
Bacteriophages fEV-1 and fD1 Infect Yersinia pestis
by
 , and
, and
Mikael Skurnik
1,2,* ,
,
Salla Jaakkola
1,
Laura Mattinen
1,
Lotta von Ossowski
3,
Ayesha Nawaz
1,
Maria I. Pajunen
1 and
Lotta J. Happonen
4 1
Department of Bacteriology and Immunology, Medicum, Human Microbiome Research Program, Faculty of Medicine, University of Helsinki, 00290 Helsinki, Finland
2
Division of Clinical Microbiology, HUSLAB, University of Helsinki and Helsinki University Hospital, 00290 Helsinki, Finland
3
Department of Medical Biochemistry, University of Turku, 20520 Turku, Finland
4
Division of Infection Medicine, Department of Clinical Sciences Lund, Lund University, 22184 Lund, Sweden
*
Author to whom correspondence should be addressed.
Viruses 2021, 13(7), 1384; https://0-doi-org.brum.beds.ac.uk/10.3390/v13071384
Submission received: 23 June 2021
/
Revised: 14 July 2021
/
Accepted: 14 July 2021
/
Published: 16 July 2021
(This article belongs to the Special Issue Phage-Host Interactions 2021)
Abstract
:Bacteriophages vB_YpeM_fEV-1 (fEV-1) and vB_YpeM_fD1 (fD1) were isolated from incoming sewage water samples in Turku, Finland, using Yersinia pestis strains EV76 and KIM D27 as enrichment hosts, respectively. Genomic analysis and transmission electron microscopy established that fEV-1 is a novel type of dwarf myovirus, while fD1 is a T4-like myovirus. The genome sizes are 38 and 167 kb, respectively. To date, the morphology and genome sequences of some dwarf myoviruses have been described; however, a proteome characterization such as the one presented here, has currently been lacking for this group of viruses. Notably, fEV-1 is the first dwarf myovirus described for Y. pestis. The host range of fEV-1 was restricted strictly to Y. pestis strains, while that of fD1 also included other members of Enterobacterales such as Escherichia coli and Yersinia pseudotuberculosis. In this study, we present the life cycles, genomes, and proteomes of two Yersinia myoviruses, fEV-1 and fD1.
1. Introduction
Yersinia pestis, the causative agent of bubonic and pneumonic plague, is a zoonotic bacterium spread by fleas in small rodents [1]. Around 2000 cases of human plague are reported to WHO every year, Madagascar leading the statistics with hundreds of cases per year with many leading to death [2,3]. A closely related bacterium, Yersinia pseudotuberculosis and the more distant Yersinia enterocolitica, cause gastroenteritis and mesenteric lymphadenitis in humans. Antibiotic resistant strains of Y. pestis and Y. pseudotuberculosis are an emerging threat, and concerns for biological warfare have also been voiced [4]. Due to these threats, managing bacterial infections using bacteriophages, i.e., phage therapy, has gained serious interest lately, also in the case of Y. pestis [5]. Several phages that infect Y. pestis have been described throughout the years and, recently, these have been reviewed [6]. Indeed, Felix d’Herelle used phage therapy against bubonic plague in the 1920s [7].
Y. pestis specific phages, such as ΦA1122 [8] and L-413C [9], have been used in Y. pestis diagnostics and identification, and the potential of phage therapy was also studied [5]. For therapeutic use, phages need to be both lytic and nontoxic to humans. Reported lytic Yersinia phages are mostly podoviruses [6], but myoviruses, such as PST [10], ΦJA1 [11], PY100 [12], and JC221 [13] have also been described. In addition to infectious “live” phage particles, certain phage proteins can also act as antimicrobials [14].
Members of the Myoviruses are typically dsDNA viruses with isometric heads and contractile tails. Escherichia virus T4 is the type species in genus Tequatrovirus of the subfamily Tevenvirinae that belongs in the classification of the International Committee for the Taxonomy of Viruses (ICTV) to the branch Duplodnaviria > Heunggongvirae > Uroviricota > Caudoviricetes > Caudovirales > Myoviridae (https://talk.ictvonline.org/taxonomy/ 22 April 2021). Tequatroviruses are lytic and infect Gram-negative bacteria. They typically have elongated heads around 120 × 86 nm in size, an average tail length of approximately 140 nm, and their genomes are fairly large, around 160–250 kb in size [15]. Currently, ICTV classifies two sequenced Y. pestis infecting phages, PST and fD1, as their own species in genus Tequatrovirus [16].
Myoviruses with small-sized heads and genome sizes below 50 kb have been called “dwarf” myoviruses [17,18]. Their head diameters vary from 55 to 75 nm, and most have tails from 55 to 85 nm, although Aggregatibacter phages with 112–115 nm tails have also been included in this group [17]. Some of these viruses share sequence homology and genome synteny, and two different genera have been suggested. The ΦPLPE-like viruses [17] include both lytic and temperate phages, and they infect a wide range of Gram-negative bacteria, whereas the proposed phiMMP04likevirus consists of phages infecting Clostridium difficile [19]. There are only two dwarf myoviruses infecting Yersinia, i.e., PY100 and fEV-1, that have been isolated so far [6,12].
In this study we present detailed characterization of two phages that infect Y. pestis. The phages fEV-1 and fD1 were isolated in 1999 from a Turku City sewage water sample. We originally named the phages ΦEV-1 and ΦD1 or phiD1, but to avoid using the Greek letters, hereafter, we use the names fEV-1 and fD1. Their annotated genome sequences were submitted to sequence databases under accession numbers LT992259 and HE956711, respectively.
2. Materials and Methods
2.1. Bacterial Strains, Phages, and Media
Bacterial strains and phages used in this study are listed in Table 1. Yersinia cultures were grown at room temperature (RT, 22 °C) in tryptone soy broth (TSB, Lab M) and E. coli strains at 37 °C in lysogeny broth (LB). For plating, brain heart infusion agar (BHI, Becton Dickinson Co., Oxford, UK) was used for Yersinia strains and LB agar (LA) for E. coli. For solid plates and soft agar media, 1.5% or 0.4% bacteriological agar (Lab M) was used, respectively. Kanamycin (Kan, 30 µg/mL) was added when required.
2.2. Phage Titration, Host Screening, and Efficiency of Plating
Phage titers were determined by double-layer agar method [27]. For host screening, bacteria (Table 1) were plated using the double-layer method, and drops of fD1 and fEV-1 dilutions were added on plates. Plates were incubated overnight and screened for plaques to determine the host specificity. Efficiency of plating was further determined on susceptible hosts by titration. Y. pestis D27 and EV76 were used as positive controls.
2.3. Phage Production and Purification
For phage production, overnight liquid cultures of Y. pestis D27 or Y. pestis EV76 were plated with fD1 or fEV-1, respectively, to produce semi confluent plates. The soft agar layer was mixed with SM buffer (100 mM NaCl, 10 mM MgSO4, 50 mM Tris-HCl (pH 7.5), 0.01% gelatin), centrifuged, and the supernatant was treated with chloroform to lyse any remaining bacteria. The lysate was filtered, and sucrose was added to a final concentration of 0.8% (w/v).
The phage preparations were further purified by discontinuous glycerol density gradient ultracentrifugation through 5% and 40% glycerol layers in TM buffer using the Beckman BSW55Ti rotor (Indianapolis, IN, USA) at 40,000 rpm at 4 °C for 3 h [28]. The phage pellet was resuspended in SM buffer containing 8% sucrose.
2.4. Electron Microscopy
Aliquots of phage preparate (5 μL) were adsorbed on holey-carbon film coated grids (Quantifoil R 2/2, Electron Microscopy Sciences, Hatfield, PA, USA) for 1 min prior to negative staining with 2% (w/v) uranyl acetate (pH 4.5). The specimens were imaged in a FEI Tecnai F20 field emission gun transmission electron microscope (FEI, Eindhoven, the Netherlands) operating at 200 kV. Micrographs were recorded on a Gatan UltraScan 4000 charge-coupled-device (CCD) camera (Gatan, Inc., Pleasanton, CA, USA) at nominal magnifications of 39,440× and 68,000×. The data were collected in the Biocenter Finland National Cryo-EM unit, Institute of Biotechnology, University of Helsinki (Helsinki, Finland).
2.5. Phage Genome Extraction, Sequencing, and Analysis
Phage DNA was obtained from high-titer phage preparations, as described earlier [27]. Phage fD1 DNA was sequenced using Illumina GAIIx (genome analyzer) technology at the FIMM Sequencing unit (Helsinki, Finland). The sequence assembly was performed with the NextGene (http://www.softgenetics.com (accessed on 12 December 2010)) and Staden software packages [29]. The Artemis genome-browsing and annotation tool [30] was used for genome annotation. Phage fEV-1 DNA was sequenced as above but due to low quality, complemented in addition with a second round using the sequencing service at Novogene Europe (Cambridge, UK) using Illumina HiSeq (San Diego, CA, USA) with 150 bp paired end reads.
The obtained sequence reads were assembled de novo using the A5-miseq pipeline [31]. The read coverages of the resulting contigs were checked with the Artemis software [32,33] and contaminating bacterial genomic sequences identified by their >100-fold lower read coverages (the read coverages of the phage contigs were >10,000) and BLASTN searches against the Y. pestis CO-92 genome sequence (accession no. CP009973). To verify the fidelity of the assemblies, the reads were mapped back to the de novo assembled contigs using the tools of the Geneious Prime software version 2021.0.3 (Biomatters Ltd., Auckland, New Zealand) [34]. Preliminary annotations of the phage genomes were carried out using Rapid Annotation Subsystems Technology [RAST] [35] that was manually checked and revised with the Artemis software [32,33]. The PhageTerm program was used to identify the termini of the phage genomes [36] and tRNAscan-SE v. 2.0 was used to identify tRNA genes [37,38].
The identities and functions of the predicted genes and gene products were analyzed using the PSI-BLAST (https://blast.ncbi.nlm.nih.gov/Blast.cgi (accessed on several occasions since 10 February 2012)) server. Different EMBOSS sequence analysis tools were used through the Chipster platform [39] at the Centers for Scientific Computing (https://www.csc.fi/ (accessed on several occasions since 10 February 2012)). The phylogenetic phage proteomic trees were generated by VIPTree [40]. The promoters and terminators were predicted using the BPROM and FindTerm tools [41].
2.6. PCR and Sanger Sequencing
The PCR primers (Table 2) were designed using the EMBOSS Eprimer3 tool in Chipster [39], and commercially synthetized at Metabion International AG (Steinkirchen, Germany). The PCRs were performed in 0.2 mL thin-walled PCR tubes (4titude® Ltd., Wotton, Surrey, UK), in a total volume of 25 μL containing 1 μL of DNA template, 0.2 μM of each primer (Table 2), 200 μM of dNTP mix (Thermo Fisher Scientific, Waltham, MA, USA), 2.5 μL of 10× Standard Taq Reaction Buffer, and 1.25 U of Taq DNA Polymerase (Thermo Fisher Scientific, Waltham, MA, USA). The PCR cycling included an initial denaturation at 95 °C for 3 min, followed by 34 cycles each consisting of denaturation at 95 °C for 30 s, 30 s at annealing temperature, and extension for 30 s at 72 °C. This was followed by a final extension step at 72 °C for 5 min, after which PCR products were kept on hold at 4 °C until further processing. The annealing temperatures were calculated using the on-line service at https://www.thermofisher.com/ (accessed on 8 November 2020). The PCR products were analyzed using 1% agarose gel electrophoresis and cleaned using the Nucleospin Gel extraction and PCR clean-up kit (Macherey-Nagel GmbH, Düren, Germany). The purified PCR fragments were sequenced with appropriate sequencing primers using the Sanger sequencing service at the Institute for Molecular Medicine Finland (https://www.fimm.fi/en/services/technology-centre/sequencing (accessed on 4 December 2020)).
