
Variant Analysis of SARS-CoV-2 Genomes from Belgian Military Personnel Engaged in Overseas Missions and Operations

 ,
,  , , , , , , , , add
Show full author list
, , , , , , , , add
Show full author list
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
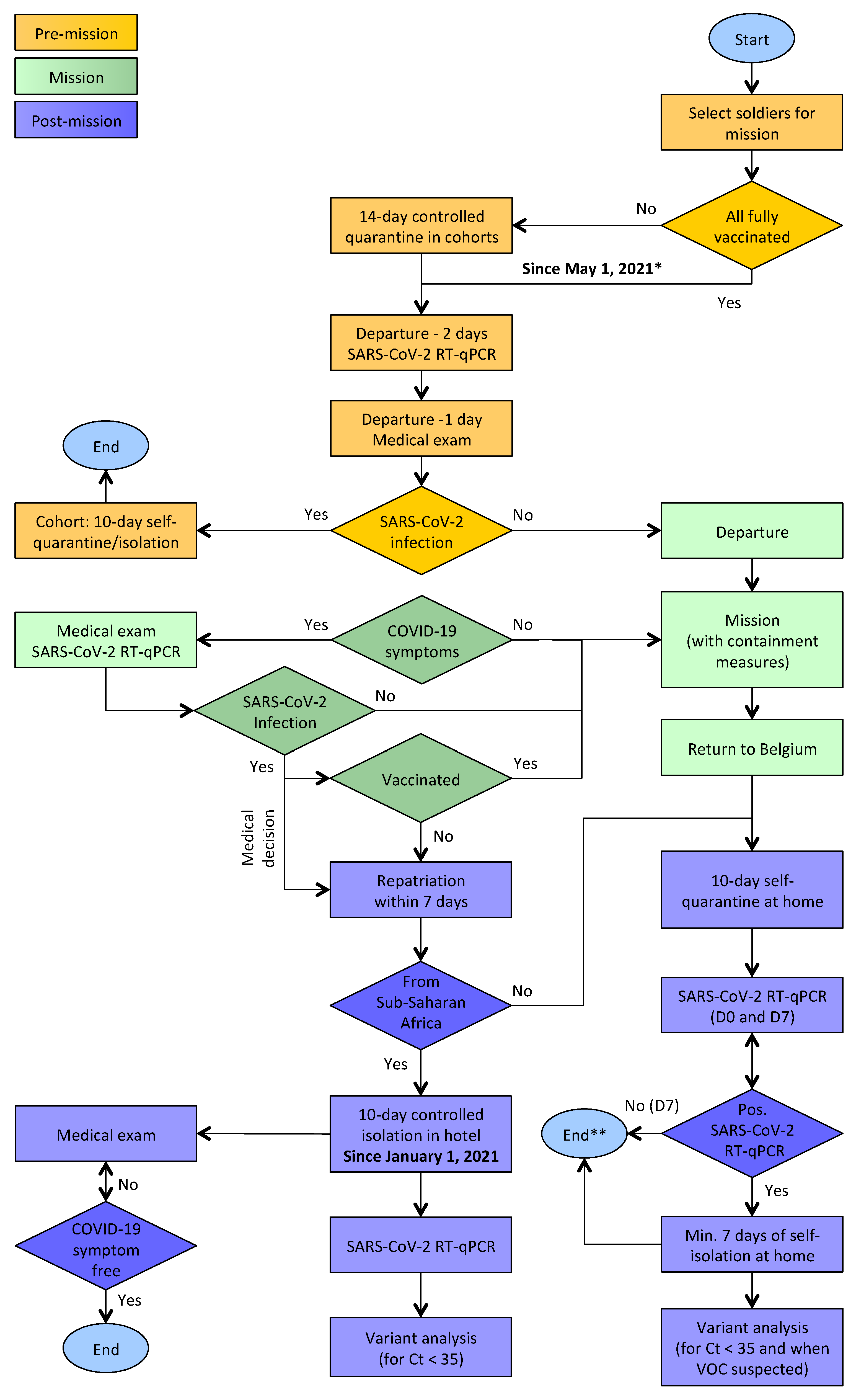
2. Materials and Methods
2.1. SARS-CoV-2 RT-qPCR
2.2. SARS-CoV-2 Genome Sequencing
