADA) for the treatment of infants and children with severe combined immunodeficiency (SCID). Biol. Targets Ther. 2009, 3, 349–358. [Google Scholar]
- Thorner, M.O. The Discovery of Growth Hormone-Releasing Hormone. J. Clin. Endocrinol. Metab. 1999, 84, 4671–4676. [Google Scholar] [CrossRef]
- Thorner, M.O.; Strasburger, C.J.; Wu, Z.; Straume, M.; Bidlingmaier, M.; Pezzoli, S.S.; Zib, K.; Scarlett, J.C.; Bennett, W.F. Growth Hormone (GH) Receptor Blockade with a PEG-Modified GH (B2036-PEG) Lowers Serum Insulin-Like Growth Factor-I but Does Not Acutely Stimulate Serum GH. J. Clin. Endocrinol. Metab. 1999, 84, 2098–2103. [Google Scholar] [CrossRef]
- Delgado, C.; Francis, G.E.; Fisher, D. The uses and properties of PEG-linked proteins. Crit. Rev. Ther. Drug Carr. Syst. 1992, 9, 249–304. [Google Scholar]
- Parveen, S.; Sahoo, S.K. Nanomedicine. Clin. Pharmacokinet. 2006, 45, 965–988. [Google Scholar] [CrossRef]
- O’Riordan, C.R.; Lachapelle, A.; Delgado, C.; Parkes, V.; Wadsworth, S.C.; Smith, A.E.; Francis, G.E. PEGylation of Adenovirus with Retention of Infectivity and Protection from Neutralizing Antibody in Vitro and in Vivo. Hum. Gene Ther. 1999, 10, 1349–1358. [Google Scholar] [CrossRef]
- Lanciotti, J.; Song, A.; Doukas, J.; Sosnowski, B.; Pierce, G.; Gregory, R.; Wadsworth, S.; O’Riordan, C. Targeting adenoviral vectors using heterofunctional polyethylene glycol FGF2 conjugates. Mol. Ther. 2003, 8, 99–107. [Google Scholar] [CrossRef]
- Hofherr, S.E.; Mok, H.; Gushiken, F.C.; Lopez, J.A.; Barry, M.A. Polyethylene Glycol Modification of Adenovirus Reduces Platelet Activation, Endothelial Cell Activation, and Thrombocytopenia. Hum. Gene Ther. 2007, 18, 837–848. [Google Scholar] [CrossRef]
- Croyle, M.A.; Chirmule, N.; Zhang, Y.; Wilson, J.M. “Stealth” Adenoviruses Blunt Cell-Mediated and Humoral Immune Responses against the Virus and Allow for Significant Gene Expression upon Readministration in the Lung. J. Virol. 2001, 75, 4792–4801. [Google Scholar] [CrossRef] [Green Version]
- Croyle, M.A.; Chirmule, N.; Zhang, Y.; Wilson, J.M. PEGylation of E1-Deleted Adenovirus Vectors Allows Significant Gene Expression on Readministration to Liver. Hum. Gene Ther. 2002, 13, 1887–1900. [Google Scholar] [CrossRef]
- Wortmann, A.; Vöhringer, S.; Engler, T.; Corjon, S.; Schirmbeck, R.; Reimann, J.; Kochanek, J.; Kreppel, F. Fully Detargeted Polyethylene Glycol-coated Adenovirus Vectors Are Potent Genetic Vaccines and Escape from Pre-existing Anti-adenovirus Antibodies. Mol. Ther. 2008, 16, 154–162. [Google Scholar] [CrossRef]
- Khare, R.; Reddy, V.S.; Nemerow, G.R.; Barry, M.A. Identification of Adenovirus Serotype 5 Hexon Regions That Interact with Scavenger Receptors. J. Virol. 2011, 86, 2293–2301. [Google Scholar] [CrossRef] [Green Version]
- Weaver, E.A.; Barry, M.A. Effects of Shielding Adenoviral Vectors with Polyethylene Glycol on Vector-Specific and Vaccine-Mediated Immune Responses. Hum. Gene Ther. 2008, 19, 1369–1382. [Google Scholar] [CrossRef]
- Tian, J.; Xu, Z.; Smith, J.S.; Hofherr, S.E.; Barry, M.A.; Byrnes, A.P. Adenovirus Activates Complement by Distinctly Different Mechanisms In Vitro and In Vivo: Indirect Complement Activation by Virions In Vivo. J. Virol. 2009, 83, 5648–5658. [Google Scholar] [CrossRef] [Green Version]
- Croyle, M.A.; Le, H.T.; Linse, K.D.; Cerullo, V.; Toietta, G.; Beaudet, A.; Pastore, L. PEGylated helper-dependent adenoviral vectors: Highly efficient vectors with an enhanced safety profile. Gene Ther. 2005, 12, 579–587. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Abbreviation | Polymer | Reactivity | Functionality | Bioresponsive |
|---|---|---|---|---|
| ONp-HPMA | 4-nitrophenoxy-poly-N-(2-hydroxypropyl) methacrylamide | –NH | polyfunctional | no |
| mal-HPMA | maleimide-poly-N-(2-hydroxypropyl) methacrylamide | –SH | polyfunctional | no |
| OPSS-HPMA | orthopyridyl-disulfide-poly-N-(2-hydroxypropyl) methacrylamide | –SH | polyfunctional | yes |
| Next Generation HPMA | EC208 | –NH | polyfunctional | yes |
| T-MPEG | tresyl-monomethoxypolyethylene glycol | –NH | monofunctional | no |
| CC-MPEG | cyanuric chloride monomethoxypolyethylene glycol | –NH | monofunctional | no |
| SS-MPEG | succinimidyl succinate monomethoxypolyethylene glycol | –NH | monofunctional | no |
| T-MPEG-mal | tresyl-polyethylene-glycol-maleimide | –NH, –SH | heterobifunctional | no |
| mal-PEG | maleimide-polyethylene glycol | –SH | monofunctional | no |
| SPA-PEG | succinimidyl propionate polyethylene glycol | –NH | monofunctional | no |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Weklak, D.; Pembaur, D.; Koukou, G.; Jönsson, F.; Hagedorn, C.; Kreppel, F. Genetic and Chemical Capsid Modifications of Adenovirus Vectors to Modulate Vector–Host Interactions. Viruses 2021, 13, 1300. https://0-doi-org.brum.beds.ac.uk/10.3390/v13071300
Weklak D, Pembaur D, Koukou G, Jönsson F, Hagedorn C, Kreppel F. Genetic and Chemical Capsid Modifications of Adenovirus Vectors to Modulate Vector–Host Interactions. Viruses. 2021; 13(7):1300. https://0-doi-org.brum.beds.ac.uk/10.3390/v13071300
Chicago/Turabian StyleWeklak, Denice, Daniel Pembaur, Georgia Koukou, Franziska Jönsson, Claudia Hagedorn, and Florian Kreppel. 2021. "Genetic and Chemical Capsid Modifications of Adenovirus Vectors to Modulate Vector–Host Interactions" Viruses 13, no. 7: 1300. https://0-doi-org.brum.beds.ac.uk/10.3390/v13071300