2.7. Restriction Endonuclease Analysis
Restriction digestions were carried out using the restriction enzymes EcoRI, HincII, NsiI, PstI, SalI, ScaI, SexAI, and SpeI (Thermo Fischer Scientific). These enzymes gave several well separated bands when in silico digested by the NEBcutter (http://nc2.neb.com/NEBcutter2/ (accessed on 8 May 2021)). The digestions were carried out in a final volume of 10 µL, containing DNA (ca. 300 ng), 0.5 µL of enzyme, and 1 µL of Fast digest green buffer (10×, Thermo Fisher Scientific). After 2–16 h incubation at 37 °C, the restriction fragments along with undigested DNA and GeneRuler 1 kb plus DNA Ladder (Thermo Fisher Scientific) were separated on 1% (w/v) agarose gel and detected with ethidium bromide staining using UV transillumination. Images were recorded using the BioRad GelDoc XR+ imaging system.
2.8. Sample Preparation for Mass Spectrometry
For the proteome analyses, the phages fEV-1 and fD1 were purified as described above, except that the final resuspension after ultracentrifugation was done in SM buffer without sucrose. The purified fEV-1 phage preparation was digested for mass spectrometry in triplicates, whereas the fD1 dataset consisted of two different phage preparations both as triplicate samples. Briefly, the samples were mixed with 8 M urea and 100 mM ammonium bicarbonate, and the cysteine bonds were reduced with 5 mM TCEP (37 °C for 30 min) and alkylated with 10 mM iodoacetamide (22 °C for 60 min). Samples were diluted with 100 mM ammonium bicarbonate to a final urea concentration of 1.5 M, and sequencing grade trypsin (Promega, Madison, WI, USA) was added for protein digestion (37 °C for 18 h). Samples were acidified (to a final pH 3.0) with 10% formic acid, and the peptides subsequently purified with C18 reverse phase spin columns, according to the manufacturer’s instructions (Microspin and Macrospin columns, Harvard Apparatus, Holliston, MA, USA). Peptides were dried using a Speedvac and reconstituted in 2% acetonitrile/0.2% formic acid prior to mass spectrometric analyses.
2.9. Sample Preparation for Mass Spectrometry
The peptide analyses were performed on a Q Exactive Plus (fEV-1 and fD1) or Q Exactive HFX (fD1) mass spectrometer (Thermo Scientific). The Q Exactive Plus mass spectrometer was connected to an EASY-nLC 1000 ultra-high-performance liquid chromatography system (Thermo Scientific). The peptides were separated on an EASY-Spray column (Thermo Scientific) ID 75 μm × 25 cm, column temperature at 45 °C. The column was equilibrated, and the samples loaded using a constant pressure of 600 bars. The peptides were separated using a linear gradient from 5 to 35% acetonitrile in aqueous 0.1% formic acid for 75 min at a flow rate of 300 nL min−1. One full MS scan (resolution 70,000 at 200 m/z, mass range 400–1600 m/z) was followed by MS/MS scans (resolution 17,500 at 200 m/z) of the 15 most abundant ion signals. Precursor ions were isolated with 2 m/z isolation width and fragmented using higher-energy collisional-induced dissociation (HCD) at a normalized collision energy (NCE) of 30. Charge state screening was enabled, and precursors with an unknown charge state and singly charged ions were rejected. The automatic gain control was set to 1 × 106 for both MS and MS/MS with ion accumulation times of 100 and 60 ms, respectively. The intensity threshold for precursor ion selection was set to 1.7 × 104.
The Q Exactive HFX mass spectrometer was connected to an EASY-nLC 1200 ultra-high-performance liquid chromatography system (Thermo Scientific). Peptides were separated on an EASY-Spray column (Thermo Scientific) ID 75 μm × 25 cm, column temperature 45 °C, operated at a constant pressure of 800 bar. A linear gradient from 5% to 35% acetonitrile in aqueous 0.1% formic acid was run for 65 min at a flow rate of 300 nL min−1. One full MS scan (resolution 60,000@200 m/z, mass range 350–1400 m/z) was followed by MS/MS scans (resolution 15,000@200 m/z) of the 15 most abundant ion signals. The precursor ions were isolated with 1.3 m/z isolation width and fragmented using HCD at an NCE of 28. Precursors with an unknown charge state, singly charged ions or with a charge state of 6 or above, were rejected. The dynamic exclusion window was set to 10 s. The automatic gain control was set to 1 × 106 for both MS and MS/MS, with ion accumulation times of 100 and 60 ms, respectively. The intensity threshold for precursor ion selection was set to 1.7 × 104.
2.10. Mass Spectrometry Data Analysis
MS raw data were converted to gzipped and Numpressed [42] mzML using the tool msconvert from ProteoWizard, v3.0.5930 suite [43]. All data analyses were stored and managed using openBIS [44]. For fEV-1, acquired spectra were analyzed using the search engine X! Tandem (2013.06.15.1-LabKey, Insilicos, ISB) [45] against an in-house compiled dataset containing the reference proteome of Y. pestis CO-92/Biovar Orientalis (UniProt proteome ID UP000000815) and that of Yersinia phage fEV-1 (UniProt proteome ID UP000274108) (both accessed on 8 June 2021), yielding a total of 3966 protein entries and an equal amount of reverse decoy sequences. We also performed an additional analysis in order to identify any expressed open reading frames (ORFs) missed in the genome annotation. For this, the genome of fEV-1 was analyzed for ORFs via the NCBI ORF finder tool (https://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/orffinder/ (accessed on 12 June 2021)) using 75 nt as the minimal ORF length, standard genetic code as code, and ATG and alternative initiation codons as ORF start codon. This approach generated 534 translated ORFs, which were used together with an equal amount of reverse decoy sequences as an alternative reference proteome.
For fD1, acquired spectra were analyzed against an in-house compiled dataset containing the Y. pestis CO-92/Biovar Orientalis and Yersinia phage fD1 reference proteomes (UniProt proteome IDs UP000000815 and UP000002906, respectively), yielding a total of 4186 protein entries and an equal amount of reverse decoy sequences. We also performed the same analysis as for fEV-1 in order to identify any expressed ORFs missed in the original genome annotation. For this, the genome of fD1 (accession number HE956711.1) was analyzed for ORFs via the NCBI ORF finder tool (https://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/orffinder/ (accessed on 12 June 2021)) using 75 nt as the minimal ORF length, standard genetic code as code, and ATG and alternative initiation codons as ORF start codon. This approach generated 505 translated ORFs, which was used together with an equal amount of reverse decoy sequences as an alternative reference proteome.
For both phages, fully tryptic digestion was used allowing one missed cleavage; Carbamidomethylation (C) was set to static and oxidation (M) to variable modifications. Mass tolerance for precursor ions was set to 20 ppm, and for fragment ions to 50 ppm. Identified peptides were processed and analyzed through the Trans-Proteomic Pipeline (TPP v4.7 POLAR VORTEX rev 0, Build 201403121010) using PeptideProphet [46]. A protein was only considered identified, if we could detect peptides in all three replicates with an average spectral count of 2 or more. The mass spectrometry data have been deposited to the ProteomeXchange [47] consortium via the MassIVE partner repository (https://massive.ucsd.edu/ (accessed on 19 June 2021)) with the dataset identifiers PXD026811 (fEV-1) and PXD026812 (fD1).
2.11. Phage Growth Curves
Liquid cultures of host bacteria (Y. pestis D27 or E. coli 538 for fD1, and Y. pestis EV76 for fEV-1) in logarithmic phase were infected with a MOI of 0.01 and incubated for 5 min at RT. The remaining free virions were removed by centrifugation (1923× g for 3 min) and rinsing the pellet with TSB, and infected cells were resuspended to original volume. Phage titer was determined at different time points to determine the phage latency period and burst size. The measurements were repeated at least three times.
3. Results and Discussion
3.1. Electron Microscopy
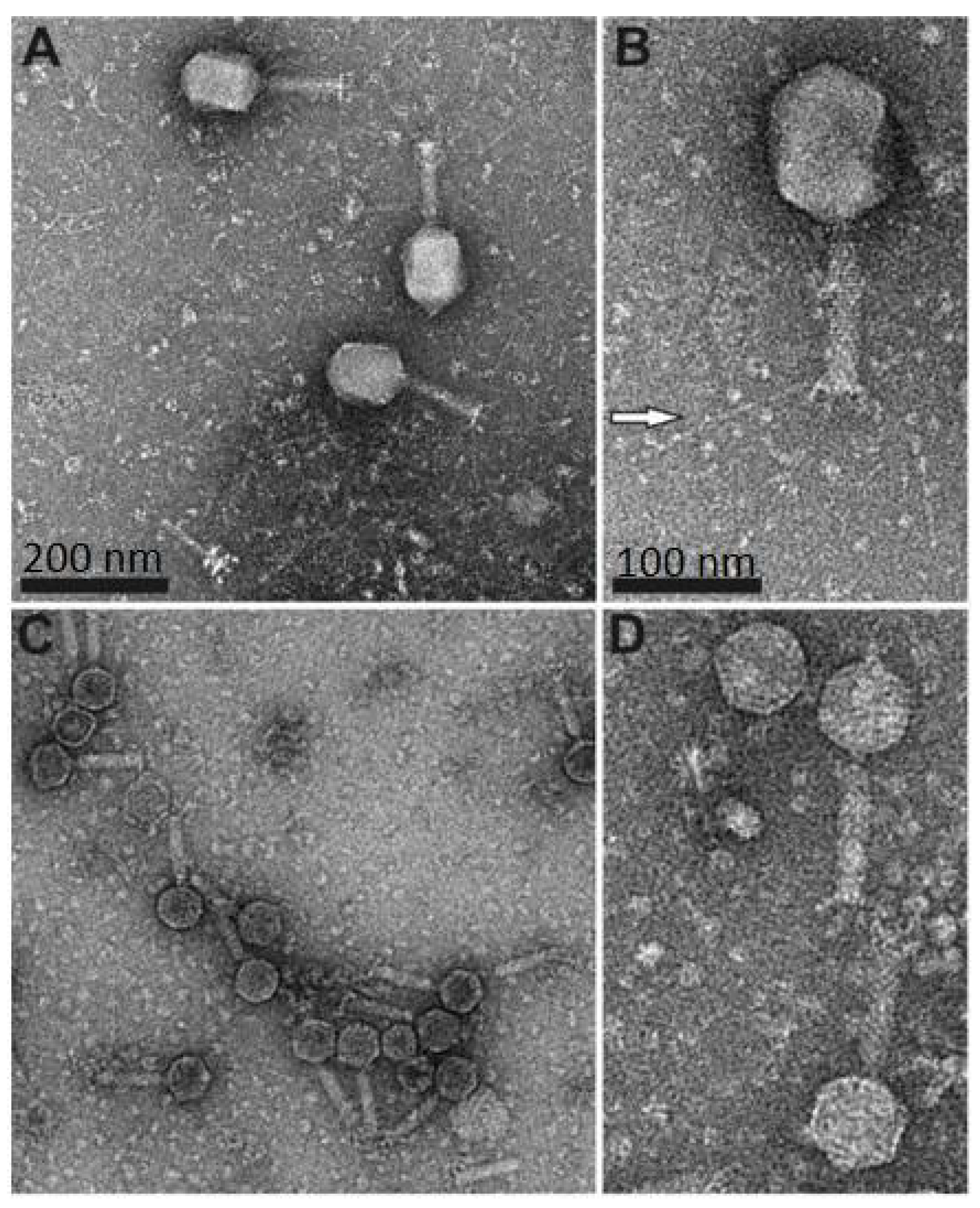
Electron microscopy revealed that both fD1 and fEV-1 were tailed phages with a myovirus morphotype. The isometric head of fEV-1 is 65 ± 3 nm vertex-to-vertex, 59 ± 4 nm edge-to-edge, and the tail has an average length of 92 ± 5 nm (n = 8 for all measurements). The head measures of fEV-1, hence, align well with the head sizes (55–75 nm) of other dwarf myoviruses, whereas the tail of fEV-1 is slightly longer than the reported 55–85 nm [17,18,48,49]. We do not see any apparent long tail fibers in our fEV-1 preparation; however, several thick and short fibers are clearly visible (Figure 1). The prolate head of fD1 is 111 ± 5 nm long and 83 ± 3 nm wide. The tail measures 112 ± 2 nm with extended long tail fibers of an average length of 135 ± 13 nm (for all fD1 measurements, n = 5). These values are very close to those of T4, with a 120 nm long and 86 nm wide prolate head [50], and a 140 nm long contractile tail with six long tail fibers [51].