2.3. Genomic Epidemiology
2.4. Phylogenetic Inference
2.5. Ethics Statement
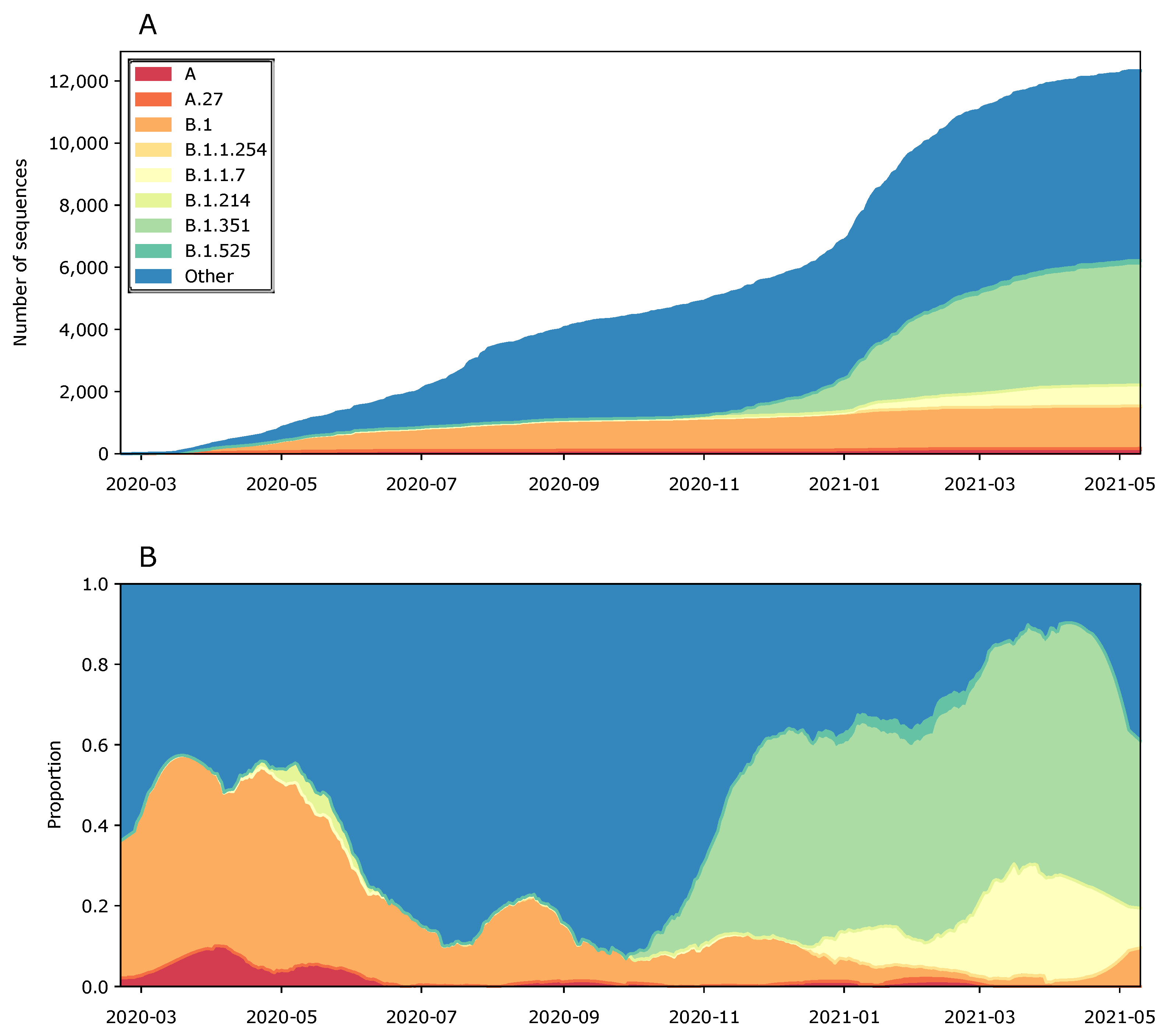
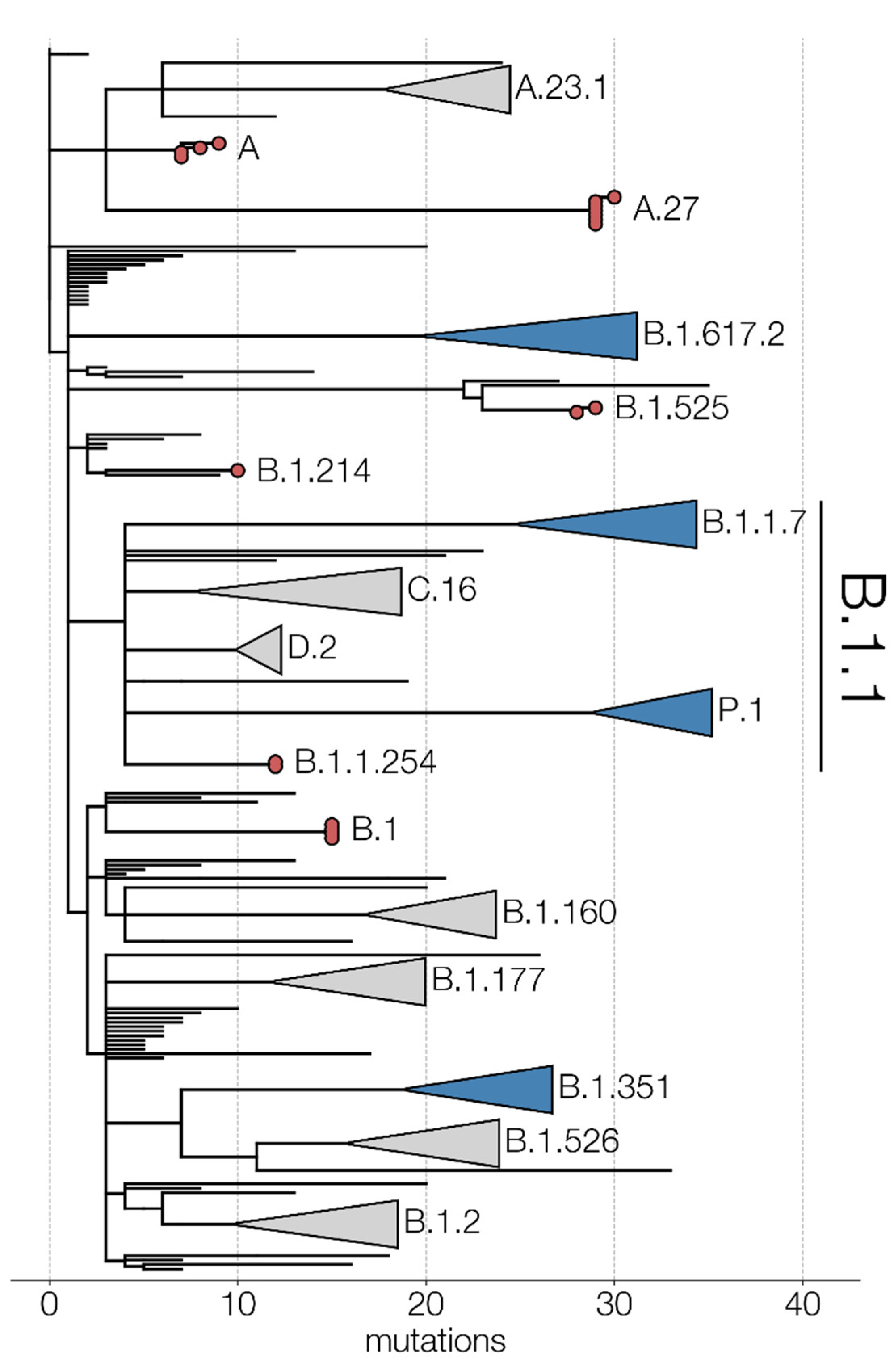
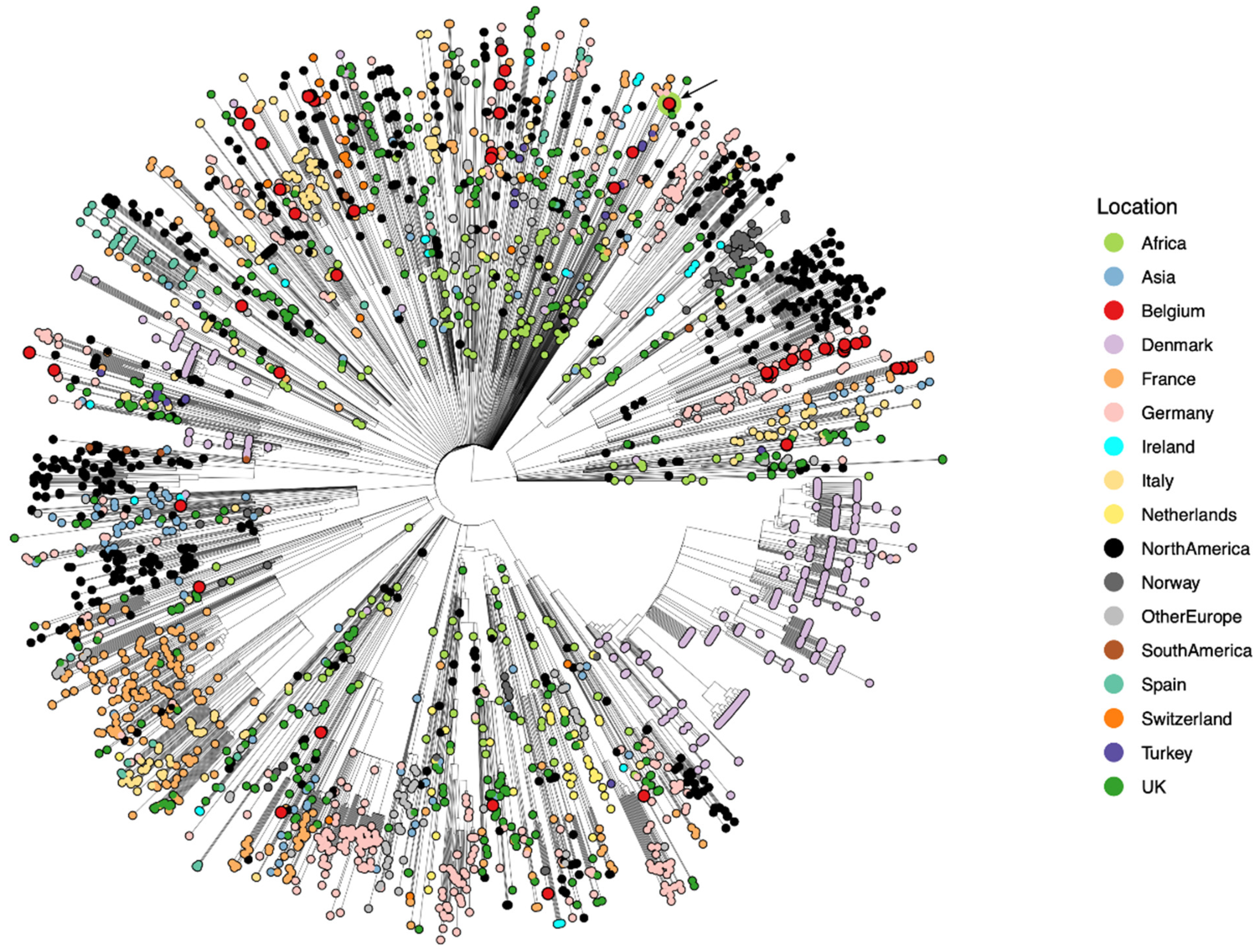
3. Results and Discussion
3.1. Niger 1–3 (Maradi, 19 May 2020)
3.2. DRC 1 (Kinshasa, 2 June 2020)
3.3. Afghanistan 1 (Kabul, 30 June 2020)
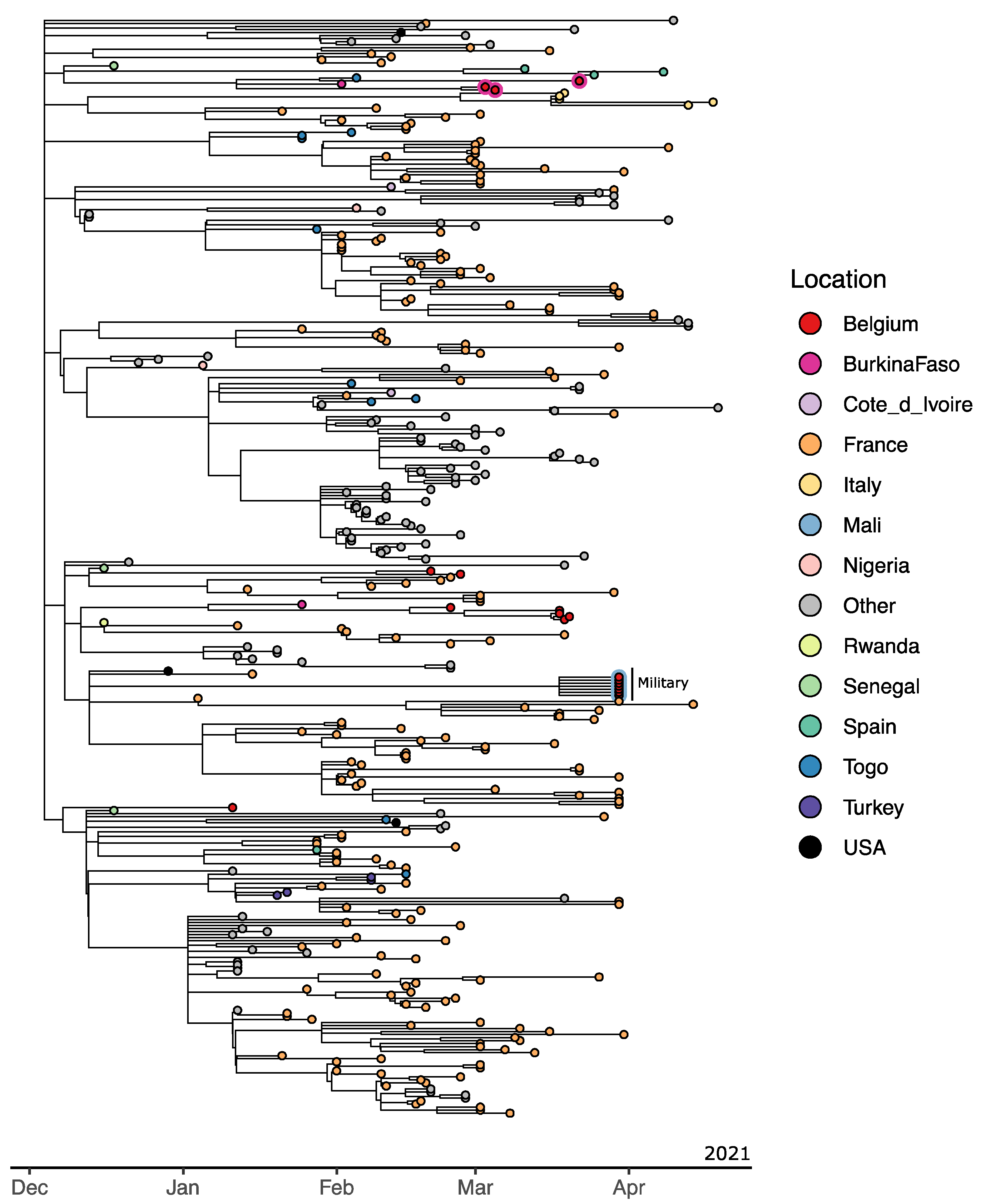
3.4. Mali 1 (Bamako, 8 November 2020)
3.5. Mali 2 and 3 (Gao, 1 April 2021)
3.6. Mali 4 (Bamako, 12 April 2021)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.M.; Wang, W.; Song, Z.G.; Hu, Y.; Tao, Z.W.; Tian, J.H.; Pei, Y.Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruzzenenti, G.; Maloberti, A.; Giani, V.; Biolcati, M.; Leidi, F.; Monticelli, M.; Grasso, E.; Cartella, I.; Palazzini, M.; Garatti, L.; et al. Covid and Cardiovascular Diseases: Direct and Indirect Damages and Future Perspective. High Blood Press. Cardiovasc. Prev. 2021, 26, 1–7. [Google Scholar]
- World Health Organization (WHO). WHO Coronavirus Disease (COVID-19) Dashboard. Available online: https://covid19.who.int/?gclid=EAIaIQobChMItf_K_u6s7gIVja3tCh1JTgrSEAAYASAAEgK7R_D_BwE (accessed on 16 June 2021).