3.2. Phage Host Specificity and Efficiency of Plating
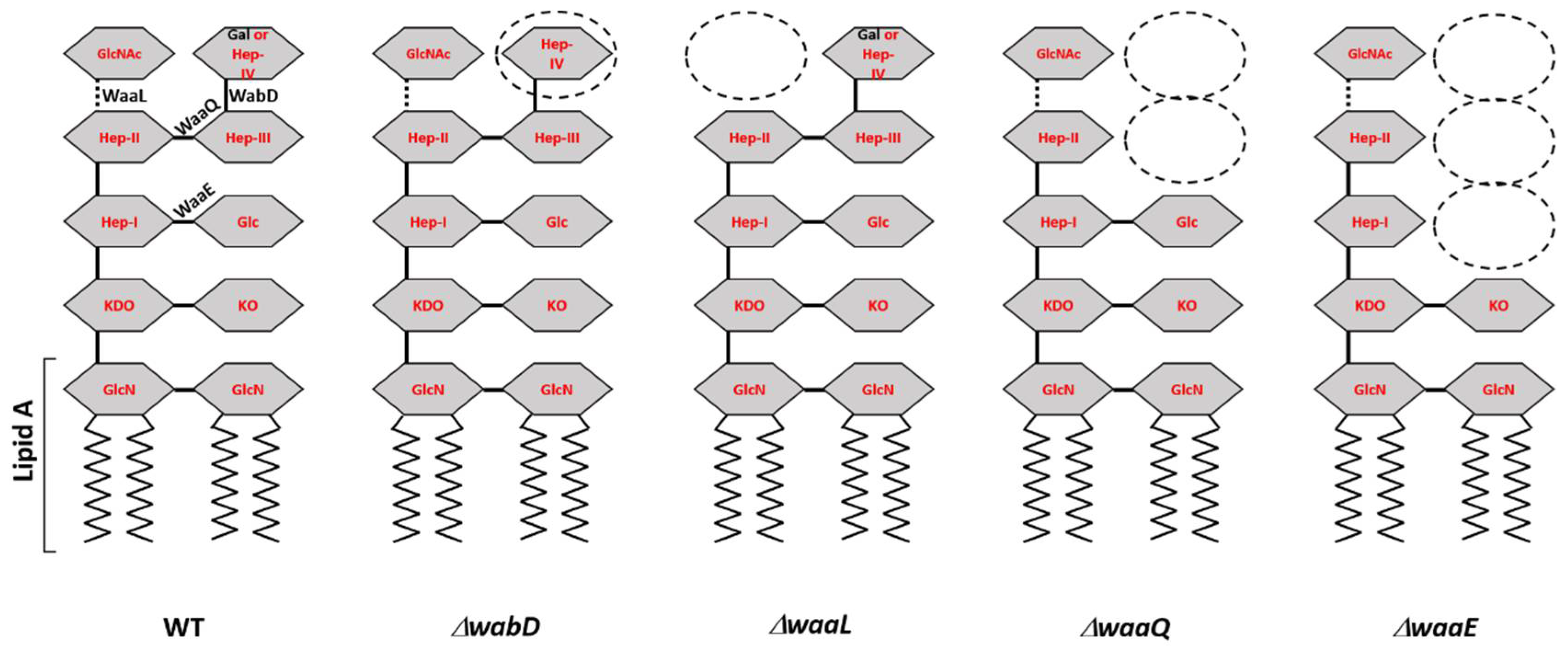
All Y. pestis laboratory strains, and some clinical strains of E. coli (storage #s 6588, 6589, and 6590) and Y. pseudotuberculosis (#s 2069, 2070, and 2649) were susceptible to fD1 (Table 1). Of the tested strains, the fD1 efficiency of plating (Table 1) was highest on E. coli strain 6588, which was later used in the growth curve experiment (see below). While fD1 infected equally all the different Y. pestis D27 LPS-mutants (Figure 2) with progressively truncated LPS core structures [21], for fEV-1, there were some differences in plating efficiencies (Table 1). The EOP of fEV-1 on the ΔwaaQ mutant was 4 logs lower than in LPS wild type strain indicating that the heptose III and IV residues, missing in the mutant, were essential components of the fEV-1 receptor. Furthermore, the additional truncation of the structure by the heptose I -linked glucose in the ΔwaaE mutant decreased the EOP more, altogether 5 logs, indicating that the glucose residue is also an essential component of the receptor. In the ΔwaaE mutant, fEV-1 formed small and turbid plaques that were difficult to see. Interestingly, the distal GlcNAc residue eliminated in the ΔwaaL mutant appeared to play no role for the fEV-1 receptor.
3.3. Phage Growth Curves
In the one step growth curve of phage fEV-1, grown on Y. pestis EV76 at RT, the latency period was exceptionally long, i.e., 185 min, with a burst size of >180 plaque forming units (PFU) per infected cell (Figure 3a). In comparison, the dwarf myovirus ΦPLPE has a 90 min latent period and a burst size of 100 particles [52]. The fEV-1 latency period was twice as long, and the burst size bigger. It is possible that dwarf myoviruses, in general, have long latency periods, but very scarce data has been published on that topic. Moreover, for ΦPLPE, a long latency due to the host being lysogenized by the phage or already harboring a temperate ϕPLPE giving the host superinfection immunity, has been ruled out [52]. In Y. pestis D27 grown at RT, fD1 had a latent period of 35–40 min and had a burst size of 20–30 PFU per infected cell at 50–60 min. On the contrary, when grown in E. coli # 6588 at 37 °C, the growth was significantly more vigorous with a short 20 min latency period and a burst size of >500 PFU per infected cell at 35 min after infection (Figure 3b). The phages both appeared to be efficient in killing Y. pestis (Figure 4). In the fEV-1 infected cultures, regrowth of bacteria, indicating appearance of phage-insensitive mutants, started after 6–7 h of incubation. Similar phenomenon was not apparent for fD1-infected cultures.
3.4. The Genome and Taxonomic Position of fEV-1
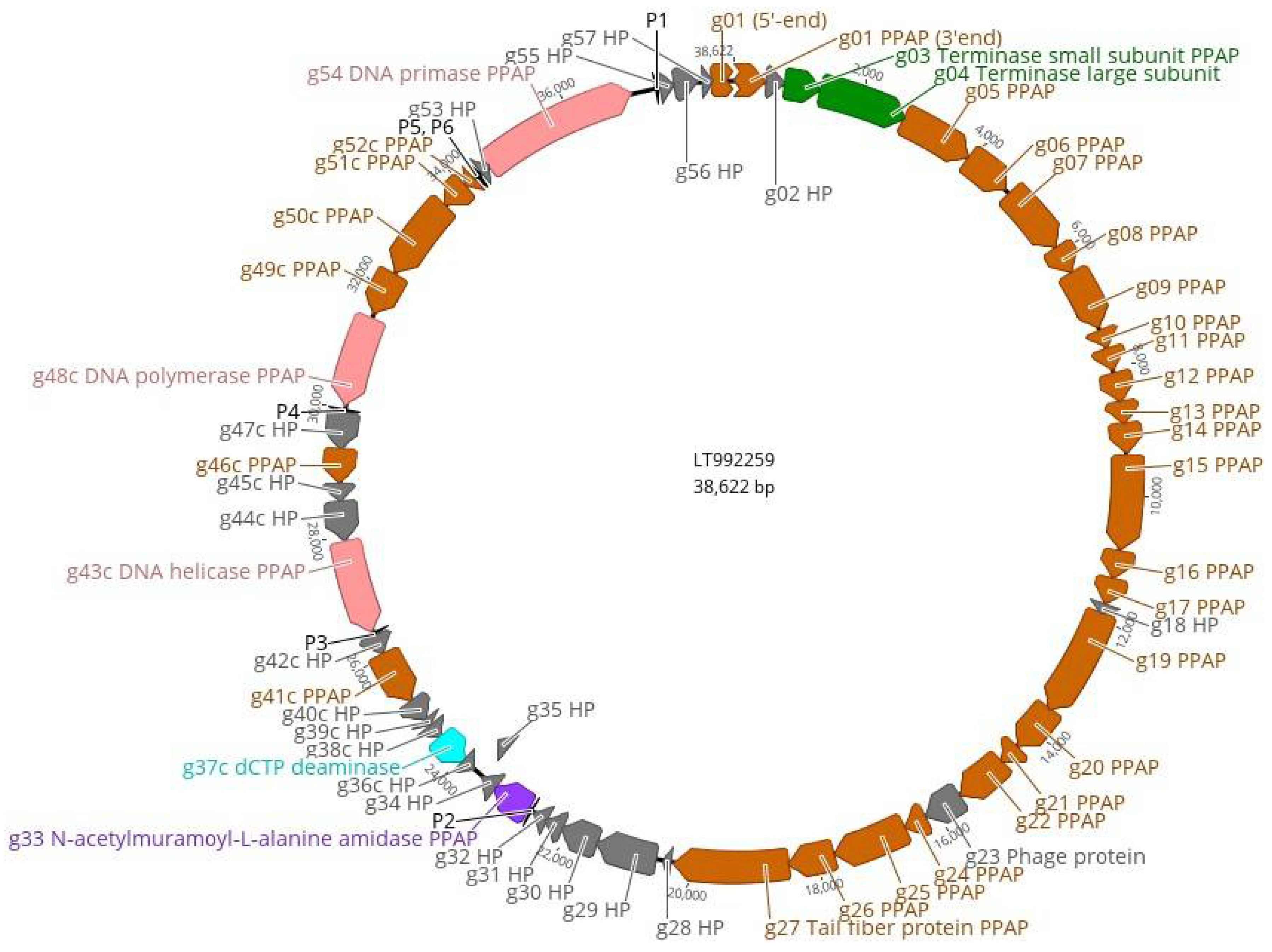
The genome of fEV-1 is small, only 38,622 bp in size; smaller than that of several other dwarf myoviruses [17]. Altogether, four, eleven, seven, and seven restriction sites for AvaI, EcoRV, NcoI, and SexAI, were identified from the sequence, respectively. Restriction digestions with these enzymes produced the in silico predicted restriction fragments, but did not allow unambiguous identification of the physical termini of the genome (Figure S1). PhageTerm analysis could neither identify any fixed termini (Figure S2). Inspection of the sequences at the predicted ends of the genome revealed the presence of a long repeat region that the NGS approach failed to assemble correctly. Several primers were designed on both sides outside the repeat region (Table 2) and the sequences of the fragments amplified by PCR were determined by Sanger sequencing. The results showed that the repeat region was 518 bp long and contained, altogether, 34.5 repeats of the 15 bp sequence 5′-C(g/c)GCGCAAATCTG(g/t/a)(a/c)-3′ that spanned the repeat region. At that point, we had annotated the nucleotide 1 inside the repeat region (Figure 5), but the proteome analysis carried out later revealed that the repeat region was within a coding sequence encoding a pentapeptide-repeat-containing protein (see Section 3.5). Altogether, these findings suggested that the genome might be circularly permuted. This conclusion was supported by an in silico prediction of the restriction fragments, assuming that the genome is circular (Figure S1), which perfectly matched the experimental data.