- Laamarti, M.; Alouane, T.; Kartti, S.; Chemao-Elfihri, M.W.; Hakmi, M.; Essabbar, A.; Laamarti, M.; Hlali, H.; Bendani, H.; Boumajdi, N.; et al. Large scale genomic analysis of 3067 SARS-CoV-2 genomes reveals a clonal geo-distribution and a rich genetic variations of hotspots mutations. PLoS ONE 2020, 15, e0240345. [Google Scholar] [CrossRef]
- Koyama, T.; Platt, D.; Parida, L. Variant analysis of SARS-CoV-2 genomes. Bull. World Health Organ. 2020, 98, 495–504. [Google Scholar] [CrossRef]
- Mahase, E. Covid-19: What have we learnt about the new variant in the UK? BMJ 2020, 371, m4944. [Google Scholar] [CrossRef] [PubMed]
- European Centre for Disease Prevention and Control (ECDC). Threat Assessment Brief: Rapid Increase of a SARS-CoV-2 Variant with Multiple Spike Protein Mutations Observed in the United Kingdom. Available online: https://www.ecdc.europa.eu/en/publications-data/threat-assessment-brief-rapid-increase-sars-cov-2-variant-united-kingdom (accessed on 17 April 2021).
- Davies, N.G.; Jarvis, C.I.; Edmunds, W.J.; Jewell, N.P.; Diaz-Ordaz, K.; Keogh, R.H.; CMMID COVID-19 Working Group. Increased mortality in community-tested cases of SARS-CoV-2 lineage B.1.1.7. Nature 2021, 593, 270–274. [Google Scholar] [CrossRef]
- Volz, E.; Hill, V.; McCrone, J.T.; Price, A.; Jorgensen, D.; O’Toole, Á.; Southgate, J.; Johnson, R.; Jackson, B.; Nascimento, F.F.; et al. Evaluating the Effects of SARS-CoV-2 Spike Mutation D614G on Transmissibility and Pathogenicity. Cell 2021, 184, 64–75.e11. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.W.; Toovey, O.T.R.; Harvey, K.N.; Hui, D.D.S. Introduction of the South African SARS-CoV-2 variant 501Y.V2 into the UK. J. Infect. 2021, 82, e8–e10. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Nair, M.S.; Liu, L.; Iketani, S.; Luo, Y.; Guo, Y.; Wang, M.; Yu, J.; Zhang, B.; Kwong, P.D.; et al. Antibody resistance of SARS-CoV-2 variants B.1.351 and B.1.1.7. Nature 2021, 593, 130–135. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Casner, R.G.; Nair, M.S.; Wang, M.; Yu, J.; Cerutti, G.; Liu, L.; Kwong, P.D.; Huang, Y.; Shapiro, L.; et al. Increased resistance of SARS-CoV-2 variant P.1 to antibody neutralization. Cell Host Microbe 2021, 29, 747–751.e4. [Google Scholar] [CrossRef]
- Conti, P.; Caraffa, A.; Gallenga, C.E.; Kritas, S.K.; Frydas, I.; Younes, A.; Di Emidio, P.; Tetè, G.; Pregliasco, F.; Ronconi, G. The British variant of the new coronavirus-19 (Sars-Cov-2) should not create a vaccine problem. J. Biol. Regul. Homeost. Agents 2021, 35, 1–4. [Google Scholar] [PubMed]
- Segal, D.; Rotschield, J.; Ankory, R.; Kutikov, S.; Moaddi, B.; Verhovsky, G.; Benov, A.; Twig, G.; Glassberg, E.; Fink, N.; et al. Measures to Limit COVID-19 Outbreak Effects Among Military Personnel: Preliminary Data. Mil. Med. 2020, 185, e1624–e1631. [Google Scholar] [CrossRef] [PubMed]
- Baettig, S.J.; Parini, A.; Cardona, I.; Morand, G.B. Case series of coronavirus (SARS-CoV-2) in a military recruit school: Clinical, sanitary and logistical implications. BMJ Mil. Health 2020. [Google Scholar] [CrossRef] [Green Version]
- Pirnay, J.P.; Selhorst, P.; Cochez, C.; Petrillo, M.; Claes, V.; Van der Beken, Y.; Verbeken, G.; Degueldre, J.; T’Sas, F.; Van den Eede, G.; et al. Study of a SARS-CoV-2 Outbreak in a Belgian Military Education and Training Center in Maradi, Niger. Viruses 2020, 12, 949. [Google Scholar] [CrossRef] [PubMed]
- Quick, J.; Grubaugh, N.D.; Pullan, S.T.; Claro, I.M.; Smith, A.D.; Gangavarapu, K.; Oliveira, G.; Robles-Sikisaka, R.; Rogers, T.F.; Beutler, N.A.; et al. Multiplex PCR method for MinION and Illumina sequencing of zika and other virus genomes directly from clinical samples. Nat. Protoc. 2017, 12, 1261–1266. [Google Scholar] [CrossRef] [Green Version]
- Colin, A. Nanopore Pipeline Wrapper. Available online: https://github.com/ColinAnthony/nanopore_pipeline_wrapper (accessed on 17 April 2021).