Another repeat region of 198 bp was located at 33 kb of the genomic map within the gene g51c (Figure 6). It contained 22 repeats of the 9 bp sequence 5′-TGCTGGTGC-3′, thereby, encoding a similar number of Pro-Ala-Ala repeats for Gp51c. In the proteome analysis (see below), Gp51c was detected as a phage particle associated protein (PPAP). However, the role of the Pro-Ala-Ala repeats for Gp51c remains unknown.
These results allowed us to finalize and annotate the genomic nucleotide sequence of fEV-1. The data were complemented by the proteomics data to identify the PPAPs (Section 3.5), and by the prediction of six sigma70-like promoters (P1–P6, Table S1). The overall organization of the genome is presented in Figure 7.
Altogether, 57 predicted genes were identified from the genome with 40 genes in the forward and 17 genes in the reverse strand organized in three different blocks. The promoters P1, P2, and P6 initiate transcription in the forward direction, and the promoters P3, P4, and P5 initiate the transcription of the reverse strand (Figure 7). According to the LC-MS/MS analysis (see below), 33 gene products were identified as PPAPs and 24 as hypothetical proteins (HPs). The nonstructural gene products with a predicted function included the DNA polymerase (Gp48c), DNA primase (Gp54) and DNA helicase (Gp43c), the small and large terminase subunits (Gp03 and Gp04), N-acetylmuramoyl-L-alanine amidase (Gp33), and deoxycytidine triphosphate deaminase (Gp37).
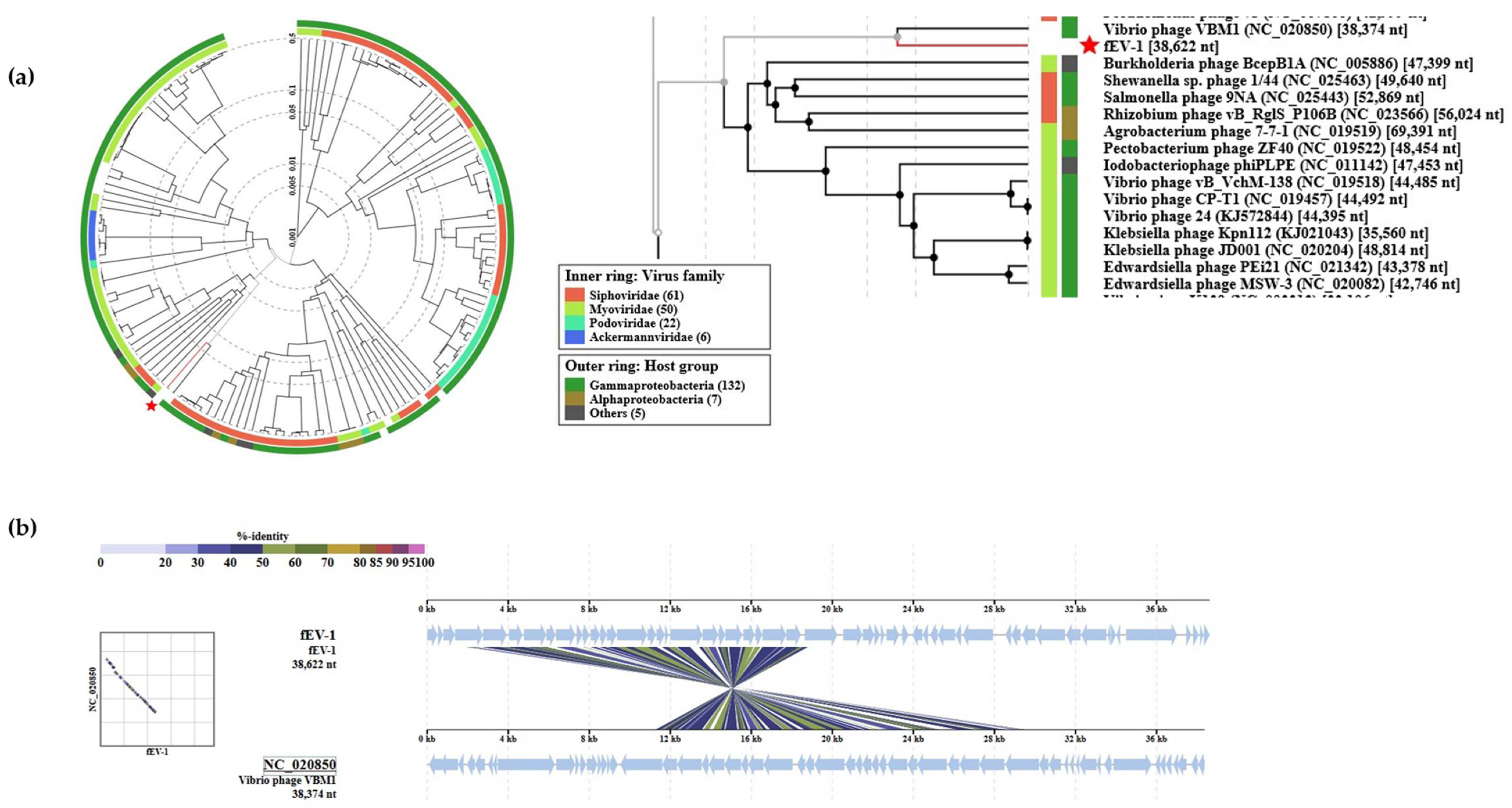
The VIPtree analysis was carried out for fEV-1 and the results are shown in Figure 8. The analysis positioned fEV-1 next to the Vibrio phage VBM1 (accession no. NC_020850) and close to the number of Myoviruses with 35–70 kb genomes (Figure 8a). The similarity with VBM1 spans a 16 kb region and is not very high (Figure 8b), indicating that fEV-1 clearly represents a new dwarf myovirus type, which, at present, is classified as an unclassified myovirus in the taxonomical branch Duplodnaviria > Heunggongvirae > Uroviricota > Caudoviricetes > Caudovirales > Myoviridae.
3.5. The Proteome of fEV-1
We used in-solution tryptic digestion of fEV-1 virions purified by ultracentrifugation to identify expressed viral proteins associated with the virion. By using label-free data dependent acquisition (DDA) quantification, and two different in-house generated datasets for peptide matching, altogether, we identified 32 viral and 240 host-derived proteins associated with the virion at a false discovery rate (FDR) of <1% (ProteomeXchange dataset PXD026811). The identified fEV-1 proteins constitute 56% of the 57 gene products predicted for fEV-1 (Table S2). According to the label-free quantification, the most abundant PPAP was Gp09, which is closely related to the major capsid protein of the Vibrio phage 1.052.A._10N.286.46.C3 (Table S2) [53]. Five fEV-1 predicted gene products had no known homologs in the databases based on PSI-BLAST analysis (Table S2).
One of the in-house generated datasets contained the fEV-1 and the Y. pestis CO-92/Biovar Orientalis proteomes, and the other dataset contained all six frame translations of the fEV-1 genome, for a total of 534 translated ORFs. This approach allowed us to detect that we had missed the 5′-end of the gene g01 overlapping the predicted physical ends of the genome and that the predicted gene started at nucleotide 38297. The revised g01 is 837 bp in size, encoding a 279 amino acids long gene product homologous to many bacterial pentapeptide repeats containing proteins of Pseudomonas, Salmonella, Raoultella, Cronobacter, and Rhizobium including the type III secretion system effector protein PipB2 of Salmonella [54] and the Caulobacter podovirus Jess A (GenBank QCW21951.1). This raised the number of PPAPs to 33. Importantly, our results confirm the expression of more than 20 uncharacterized PPAPs (Table S2), warranting further biochemical characterization of their functions and role in virus replication and assembly.
3.6. The Genome and Taxonomic Position of fD1
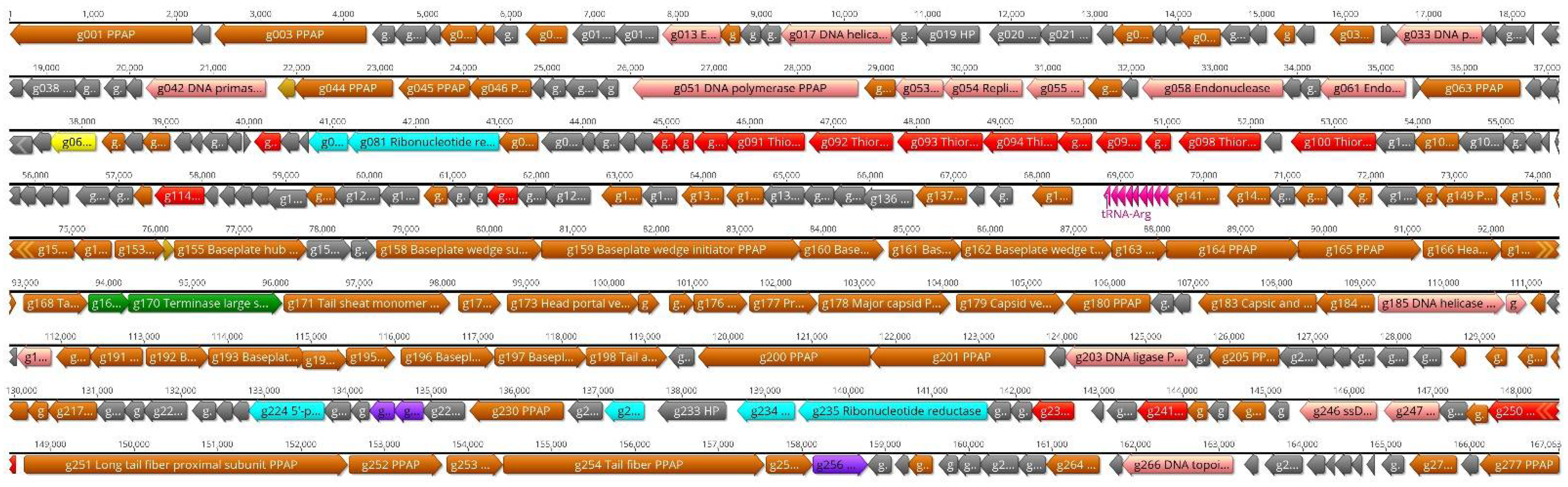
The genome of fD1 is 167,063 bp in size and contains 277 predicted protein coding and 9 tRNA genes (Figure 9).
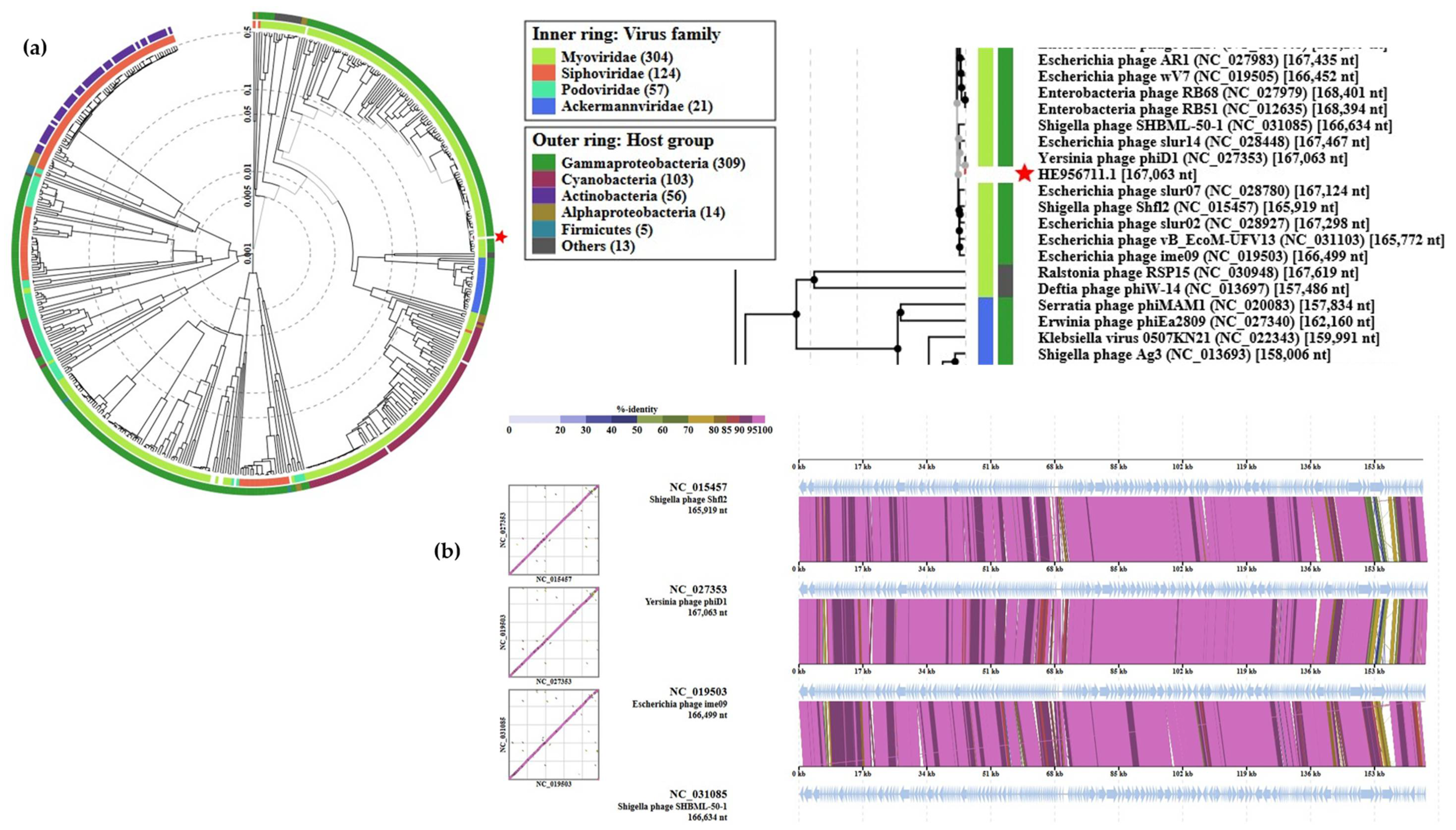
The phages closest related to fD1 were found among several closely related Escherichia and Shigella phages (Figure 10). fD1 shows a maximum of 91% overall identity with Shigella phage Shfl2 (NC_015457). ICTV classifies fD1 as the only representative of phage species Yersinia virus D1 that belongs in the taxonomy to the Duplodnaviria > Heunggongvirae > Uroviricota > Caudoviricetes > Caudovirales > Myoviridae > Tevenvirinae > Tequatrovirus branch.
3.7. The Proteome of fD1
We used in-solution tryptic digestion of fD1 virions purified by ultracentrifugation to identify expressed viral proteins associated with the virion from two different virus preparations. By using label-free DDA quantification and two different in-house generated datasets for peptide matching, we identified several viral and host-derived proteins associated with the virion at a false discovery rate (FDR) of <1% (ProteomeXchange dataset PXD026812). Altogether 113 fD1 gene products were identified as PPAPs (Table S3) using a threshold of two or more peptides identified per protein, at an average spectral count of two or more. A total of 70 proteins were identified in both preparations, whereas 20 and 23 proteins were identified in only one of the preparations, respectively. Importantly, one of our in-house generated datasets contained all six frame translations of the fD1 genome, yielding 505 translated ORFs. This approach allowed us to detect a total of nine coding regions originally missed in the original annotation of the fD1 genome. These predicted genes encode for the gene products Gp8, Gp14, Gp29, Gp31, Gp45, Gp51, Gp55, Gp56, and Gp63. If we include identified proteins falling below the stricter threshold above (see Table S3 for details), there are another 17 PPAPs. Homologues to some of these gene products (Gp147, Gp160, Gp167, Gp169, Gp191, Gp199, Gp202, Gp215, and Gp233) are expressed by Escherichia phage T4, indicating that they might be true PPAPs, despite falling below our stricter thresholds. Other PPAPs falling below the threshold are located within an operon (Table S3), again, indicating that they most likely are true PPAPs. Notably, for fEV-1, these same less strict settings did not yield additional identifications. If we include all 137 identified fD1 proteins, they constitute 49% of the 277 gene products predicted for fD1. In addition to expressed fD1 gene products, we identified 672 co-purifying host proteins, 372 of which were identified in both virus preparations, and the remaining in only either preparation. Importantly, our results confirm the expression of close to 40 uncharacterized PPAPs (Table S3) warranting further biochemical characterization of their functions and role in virus replication and assembly.
We recently characterized three T4-like Yersinia phages, i.e., fPS-2, fPS-65, and fPS-90 [55], and, as phage fD1 is very closely related (87% genome identity) to them, we wanted to compare their long tail fiber (LTF) proteins as they are the ones that first recognize the bacterial host surface [56]. In T4-like phages, the LTFs are typically composed of four subunits, identified as Gp34–Gp37 for T4. Additionally, in T4, Gp38 functions as a chaperone that guides the Gp37 folding and trimerization, and does not remain associated with the LTF after its assembly [57]. In the LTF structure, Gp34 is proximal to the tail baseplate, followed by Gp35 and Gp36 that form the hinge to which is attached the trimeric Gp37. In phage T4, the distal tip of Gp37 is the adhesin, while in T2 type phages, the adhesin is the Gp38 chaperone that after helping the Gp37 trimerization remains associated to the tip of LTF [57].
We identified the fD1 genetic locus that encompasses genes g251–g255, as those encoding for the long tail fiber subunits. Comparison of the fD1 locus to those of the fPS phages revealed that they shared practically no similarity. As the fPS phages were proposed to utilize, similar to Salmonella phage S16 [57], the phage T2 type Gp38 chaperone homolog as the receptor binding protein [56], we carried out BLAST searches to identify closest related homologs for the Gp251–Gp255 of fD1.
The fD1 genes revealed extensive similarity to the corresponding genes of Escherichia phages Mobillu (accession no. MN850622.1), vB_EcoM_WFbE185 (MK373778.1), vB_EcoM_KAW3E185 (MK373782.1), vB_EcoM_MM02 (MK373784.1), and Shigella phage SHSML-52-1 (KX130865.1), among others. Among the long tail fiber subunits, Gp251, the predicted proximal subunit of the long tail fiber, was 97–99% identical to the homologs of closest Escherichia, Citrobacter, Enterobacteria, Salmonella and Shigella phages. Gp252, the putative proximal hinge connector protein, and Gp253, the putative distal hinge connector protein of long tail fiber, were both >99% identical to those of several Escherichia and Shigella phages. Finally, Gp255, the putative tail fiber assembly protein, was 100% identical to homologs of several Escherichia phages, indicating that it would not function as a receptor binding protein. On the contrary, Gp254, a 1038 amino-acid-residue LTF distal subunit protein, was significantly less similar to its homologs, showing at best 93% identity to several Escherichia phage counterparts, including Mobillu, vB_EcoM_WFbE185, Shigella phage SHSML-52-1, among others. Multiple alignment of the sequences revealed that the N-terminal end of the protein was highly conserved between the phages, whereas there were more differences among amino acid residues from 520 to 700. This was followed by a high identity range (residues 701–922), where the identity abruptly broke for about 40 residues followed by 100% identical last 70 C-terminal residues. It is very likely that the last 110 residues make up an intramolecular chaperone (IMC) that is auto cleaved away after the trimerization of the fiber protein [57], and that exposes the adhesin surface at the tip of the LTF. Significantly, the Gp251-Gp254 proteins were all identified as PPAPs, i.e., present in the phage particle, whereas the assembly chaperone, Gp255, was not (Table S3). Therefore, it is very likely that Gp254 is the receptor binding protein of fD1, and that amino acid residues from 520 to 700 are important to the binding specificity.
4. Conclusions
Here, we have characterized two phages isolated simultaneously against Y. pestis strains KIM D27 and EV76 from an incoming sewage water sample obtained from the City of Turku sewage treatment plant (Turku, Finland). Using the Y. pestis strains as enrichment hosts was somewhat risky as the last bubonic or pneumonic plague infections had occurred in Turku almost 300 years earlier, during the outbreak in 1711. However, it has been reported that many Yersinia phages can also infect other members of Enterobacteriaceae, such as E. coli, Shigella, and Salmonella [9]. For example, the Yersinia phage L-413C has been suggested to be an E. coli phage. Therefore, although fD1 and fEV-1 were isolated using Y. pestis, it was not a likely candidate for a natural host for these phages, since Y. pestis has not been encountered in Finland for centuries. On the one hand, alternative hosts could be Y. pseudotuberculosis strains or strains of other Enterobacterales. Therefore, it was not surprising that we could find several E. coli hosts for fD1, especially among laboratory strains. In addition to that, the phage appeared to reproduce much more efficiently in E. coli strains than in Y. pestis (Figure 3). On the other hand, of the hosts we used for screening, fEV-1 could only infect Y. pestis strains. This might be a positive feature, as the phages to be used in phage therapy should only be specific for the target microbes, and not infect the patient’s normal flora.
Lipopolysaccharide (LPS) is an important virulence factor of the pathogenic Gram-negative bacteria including pathogenic members of genus Yersinia [58,59,60,61,62,63,64]. It also provides receptor sites for many phages, and changes in LPS structure can affect the sensitivity of a bacterium to a phage. While we were not able to identify the exact nature of the receptors for phages fEV-1 and fD1, the EOP of fEV-1 was significantly lowered when tested against LPS mutants (Table 1 and Figure 2), suggesting that the LPS core heptose and glucose residues are essential constituents of the phage receptor.
On the basis of the morphological characterization, fEV-1 is a dwarf myovirus. Currently, this group of viruses is understudied. Here, we add information on these viruses by determining the host range, latency period, and burst size of fEV-1. The latency period of both fEV-1 and ΦPLPE has been determined to be long. It is possible that dwarf myoviruses, in general, have long latency periods, but current data is very scarce. In addition to characterizing the infection of a novel dwarf myovirus, we provide a comprehensive and quantitative proteomic analysis of such a virus, as such data has been missing to date. Here, we confirm the expression of 33 out of 57 predicted gene products for fEV-1; 20 of which are uncharacterized PPAPs. A more in-depth analysis of these is required to verify their homologues in other dwarf myoviruses, with a potential extension to possibly identifying a protein responsible for causing a long latency. Moreover, two out of five genes lacking homologues in sequence databases are expressed PPAPs. The structure and function of these warrant future studies to verify their role in infection, replication, and virus assembly.
Supplementary Materials
The following are available online at https://0-www-mdpi-com.brum.beds.ac.uk/article/10.3390/v13071384/s1, Figure S1: Restriction digestion analysis of the fEV-1 genomic DNA, Figure S2: Determination of the physical termini of the phage fEV-1 genome, Table S1: The predicted sigma70 promoters of phage fEV-1, Table S2: Annotation of the fEV-1 genome and PSI-BLAST results for fEV-1 gene products, Table S3: Annotation of the fD1 genome and PSI-BLAST results for fD1 gene products.