- Loman, N.; Rowe, W.; Rambaut, A. nCoV-2019 Novel Coronavirus Bioinformatics Protocol. Available online: https://artic.network/ncov-2019/ncov2019-bioinformatics-sop.html (accessed on 17 April 2021).
- Simpson, J. Nanopolish. Available online: https://github.com/jts/nanopolish (accessed on 17 April 2021).
- Elbe, S.; Buckland-Merrett, G. Data, disease and diplomacy: GISAID’s innovative contribution to global health. Glob. Chall. 2017, 1, 33–46. [Google Scholar] [CrossRef] [Green Version]
- Henikoff, S.; Henikoff, J.G. Amino acid substitution matrices from protein blocks. Proc. Natl. Acad. Sci. USA 1992, 89, 10915–10919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemey, P.; Hong, S.L.; Hill, V.; Baele, G.; Poletto, C.; Colizza, V.; O’Toole, Á.; McCrone, J.T.; Andersen, K.G.; Worobey, M.; et al. Accommodating individual travel history and unsampled diversity in Bayesian phylogeographic inference of SARS-CoV-2. Nat. Commun. 2020, 11, 5110. [Google Scholar] [CrossRef]
- Hadfield, J.; Megill, C.; Bell, S.M.; Huddleston, J.; Potter, B.; Callender, C.; Sagulenko, P.; Bedford, T.; Neher, R.A. Nextstrain: Real-time tracking of pathogen evolution. Bioinformatics 2018, 34, 4121–4123. [Google Scholar] [CrossRef] [PubMed]
- Savitzky, A.; Golay, M.J.E. Smoothing and Differentiation of Data by Simplified Least Squares Procedures. Anal. Chem. 1964, 36, 1627–1639. [Google Scholar] [CrossRef]
- Virtanen, P.; Gommers, R.; Oliphant, T.E.; Haberland, M.; Reddy, T.; Cournapeau, D.; Burovski, E.; Peterson, P.; Weckesser, W.; Bright, J.; et al. SciPy 1.0: Fundamental algorithms for scientific computing in Python. Nat. Methods 2020, 17, 261–272. [Google Scholar] [CrossRef] [Green Version]
- Rambaut, A.; Holmes, E.C.; Hill, V.; O’Toole, Á.; McCrone, J.T.; Ruis, C.; du Plessis, L.; Pybus, O.G. A dynamic nomenclature proposal for SARS-CoV-2 to assist genomic epidemiology. Nat. Microbiol. 2020, 5, 1403–1407. [Google Scholar] [CrossRef] [PubMed]
- Aksamentov, I.; Neher, R. Nextclade. Available online: https://clades.nextstrain.org (accessed on 13 May 2021).
- Hodcroft, E. CoVariants. Available online: https://covariants.org (accessed on 17 April 2021).
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rambaut, A.; Lam, T.T.; Max Carvalho, L.; Pybus, O.G. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol. 2016, 2, vew007. [Google Scholar] [CrossRef] [Green Version]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [Green Version]
- Sagulenko, P.; Puller, V.; Neher, R.A. TreeTime: Maximum-likelihood phylodynamic analysis. Virus Evol. 2018, 4, vex042. [Google Scholar] [CrossRef] [PubMed]
- Yu, G. Using ggtree to Visualize Data on Tree-Like Structures. Curr. Protoc. Bioinformatics 2020, 69, e96. [Google Scholar] [CrossRef]
- Mercatelli, D.; Giorgi, F.M. Geographic and Genomic Distribution of SARS-CoV-2 Mutations. Front. Microbiol. 2020, 11, 1800. [Google Scholar] [CrossRef] [PubMed]
- Coppée, F.; Lechien, J.R.; Declèves, A.E.; Tafforeau, L.; Saussez, S. Severe acute respiratory syndrome coronavirus 2: Virus mutations in specific European populations. New Microbes New Infect. 2020, 36, 100696. [Google Scholar] [CrossRef]
- Bianchi, M.; Borsetti, A.; Ciccozzi, M.; Pascarella, S. SARS-Cov-2 ORF3a: Mutability and function. Int. J. Biol. Macromol. 2021, 170, 820–826. [Google Scholar] [CrossRef]
- Issa, E.; Merhi, G.; Panossian, B.; Salloum, T.; Tokajian, S. SARS-CoV-2 and ORF3a: Nonsynonymous Mutations, Functional Domains, and Viral Pathogenesis. mSystems 2020, 5, e00266-20. [Google Scholar] [CrossRef] [PubMed]
- Flores-Alanis, A.; Cruz-Rangel, A.; Rodríguez-Gómez, F.; González, J.; Torres-Guerrero, C.A.; Delgado, G.; Cravioto, A.; Morales-Espinosa, R. Molecular Epidemiology Surveillance of SARS-CoV-2: Mutations and Genetic Diversity One Year after Emerging. Pathogens 2021, 10, 184. [Google Scholar] [CrossRef] [PubMed]
- Hillen, H.S.; Kokic, G.; Farnung, L.; Dienemann, C.; Tegunov, D.; Cramer, P. Structure of replicating SARS-CoV-2 polymerase. Nature 2020, 584, 154–156. [Google Scholar] [CrossRef]