Author Contributions
Conceptualization, M.S., M.I.P., and L.J.H.; methodology, M.S., M.I.P., L.J.H., L.v.O., A.N., L.M., and S.J.; validation, M.S., M.I.P., and L.J.H.; formal analysis, M.S., M.I.P., L.J.H., L.v.O., L.M., and S.J.; investigation, M.S., M.I.P., A.N., L.J.H., L.v.O., L.M., and S.J.; resources, M.S. and L.J.H.; writing—original draft preparation, M.S., M.I.P., L.J.H., and S.J.; writing—review and editing, M.S., M.I.P., L.J.H., L.v.O., A.N., L.M., and S.J.; data curation, M.S., M.I.P., and L.J.H.; visualization, M.S., M.I.P., L.J.H., and S.J.; supervision, M.S., M.I.P., and L.J.H.; project administration, M.S.; funding acquisition, M.S., M.I.P., and L.J.H. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Academy of Finland grant number 288701 to M.S. Open access funding provided by University of Helsinki.
Data Availability Statement
The genome sequences of the Yersinia phages fEV-1 and fD1 are available in Genbank under the accession numbers LT992259 and HE956711, respectively. The mass spectrometry data have been deposited to the ProteomeXchange [47] consortium via the MassIVE partner repository (https://massive.ucsd.edu/ (accessed on 14 July 2021)) with the dataset identifiers PXD026811 (fEV-1) and PXD026812 (fD1).
Acknowledgments
Milla Maaria Trivedi is thanked for help with the restriction digestion analysis of fEV-1 shown in Figure S1.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- Perry, R.D.; Fetherston, J.D. Yersinia pestis—Etiologic agent of plague. Clin. Microbiol. Rev. 1997, 10, 35–66. [Google Scholar] [CrossRef]
- Mead, P.S. Plague in Madagascar—A Tragic Opportunity for Improving Public Health. N. Engl. J. Med. 2018, 378, 106–108. [Google Scholar] [CrossRef]
- Nguyen, V.K.; Parra-Rojas, C.; Hernandez-Vargas, E.A. The 2017 plague outbreak in Madagascar: Data descriptions and epidemic modelling. Epidemics 2018, 25, 20–25. [Google Scholar] [CrossRef] [PubMed]
- Hinnebusch, B.J.; Rosso, M.L.; Schwan, T.G.; Carniel, E. High-frequency conjugative transfer of antibiotic resistance genes to Yersinia pestis in the flea midgut. Mol. Microbiol. 2002, 46, 349–354. [Google Scholar] [CrossRef]
- Filippov, A.A.; Sergueev, K.V.; He, Y.; Huang, X.-Z.; Gnade, B.T.; Mueller, A.J.; Fernandez-Prada, C.M.; Nikolich, M.P. Bacteriophage Therapy of Experimental Bubonic Plague in Mice. Adv. Exp. Med. Biol. 2012, 954, 337–348. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Skurnik, M. Bacteriophages of Yersinia pestis. Adv. Exp. Med. Biol. 2016, 918, 361–375. [Google Scholar] [CrossRef] [PubMed]
- Summers, W.C. Cholera and plague in India: The bacteriophage inquiry of 1927–1936. J. Hist. Med. Allied Sci. 1993, 48, 275–301. [Google Scholar] [CrossRef] [Green Version]
- Garcia, E.; Elliott, J.M.; Ramanculov, E.; Chain, P.S.; Chu, M.C.; Molineux, I.J. The genome sequence of Yersinia pestis bacteriophage fA1122 reveals an intimate history with the coliphage T3 and T7 genomes. J. Bacteriol. 2003, 185, 5248–5262. [Google Scholar] [CrossRef] [Green Version]
- Filippov, A.A.; Sergueev, K.V.; He, Y.; Nikolich, M.P. Bacteriophages Capable of Lysing Yersinia pestis and Yersinia pseudotuberculosis: Efficiency of Plating Tests and Identification of Receptors in Escherichia coli K-12. Adv. Exp. Med. Biol. 2012, 954, 123–134. [Google Scholar] [CrossRef]
- Knapp, W. On the varying behavior of Pasteurella phages. Zent. Bakteriol. Orig. 1963, 190, 39–46. [Google Scholar]
- Filippov, A.A.; Sergueev, K.V.; He, Y.; Huang, X.-Z.; Gnade, B.T.; Mueller, A.J.; Fernandez-Prada, C.M.; Nikolich, M.P. Bacteriophage-Resistant Mutants in Yersinia pestis: Identification of Phage Receptors and Attenuation for Mice. PLoS ONE 2011, 6, e25486. [Google Scholar] [CrossRef] [Green Version]
- Schwudke, D.; Ergin, A.; Michael, K.; Volkmar, S.; Appel, B.; Knabner, D.; Konietzny, A.; Strauch, E. Broad-Host-Range Yersinia Phage PY100: Genome Sequence, Proteome Analysis of Virions, and DNA Packaging Strategy. J. Bacteriol. 2008, 190, 332–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, Y.; Xi, H.; Dai, J.; Zhong, Y.; Lu, S.; Wang, T.; Yang, L.; Guan, Y.; Wang, P. The characteristics and genome analysis of the novel Y. pestis phage JC221. Virus Res. 2020, 283, 197982. [Google Scholar] [CrossRef] [PubMed]
- Schmelcher, M.; Donovan, D.M.; Loessner, M.J. Bacteriophage endolysins as novel antimicrobials. Future Microbiol. 2012, 7, 1147–1171. [Google Scholar] [CrossRef] [Green Version]
- Nolan, J.M.; Petrov, V.; Bertrand, C.; Krisch, H.M.; Karam, J.D. Genetic diversity among five T4-like bacteriophages. Virol. J. 2006, 3, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, M.J.; Lefkowitz, E.J.; King, A.M.Q.; Harrach, B.; Harrison, R.L.; Knowles, N.J.; Kropinski, A.M.; Krupovic, M.; Kuhn, J.H.; Mushegian, A.R.; et al. Ratification vote on taxonomic proposals to the International Committee on Taxonomy of Viruses (2016). Arch. Virol. 2016, 161, 2921–2949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Comeau, A.M.; Tremblay, D.; Moineau, S.; Rattei, T.; Kushkina, A.I.; Tovkach, F.I.; Krisch, H.M.; Ackermann, H.-W. Phage Morphology Recapitulates Phylogeny: The Comparative Genomics of a New Group of Myoviruses. PLoS ONE 2012, 7, e40102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yasuike, M.; Nishiki, I.; Iwasaki, Y.; Nakamura, Y.; Fujiwara, A.; Sugaya, E.; Kawato, Y.; Nagai, S.; Kobayashi, T.; Ototake, M.; et al. Full-genome sequence of a novel myovirus, GF-2, infecting Edwardsiella tarda: Comparison with other Edwardsiella myoviral genomes. Arch. Virol. 2015, 160, 2129–2133. [Google Scholar] [CrossRef]
- Hargreaves, K.R.; Clokie, M.R.J. A Taxonomic Review of Clostridium difficile Phages and Proposal of a Novel Genus, “Phimmp04likevirus”. Viruses 2015, 7, 2534–2541. [Google Scholar] [CrossRef] [Green Version]
- Garcia, E.; Nedialkov, Y.A.; Elliott, J.; Motin, V.; Brubaker, R.R. Molecular Characterization of KatY (Antigen 5), a Thermoregulated Chromosomally Encoded Catalase-Peroxidase of Yersinia pestis. J. Bacteriol. 1999, 181, 3114–3122. [Google Scholar] [CrossRef] [Green Version]
- Kiljunen, S.; Datta, N.; Dentovskaya, S.V.; Anisimov, A.P.; Knirel, Y.A.; Bengoechea, J.A.; Holst, O.; Skurnik, M. Identification of the lipopolysaccharide core of Yersinia pestis and Yersinia pseudotuberculosis as the receptor for bacteriophage fA1122. J. Bacteriol. 2011, 193, 4963–4972. [Google Scholar] [CrossRef] [Green Version]
- Portnoy, D.A.; Falkow, S. Virulence-associated plasmids from Yersinia enterocolitica and Yersinia pestis. J. Bacteriol. 1981, 148, 877–883. [Google Scholar] [CrossRef] [Green Version]
- Tsubokura, M.; Aleksić, S. A simplified antigenic scheme for serotyping of Yersinia pseudotuberculosis: Phenotypic characterization of reference strains and preparation of O and H factor sera. Contrib. Microbiol. Immunol. 1995, 13, 99–105. [Google Scholar]
- Kenyon, J.J.; Duda, K.A.; De Felice, A.; Cunneen, M.M.; Molinaro, A.; Laitinen, J.; Skurnik, M.; Holst, O.; Reeves, P.; De Castro, C. Serotype O:8 isolates in the Yersinia pseudotuberculosis complex have different O-antigen gene clusters and produce various forms of rough LPS. Innate Immun. 2016, 22, 205–217. [Google Scholar] [CrossRef] [Green Version]
- Skurnik, M. Studies on the Virulence Plasmids of Yersinia Species. Ph.D. Thesis, University of Oulu, Oulu, Finland, 1985. [Google Scholar]
- Macrina, F.L.; Kopecko, D.J.; Jones, K.R.; Ayers, D.J.; McCowen, S.M. A multiple plasmid-containing Escherichia coli strain: Convenient source of size reference plasmid molecules. Plasmid 1978, 1, 417–420. [Google Scholar] [CrossRef]
- Sambrook, J.; Russell, D.W. Molecular Cloning: A laboratory Manual, 3rd ed.; Cold Spring Harbor Laboratory: Cold Spring Harbor, NY, USA, 2001. [Google Scholar]
- Sambrook, J.; Russell, D.W. Purification of bacteriophage λ particles by centrifugation through a glycerol step gradient. CSH Protoc. 2006. [Google Scholar] [CrossRef]
- Staden, R.; Judge, D.P.; Bonfield, J.K. Managing sequencing projects in the GAP4 environment. In Introduction to Bioinformatics. A Theoretical and Practical Approach; Krawetz, S.A., Womble, D.D., Eds.; Humana Press: Totowa, NJ, USA, 2003; pp. 327–344. [Google Scholar]
- Rutherford, K.; Parkhill, J.; Crook, J.; Horsnell, T.; Rice, P.; Rajandream, M.-A.; Barrell, B. Artemis: Sequence visualization and annotation. Bioinformatics 2000, 16, 944–945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coil, D.; Jospin, G.; Darling, A.E. A5-miseq: An updated pipeline to assemble microbial genomes from Illumina MiSeq data. Bioinformatics 2015, 31, 587–589. [Google Scholar] [CrossRef] [PubMed]
- Carver, T.J.; Berriman, M.; Tivey, A.; Patel, C.; Böhme, U.; Barrell, B.G.; Parkhill, J.; Rajandream, M.-A. Artemis and ACT: Viewing, annotating and comparing sequences stored in a relational database. Bioinformatics 2008, 24, 2672–2676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carver, T.; Harris, S.; Berriman, M.; Parkhill, J.; McQuillan, J.A. Artemis: An integrated platform for visualization and analysis of high-throughput sequence-based experimental data. Bioinformatics 2011, 28, 464–469. [Google Scholar] [CrossRef] [Green Version]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Aziz, R.K.; Bartels, D.; Best, A.A.; DeJongh, M.; Disz, T.; Edwards, R.A.; Formsma, K.; Gerdes, S.; Glass, E.M.; Kubal, M.; et al. The RAST Server: Rapid annotations using subsystems technology. BMC Genom. 2008, 9, 75. [Google Scholar] [CrossRef] [Green Version]
- Garneau, J.R.; Depardieu, F.; Fortier, L.C.; Bikard, D.; Monot, M. PhageTerm: A tool for fast and accurate determination of phage termini and packaging mechanism using next-generation sequencing data. Sci. Rep. 2017, 7, 8292. [Google Scholar] [CrossRef]