- Korber, B.; Fischer, W.M.; Gnanakaran, S.; Yoon, H.; Theiler, J.; Abfalterer, W.; Hengartner, N.; Giorgi, E.E.; Bhattacharya, T.; Foley, B.; et al. Tracking Changes in SARS-CoV-2 Spike: Evidence that D614G Increases Infectivity of the COVID-19 Virus. Cell 2020, 182, 812–827.e19. [Google Scholar] [CrossRef]
- Long, S.W.; Olsen, R.J.; Christensen, P.A.; Bernard, D.W.; Davis, J.J.; Shukla, M.; Nguyen, M.; Saavedra, M.O.; Yerramilli, P.; Pruitt, L.; et al. Molecular Architecture of Early Dissemination and Massive Second Wave of the SARS-CoV-2 Virus in a Major Metropolitan Area. mBio 2020, 11, e02707-20. [Google Scholar] [CrossRef] [PubMed]
- Dearlove, B.; Lewitus, E.; Bai, H.; Li, Y.; Reeves, D.B.; Joyce, M.G.; Scott, P.T.; Amare, M.F.; Vasan, S.; Michael, N.L.; et al. A SARS-CoV-2 vaccine candidate would likely match all currently circulating variants. Proc. Natl. Acad. Sci. USA 2020, 117, 23652–23662. [Google Scholar] [CrossRef] [PubMed]
- Vogels, C.B.F.; Brito, A.F.; Wyllie, A.L.; Fauver, J.R.; Ott, I.M.; Kalinich, C.C.; Petrone, M.E.; Casanovas-Massana, A.; Catherine Muenker, M.; Moore, A.J.; et al. Analytical sensitivity and efficiency comparisons of SARS-CoV-2 RT-qPCR primer-probe sets. Nat. Microbiol. 2020, 5, 1299–1305. [Google Scholar] [CrossRef] [PubMed]
- Chinese Centers for Disease Control and Prevention (China CDC). Specific Primers and Probes for Detection 2019 Novel Coronavirus. Available online: http://ivdc.chinacdc.cn/kyjz/202001/t20200121_211337.html (accessed on 17 April 2021).
- Islam, M.R.; Hoque, M.N.; Rahman, M.S.; Alam, A.S.M.R.U.; Akther, M.; Puspo, J.A.; Akter, S.; Sultana, M.; Crandall, K.A.; Hossain, M.A. Genome-wide analysis of SARS-CoV-2 virus strains circulating worldwide implicates heterogeneity. Sci. Rep. 2020, 10, 14004. [Google Scholar] [CrossRef] [PubMed]
- Rockett, R.J.; Arnott, A.; Lam, C.; Sadsad, R.; Timms, V.; Gray, K.A.; Eden, J.S.; Chang, S.; Gall, M.; Draper, J.; et al. Revealing COVID-19 transmission in Australia by SARS-CoV-2 genome sequencing and agent-based modeling. Nat. Med. 2020, 26, 1398–1404. [Google Scholar] [CrossRef] [PubMed]
- Grabowski, F.; Preibisch, G.; Giziński, S.; Kochańczyk, M.; Lipniacki, T. SARS-CoV-2 Variant of Concern 202012/01 Has about Twofold Replicative Advantage and Acquires Concerning Mutations. Viruses 2021, 13, 392. [Google Scholar] [CrossRef] [PubMed]
- European Centre for Disease Prevention and Control (ECDC). ECDC PrimerScan. Available online: https://primerscan.ecdc.europa.eu/ (accessed on 17 April 2021).
- Zheng, Y.; Zhuang, M.W.; Han, L.; Zhang, J.; Nan, M.L.; Zhan, P.; Kang, D.; Liu, X.; Gao, C.; Wang, P.H. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) membrane (M) protein inhibits type I and III interferon production by targeting RIG-I/MDA-5 signaling. Signal Transduct. Target Ther. 2020, 5, 299. [Google Scholar] [CrossRef] [PubMed]
- Centre for Virus Research. CoV-GLUE. M Replacement L13F. Available online: http://cov-glue.cvr.gla.ac.uk/#/project/replacement/M:L:13:F (accessed on 29 May 2021).
- Nelson, G.; Buzko, O.; Spilman, P.R.; Niazi, K.; Rabizadeh, S.; Soon-Shiong, P.R. Molecular dynamic simulation reveals E484K mutation enhances spike RBD-ACE2 affinity and the combination of E484K, K417N and N501Y mutations (501Y.V2 variant) induces conformational change greater than N501Y mutant alone, potentially resulting in an escape mutant. bioRxiv 2021. [Google Scholar] [CrossRef]
- Tchesnokova, V.; Kulakesara, H.; Larson, L.; Bowers, V.; Rechkina, E.; Kisiela, D.; Sledneva, Y.; Choudhury, D.; Maslova, I.; Deng, K.; et al. Acquisition of the L452R mutation in the ACE2-binding interface of Spike protein triggers recent massive expansion of SARS-Cov-2 variants. bioRxiv 2021. [Google Scholar] [CrossRef]
- Deng, X.; Garcia-Knight, M.A.; Khalid, M.M.; Servellita, V.; Wang, C.; Morris, M.K.; Sotomayor-González, A.; Glasner, D.R.; Reyes, K.R.; Gliwa, A.S.; et al. Transmission, infectivity, and antibody neutralization of an emerging SARS-CoV-2 variant in California carrying a L452R spike protein mutation. medRxiv 2021. [Google Scholar] [CrossRef]
- European Centre for Disease Prevention and Control (ECDC). SARS-CoV-2 Variants of Concern as of 11 May 2021. Available online: https://www.ecdc.europa.eu/en/covid-19/variants-concern (accessed on 14 May 2021).