- Chan, P.P.; Lowe, T.M. tRNAscan-SE: Searching for tRNA Genes in Genomic Sequences. Methods Mol. Biol. 2019, 1962, 1–14. [Google Scholar] [CrossRef]
- Lowe, T.M.; Chan, P.P. tRNAscan-SE On-line: Integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 2016, 44, W54–W57. [Google Scholar] [CrossRef] [PubMed]
- Kallio, M.A.; Tuimala, J.T.; Hupponen, T.; Klemelä, P.; Gentile, M.; Scheinin, I.; Koski, M.; Käki, J.; Korpelainen, E.I. Chipster: User-friendly analysis software for microarray and other high-throughput data. BMC Genom. 2011, 12, 507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishimura, Y.; Yoshida, T.; Kuronishi, M.; Uehara, H.; Ogata, H.; Goto, S. ViPTree: The viral proteomic tree server. Bioinformatics 2017, 33, 2379–2380. [Google Scholar] [CrossRef] [PubMed]
- Solovyev, V.; Asaf, S. Automatic annotation of microbial genomes and metagenomic sequences. Metagenomics and its applications. In Agriculture, Biomedicine and Environmental Studies; Li, R.W., Ed.; Nova Science Publishers: Hauppauge, NY, USA, 2010; pp. 61–78. [Google Scholar]
- Teleman, J.; Dowsey, A.; Gonzalez-Galarza, F.F.; Perkins, S.; Pratt, B.; Röst, H.; Malmström, L.; Malmström, J.; Jones, A.; Deutsch, E.W.; et al. Numerical Compression Schemes for Proteomics Mass Spectrometry Data. Mol. Cell. Proteom. 2014, 13, 1537–1542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chambers, M.C.; Maclean, B.; Burke, R.; Amodei, D.; Ruderman, D.L.; Neumann, S.; Gatto, L.; Fischer, B.; Pratt, B.; Egertson, J.; et al. A cross-platform toolkit for mass spectrometry and proteomics. Nat. Biotechnol. 2012, 30, 918–920. [Google Scholar] [CrossRef]
- Bauch, A.; Adamczyk, I.; Buczek, P.; Elmer, F.-J.; Enimanev, K.; Glyzewski, P.; Kohler, M.; Pylak, T.; Quandt, A.; Ramakrishnan, C.; et al. openBIS: A flexible framework for managing and analyzing complex data in biology research. BMC Bioinform. 2011, 12, 468. [Google Scholar] [CrossRef] [Green Version]
- Craig, R.; Beavis, R.C. A method for reducing the time required to match protein sequences with tandem mass spectra. Rapid Commun. Mass Spectrom. 2003, 17, 2310–2316. [Google Scholar] [CrossRef]
- Keller, A.; Nesvizhskii, A.; Kolker, E.; Aebersold, R. Empirical Statistical Model to Estimate the Accuracy of Peptide Identifications Made by MS/MS and Database Search. Anal. Chem. 2002, 74, 5383–5392. [Google Scholar] [CrossRef]
- Vizcaino, J.A.; Deutsch, E.; Wang, R.; Csordas, A.; Reisinger, F.; Ríos, D.; Dianes, J.A.; Sun, Z.; Farrah, T.; Bandeira, N.; et al. ProteomeXchange provides globally coordinated proteomics data submission and dissemination. Nat. Biotechnol. 2014, 32, 223–226. [Google Scholar] [CrossRef]
- Ackermann, H.W.; Krisch, H.M.; Comeau, A.M. Morphology and genome sequence of phage φ1402: A dwarf myovirus of the predatory bacterium Bdellovibrio bacteriovorus. Bacteriophage 2011, 1, 138–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yasuike, M.; Sugaya, E.; Nakamura, Y.; Shigenobu, Y.; Kawato, Y.; Kai, W.; Nagai, S.; Fujiwara, A.; Sano, M.; Kobayashi, T.; et al. Complete Genome Sequence of a Novel Myovirus which Infects Atypical Strains of Edwardsiella tarda. Genome Announc. 2013, 1, e00248-12. [Google Scholar] [CrossRef] [Green Version]
- Fokine, A.; Chipman, P.R.; Leiman, P.; Mesyanzhinov, V.V.; Rao, V.; Rossmann, M.G. Molecular architecture of the prolate head of bacteriophage T4. Proc. Natl. Acad. Sci. USA 2004, 101, 6003–6008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kostyuchenko, V.A.; Chipman, P.R.; Leiman, P.G.; Arisaka, F.; Mesyanzhinov, V.V.; Rossmann, M.G. The tail structure of bacteriophage T4 and its mechanism of contraction. Nat. Struct. Mol. Biol. 2005, 12, 810–813. [Google Scholar] [CrossRef] [PubMed]
- Leblanc, C.; Caumont-Sarcos, A.; Comeau, A.M.; Krisch, H.M. Isolation and genomic characterization of the first phage infecting Iodobacteria: ϕPLPE, a myovirus having a novel set of features. Environ. Microbiol. Rep. 2009, 1, 499–509. [Google Scholar] [CrossRef] [PubMed]
- Kauffman, K.M.; Brown, J.M.; Sharma, R.S.; VanInsberghe, D.; Elsherbini, J.; Polz, M.; Kelly, L. Viruses of the Nahant Collection, characterization of 251 marine Vibrionaceae viruses. Sci. Data 2018, 5, 180114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henry, T.; Couillault, C.; Rockenfeller, P.; Boucrot, E.; Dumont, A.; Schroeder, N.; Hermant, A.; Knodler, L.; Lécine, P.; Steele-Mortimer, O.; et al. The Salmonella effector protein PipB2 is a linker for kinesin-1. Proc. Natl. Acad. Sci. USA 2006, 103, 13497–13502. [Google Scholar] [CrossRef] [Green Version]
- Salem, M.; Pajunen, M.; Jun, J.; Skurnik, M. T4-like Bacteriophages Isolated from Pig Stools Infect Yersinia pseudotuberculosis and Yersinia pestis Using LPS and OmpF as Receptors. Viruses 2021, 13, 296. [Google Scholar] [CrossRef]
- Hu, B.; Margolin, W.; Molineux, I.J.; Liu, J. Structural remodeling of bacteriophage T4 and host membranes during infection initiation. Proc. Natl. Acad. Sci. USA 2015, 112, E4919–E4928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunne, M.; Denyes, J.M.; Arndt, H.; Loessner, M.J.; Leiman, P.; Klumpp, J. Salmonella Phage S16 Tail Fiber Adhesin Features a Rare Polyglycine Rich Domain for Host Recognition. Structure 2018, 26, 1573–1582.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anisimov, A.P.; Dentovskaya, S.V.; Kondakova, A.N.; Lindner, B.; Shaikhutdinova, R.Z.; Kocharova, N.A.; Senchenkova, S.N.; Knirel, Y.A. Yersinia pestis lipopolysaccharide in host-pathogen interactions. In The Challenge of Highly Pathogenic Microorganisms; Springer: Berlin/Heidelberg, Germany, 2010; pp. 77–87. [Google Scholar]
- Bengoechea, J.A.; Najdenski, H.; Skurnik, M. Lipopolysaccharide O antigen status of Yersinia enterocolitica O:8 is essential for virulence and absence of O antigen affects the expression of other Yersinia virulence factors. Mol. Microbiol. 2004, 52, 451–469. [Google Scholar] [CrossRef] [PubMed]
- Najdenski, H.; Golkocheva, E.; Vesselinova, A.; Bengoechea, J.; Skurnik, M. Proper expression of the O-antigen of lipopolysaccharide is essential for the virulence of Yersinia enterocolitica O:8 in experimental oral infection of rabbits. FEMS Immunol. Med. Microbiol. 2003, 38, 97–106. [Google Scholar] [CrossRef] [Green Version]
- Skurnik, M. Molecular genetics, biochemistry and biological role of Yersinia lipopolysaccharide. In The Genus Yersinia: Entering the Functional Genomic Era; Skurnik, M., Granfors, K., Bengoechea, J.A., Eds.; Kluwer Academic/Plenum Publishers: New York, NY, USA, 2003; pp. 187–189. [Google Scholar]
- Skurnik, M.; Venho, R.; Bengoechea, J.; Moriyon, I. The lipopolysaccharide outer core of Yersinia enterocolitica serotype O:3 is required for virulence and plays a role in outer membrane integrity. Mol. Microbiol. 1999, 31, 1443–1462. [Google Scholar] [CrossRef]
- Darwin, A.; Miller, V.L. Identification of Yersinia enterocolitica genes affecting survival in an animal host using signature-tagged transposon mutagenesis. Mol. Microbiol. 1999, 32, 51–62. [Google Scholar] [CrossRef]
- Karlyshev, A.V.; Oyston, P.C.F.; Williams, K.; Clark, G.C.; Titball, R.W.; Winzeler, E.A.; Wren, B.W. Application of High-Density Array-Based Signature-Tagged Mutagenesis to Discover Novel Yersinia Virulence-Associated Genes. Infect. Immun. 2001, 69, 7810–7819. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
Transmission electron microscopy analysis of phage fD1 (A,B) and fEV-1 (C,D) particles. The scale in (A) and (C) is 200 nm, and in (B) and (D), 100 nm. White arrow in (B), points to one of the long tail fibers of fD1.
Figure 1.
Transmission electron microscopy analysis of phage fD1 (A,B) and fEV-1 (C,D) particles. The scale in (A) and (C) is 200 nm, and in (B) and (D), 100 nm. White arrow in (B), points to one of the long tail fibers of fD1.

Figure 2.
Schematic presentation of the Y. pestis LPS mutants used in this study (Table 1). Indicated for the wild type (WT) LPS, are the glycosidic bonds formed by the eliminated glycosyltransferases of the LPS mutants and the part that forms the lipid A. The zigzag lines represent the acyl chains of the lipid A. In the mutant LPS structures, the affected sugar residues are indicated by dashed ovals. Gln, D-glucosamine; KDO, 3-deoxy-D-manno-oct-2-ulopyranosonic acid; KO, D-glycero-D-talo-oct-2-ulopyranosonic acid; Hep-I, Hep-II, and Hep-III, L-glycero-D-manno-heptopyranose; Hep-IV, D-glycero-D-manno-heptopyranose; Glc, D-glucopyranose; Gal, D-galactopyranose.
Figure 2.
Schematic presentation of the Y. pestis LPS mutants used in this study (Table 1). Indicated for the wild type (WT) LPS, are the glycosidic bonds formed by the eliminated glycosyltransferases of the LPS mutants and the part that forms the lipid A. The zigzag lines represent the acyl chains of the lipid A. In the mutant LPS structures, the affected sugar residues are indicated by dashed ovals. Gln, D-glucosamine; KDO, 3-deoxy-D-manno-oct-2-ulopyranosonic acid; KO, D-glycero-D-talo-oct-2-ulopyranosonic acid; Hep-I, Hep-II, and Hep-III, L-glycero-D-manno-heptopyranose; Hep-IV, D-glycero-D-manno-heptopyranose; Glc, D-glucopyranose; Gal, D-galactopyranose.

Figure 3.
One-step growth curves for the phages: (a) Growth curve of fEV-1 on Y. pestis EV-76 at RT; (b) growth curves of fD1 on Y pestis D27 at RT and on E. coli 6588 at 37 °C. Note the logarithmic scale in (b) that allowed the use of only the positive SD bars.
Figure 3.
One-step growth curves for the phages: (a) Growth curve of fEV-1 on Y. pestis EV-76 at RT; (b) growth curves of fD1 on Y pestis D27 at RT and on E. coli 6588 at 37 °C. Note the logarithmic scale in (b) that allowed the use of only the positive SD bars.

Figure 4.
Growth of Y. pestis strains EV-76 and D27 infected with different dilutions of phages fEV-1 and fD1, respectively, followed by OD600 measurements every 30 min for 8 h.
Figure 4.
Growth of Y. pestis strains EV-76 and D27 infected with different dilutions of phages fEV-1 and fD1, respectively, followed by OD600 measurements every 30 min for 8 h.

Figure 5.
fEV-1, repeat region spanning the nucleotide position 1. The numbers refer to the nucleotide positions in the genome sequence (accession no. LT9922259). The oversized bold A indicated the arbitrarily chosen nucleotide 1 of the sequence.
Figure 5.
fEV-1, repeat region spanning the nucleotide position 1. The numbers refer to the nucleotide positions in the genome sequence (accession no. LT9922259). The oversized bold A indicated the arbitrarily chosen nucleotide 1 of the sequence.

Figure 6.
fEV-1, the repeat region within gene g51c encoding a hypothetical protein with 22 Pro-Ala-Ala repeats. The numbers refer to the nucleotide positions in the genome sequence (accession no. LT9922259).
Figure 6.
fEV-1, the repeat region within gene g51c encoding a hypothetical protein with 22 Pro-Ala-Ala repeats. The numbers refer to the nucleotide positions in the genome sequence (accession no. LT9922259).

Figure 7.
Genomic map of phage fEV-1. The predicted genes are indicated by arrows, and different colors are used to indicate functional classes of the predicted gene products. Brown, structural and phage particle associated proteins (PPAP); turquoise, nucleotide metabolism; green, DNA packaging; violet, lysis; pink, DNA replication and repair; grey, hypothetical proteins (HP). The predicted sigma70-like promoters P1–P6 (Table S1) are indicated by black arrowheads. The figure was generated using Geneious 10.2.6 (www.geneious.com (accessed on 14 July 2021)).
Figure 7.
Genomic map of phage fEV-1. The predicted genes are indicated by arrows, and different colors are used to indicate functional classes of the predicted gene products. Brown, structural and phage particle associated proteins (PPAP); turquoise, nucleotide metabolism; green, DNA packaging; violet, lysis; pink, DNA replication and repair; grey, hypothetical proteins (HP). The predicted sigma70-like promoters P1–P6 (Table S1) are indicated by black arrowheads. The figure was generated using Geneious 10.2.6 (www.geneious.com (accessed on 14 July 2021)).

Figure 8.
Position of fEV-1 in the Phage Proteomic Tree generated by VIPTree [40]: (a) At left, a circular proteomic tree of prokaryotic dsDNA viruses colored by indicated virus families and host taxonomic groups, at right, part of the rectangular presentation of the proteomic tree showing the closest related phages to fEV-1. The location of fEV-1 in both is indicated by a red asterisk; (b) the genomic alignment of phages fEV-1 and VBM1.
Figure 8.
Position of fEV-1 in the Phage Proteomic Tree generated by VIPTree [40]: (a) At left, a circular proteomic tree of prokaryotic dsDNA viruses colored by indicated virus families and host taxonomic groups, at right, part of the rectangular presentation of the proteomic tree showing the closest related phages to fEV-1. The location of fEV-1 in both is indicated by a red asterisk; (b) the genomic alignment of phages fEV-1 and VBM1.

Figure 9.
The genomic organization of phage fD1. The predicted genes are indicated by arrows, and different colors are used to indicate functional classes of the predicted gene products. Brown, structural and phage particle associated proteins (PPAP); turquoise, nucleotide metabolism; yellow, transcription; green, DNA packaging; violet, lysis; pink, DNA replication and repair; red, auxiliary metabolism; grey, hypothetical proteins (HP); lilac, tRNA genes. The figure was generated using Geneious 10.2.6 (www.geneious.com (accessed on 14 July 2021)).
Figure 9.
The genomic organization of phage fD1. The predicted genes are indicated by arrows, and different colors are used to indicate functional classes of the predicted gene products. Brown, structural and phage particle associated proteins (PPAP); turquoise, nucleotide metabolism; yellow, transcription; green, DNA packaging; violet, lysis; pink, DNA replication and repair; red, auxiliary metabolism; grey, hypothetical proteins (HP); lilac, tRNA genes. The figure was generated using Geneious 10.2.6 (www.geneious.com (accessed on 14 July 2021)).

Figure 10.
Position of fD1 in the Phage Proteomic Tree generated by VIPTree [40]: (a) At left, a circular proteomic tree of prokaryotic dsDNA viruses colored by indicated virus families and host taxonomic groups, at right, part of the rectangular presentation of the proteomic tree showing the closest related phages to fD1. The location of fD1 in both is indicated by a red asterisk; (b) Alignment of fD1 genome with its closest related phages.
Figure 10.
Position of fD1 in the Phage Proteomic Tree generated by VIPTree [40]: (a) At left, a circular proteomic tree of prokaryotic dsDNA viruses colored by indicated virus families and host taxonomic groups, at right, part of the rectangular presentation of the proteomic tree showing the closest related phages to fD1. The location of fD1 in both is indicated by a red asterisk; (b) Alignment of fD1 genome with its closest related phages.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Bacterial strains used in this study. The phage fEV-1 and fD1 sensitivities of the strains are given as efficiency of plating (EOP) as compared with their original host strains, EV76 and KIM D27, respectively, set to 1. See Section 3.2 for details on lipopolysaccharide (LPS) structures.
Table 1.
Bacterial strains used in this study. The phage fEV-1 and fD1 sensitivities of the strains are given as efficiency of plating (EOP) as compared with their original host strains, EV76 and KIM D27, respectively, set to 1. See Section 3.2 for details on lipopolysaccharide (LPS) structures.
| Bacterial Strains | Skurnik Lab STORAGE # | EOP of fEV-1 | EOP of fD1 | Comments | Reference |
|---|---|---|---|---|---|
| Yersinia pestis | |||||
| KIM D27 | 1418 | 1 | 1 | Non-pigmented derivative of wild type strain KIM10. Lcr+ Pgm− Pst+ | [20] |
| KIM D27-ΔwaaQ | 5147 | 10−4 | 1 | ΔwaaQ::nptII KanR, deep rough LPS missing distal N-acetylglucosamine and two distal heptose residues | [21] |
| KIM D27-ΔwaaE | 5149 | 10−5 | 1 | ΔwaaE::nptII KanR, deep rough LPS missing two distal heptose residues and the proximal glucose residue | [21] |
| KIM D27-ΔwaaL | 5150 | 1 | 1 | ΔwaaL::nptII KanR, deep rough LPS missing the distal N-acetylglucosamine residue | [21] |
| KIM D27-ΔwabD | 5151 | 1 | 1 | ΔwabD::nptII KanR, rough variant LPS missing distal Gal residue | [21] |
| EV76 | 1281 | 1 | 1 | Non-pigmented derivative of wild type strain EV | [22] |
| Yersinia pseudotuberculosis | |||||
| Pa 3606 | 2061 | 0 | 0 | Serotype O:1b | [23] |
| 204 | 2069 | 1 | Serotype O:5a | [23] | |
| 197 | 2070 | 0 | 0 | Serotype O:5b | [23] |
| 151 | 2073 | 0 | 10−4 | Spontaneous rough derivative of serotype O:4a | [23,24] |
| YPIII::Δwb | 2648 | 0 | 0 | [21] | |
| PB1::Δwb | 2649 | 0 | 10−3 | [21] | |
| Escherichia coli | |||||
| ME 2881-2 | ld 536 | 0 | 0 | Clinical human isolate | |
| ME 3128 | ld 537 | 0 | 0 | Clinical human isolate | |
| ME 2886-2 | 6588 | 0 | 1 | Clinical human isolate | |
| TS 2239-1 | 6729 | 0 | 0 | Clinical human isolate | |
| ME 2861 | ld 541 | 0 | 0 | Clinical human isolate | |
| ME 2863 | ld 542 | 0 | 0 | Clinical human isolate | |
| TS 2174 | ld 543 | 0 | 0 | Clinical human isolate | |
| TS 2757 | ld 548 | 0 | 0 | Clinical human isolate | |
| ME 2671-1 | ld 558 | 0 | 0 | Clinical human isolate | |
| ME 2676-1 | ld 559 | 0 | 0 | Clinical human isolate | |
| ME 2680-1 | 6589 | 0 | 1 | Clinical human isolate | |
| ME 2683-1 | 6590 | 0 | 1 | Clinical human isolate | |
| US 1439 | 6500 | 0 | 0 | Clinical human isolate | |
| KP 1708 | 6501 | 0 | 0 | Clinical human isolate | |
| ME 1658 | 6503 | 0 | 0 | Clinical human isolate | |
| ME 1920 | 6504 | 0 | 0 | Clinical human isolate | |
| US 1769-2 | 6508 | 0 | 0 | Clinical human isolate | |
| 1100 (R1 drd-19k-1) | 251 | 0 | 1 | Laboratory strain | [25] |
| C600 su (lambda cI857) | 253 | 0 | 1 | Laboratory strain | |
| V517 | 258 | 0 | 1 | Clinical isolate | [26] |
| RY13 | 449 | 0 | 1 | Laboratory strain | |
| LE392 (P1-cml, clr100) | 629 | 0 | 1 | Laboratory strain | |
| P678-54 | 630 | 0 | 1 | Laboratory strain | |
| LE392 | 631 | 0 | 1 | Laboratory strain | |
| JM103 | 1247 | 0 | 1 | Laboratory strain | |
| PM191 | 1266 | 0 | 1 | Laboratory strain | |
| D21f2 | 1354 | 0 | 0.1 | Laboratory strain | |
| D21 | 1355 | 0 | 1 | Laboratory strain | |
| DH1 | 1378 | 0 | 1 | Laboratory strain | |
| HB101 | 1389 | 0 | 1 | Laboratory strain | |
| C600 | 1424 | 0 | 1 | Laboratory strain | |
| Klebsiella oxytoca | |||||
| TS 2752 | 547 | 0 | 0 | Clinical human isolate | |
| Shigella | |||||
| 872 | 707 | 0 | 0 | Quality control strain | |
Table 2.
Primers used in this work.
| Primer | Sequence (5′-3′) | Position in LT992259 |
|---|---|---|
| fEV1-R | CCTTGCTTGCATTCAGTTCA | 1198..1179 |
| fEV1-R2 | TGCACCTTCATTTCAAGCAG | 580..561 |
| fEV1-R3 | GCTGAAGTATCGGCTTCCAG | 449..430 |
| fEV1-F | GAAGGAGATAGTGCGCGTTC | 37351..37370 |
| fEV1-F2 | GTGAAACGCTTGATGCTGAA | 38002..38021 |
| fEV1-F3 | ACCGCACATTCAACAAAACA | 38114..38133 |
| fEV1-F4 | TCGCCTTCAGGGTATCAATC | 33590..33609 |
| fEV1-R4 | TCAAGACCCATTGCACTGAA | 34155..34136 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Skurnik, M.; Jaakkola, S.; Mattinen, L.; von Ossowski, L.; Nawaz, A.; Pajunen, M.I.; Happonen, L.J. Bacteriophages fEV-1 and fD1 Infect Yersinia pestis. Viruses 2021, 13, 1384. https://0-doi-org.brum.beds.ac.uk/10.3390/v13071384
AMA Style
Skurnik M, Jaakkola S, Mattinen L, von Ossowski L, Nawaz A, Pajunen MI, Happonen LJ. Bacteriophages fEV-1 and fD1 Infect Yersinia pestis. Viruses. 2021; 13(7):1384. https://0-doi-org.brum.beds.ac.uk/10.3390/v13071384
Chicago/Turabian StyleSkurnik, Mikael, Salla Jaakkola, Laura Mattinen, Lotta von Ossowski, Ayesha Nawaz, Maria I. Pajunen, and Lotta J. Happonen. 2021. "Bacteriophages fEV-1 and fD1 Infect Yersinia pestis" Viruses 13, no. 7: 1384. https://0-doi-org.brum.beds.ac.uk/10.3390/v13071384
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.