- Anoh, A.E.; Schubert, G.; Wayoro, O.; Pacôme, M.; Belarbi, E.; Sachse, A.; Calvignac-Spencer, S.; Leendertz, F.; Diané, B.; Akoua-Koffi, C. SARS-CoV-2 variants of concern, variants of interest and lineage A.27 are on the rise in Côte d’Ivoire. medRxiv 2021. [Google Scholar] [CrossRef]
- Calvignac-Spencer, S.; Budt, M.; Huska, M.; Richard, H.; von Kleist, M.; Kröger, S.; Semmler, T.; Wolff, T.; Hölzer, M. Emergence of SARS-CoV-2 Lineage A.27 in Germany, Expressing Viral Spike Proteins with Several Amino Acid Replacements of Interest, Including L18F, L452R, and N501Y in the Absence of D614G. Virological.org 15 May 2021. Available online: https://virological.org/t/emergence-of-sars-cov-2-lineage-a-27-in-germany-expressing-viral-spike-proteins-with-several-amino-acid-replacements-of-interest-including-l18f-l452r-and-n501y-in-the-absence-of-d614g/693/1 (accessed on 2 June 2021).
- Public Health England (PHE). Research and Analysis. Variants: Distribution of Cases Data. Available online: https://www.gov.uk/government/publications/covid-19-variants-genomically-confirmed-case-numbers/variants-distribution-of-cases-data (accessed on 14 May 2021).
- Jangra, S.; Ye, C.; Rathnasinghe, R.; Stadlbauer, D.; Krammer, F.; Simon, V.; Martinez-Sobrido, L.; García-Sastre, A.; Schotsaert, M.; Personalized Virology Initiative Study Group. SARS-CoV-2 spike E484K mutation reduces antibody neutralisation. Lancet Microbe 2021, 2, E283–E284. [Google Scholar] [CrossRef]
- Jangra, S.; Ye, C.; Rathnasinghe, R.; Stadlbauer, D.; Krammer, F.; Simon, V.; Martinez-Sobrido, L.; Garcia-Sastre, A.; Schotsaert, M. The E484K mutation in the SARS-CoV-2 spike protein reduces but does not abolish neutralizing activity of human convalescent and post-vaccination sera. medRxiv 2021. [CrossRef]
- Butera, Y.; Mukantwari, E.; Artesi, M.; Umuringa, J.D.; O’Toole, Á.N.; Hill, V.; Rooke, S.; Hong, S.L.; Dellicour, S.; Majyambere, O. Genomic Sequencing of SARS-CoV-2 in Rwanda: Evolution and regional dynamics. medRxiv 2021. [Google Scholar] [CrossRef]
- Dudas, G.; Hong, S.L.; Potter, B.; Calvignac-Spencer, S.; Niatou-Singa, F.S.; Tombolomako, T.B.; Fuh-Neba, T.; Vickos, U.; Ulrich, M.; Leendertz, F.H. Travel-driven emergence and spread of SARS-CoV-2 lineage B.1.620 with multiple VOC-like mutations and deletions in Europe. medRxiv 2021. [Google Scholar] [CrossRef]
- Worobey, M.; Pekar, J.; Larsen, B.B.; Nelson, M.I.; Hill, V.; Joy, J.B.; Rambaut, A.; Suchard, M.A.; Wertheim, J.O.; Lemey, P. The emergence of SARS-CoV-2 in Europe and North America. Science 2020, 370, 564–570. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pirnay, J.-P.; Selhorst, P.; Hong, S.L.; Cochez, C.; Potter, B.; Maes, P.; Petrillo, M.; Dudas, G.; Claes, V.; Van der Beken, Y.; et al. Variant Analysis of SARS-CoV-2 Genomes from Belgian Military Personnel Engaged in Overseas Missions and Operations. Viruses 2021, 13, 1359. https://0-doi-org.brum.beds.ac.uk/10.3390/v13071359
Pirnay J-P, Selhorst P, Hong SL, Cochez C, Potter B, Maes P, Petrillo M, Dudas G, Claes V, Van der Beken Y, et al. Variant Analysis of SARS-CoV-2 Genomes from Belgian Military Personnel Engaged in Overseas Missions and Operations. Viruses. 2021; 13(7):1359. https://0-doi-org.brum.beds.ac.uk/10.3390/v13071359
Chicago/Turabian StylePirnay, Jean-Paul, Philippe Selhorst, Samuel L. Hong, Christel Cochez, Barney Potter, Piet Maes, Mauro Petrillo, Gytis Dudas, Vincent Claes, Yolien Van der Beken, and et al. 2021. "Variant Analysis of SARS-CoV-2 Genomes from Belgian Military Personnel Engaged in Overseas Missions and Operations" Viruses 13, no. 7: 1359. https://0-doi-org.brum.beds.ac.uk/10.3390/v13071359