
Deletion of Kif5c Does Not Alter Prion Disease Tempo or Spread in Mouse Brain


 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mice
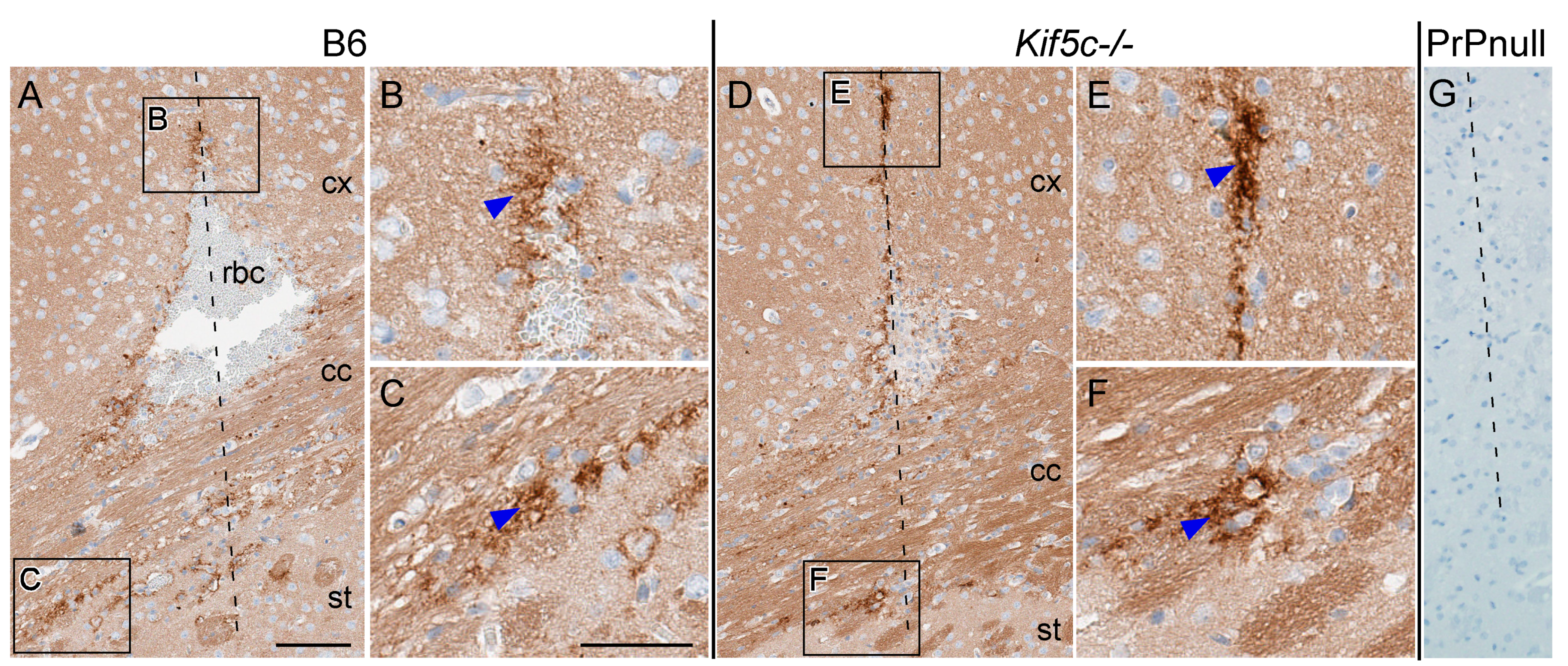
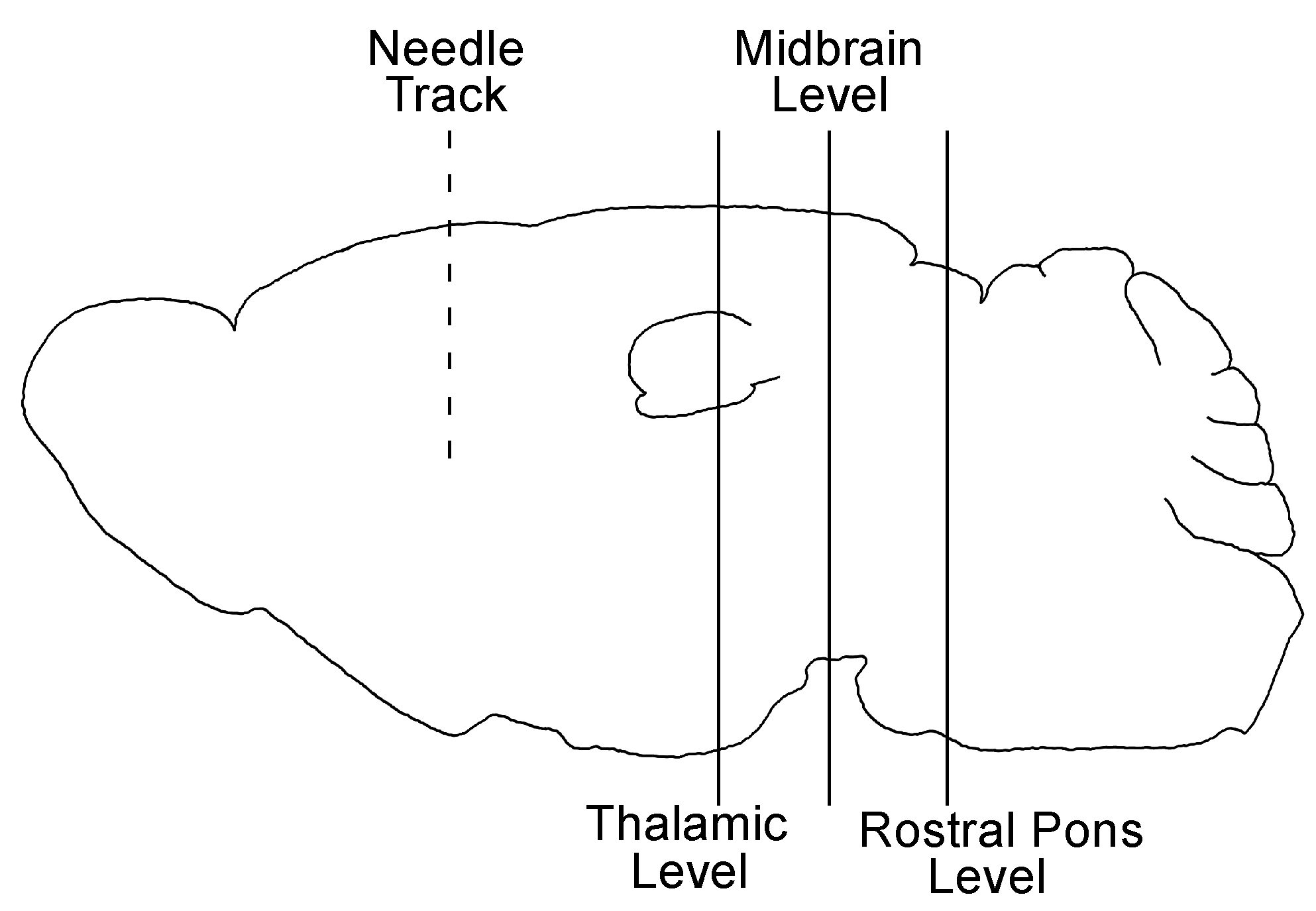
2.2. Stereotactic Surgery and Microinjection of 22 L and ME7 Prions
2.3. Kinetic Experiments
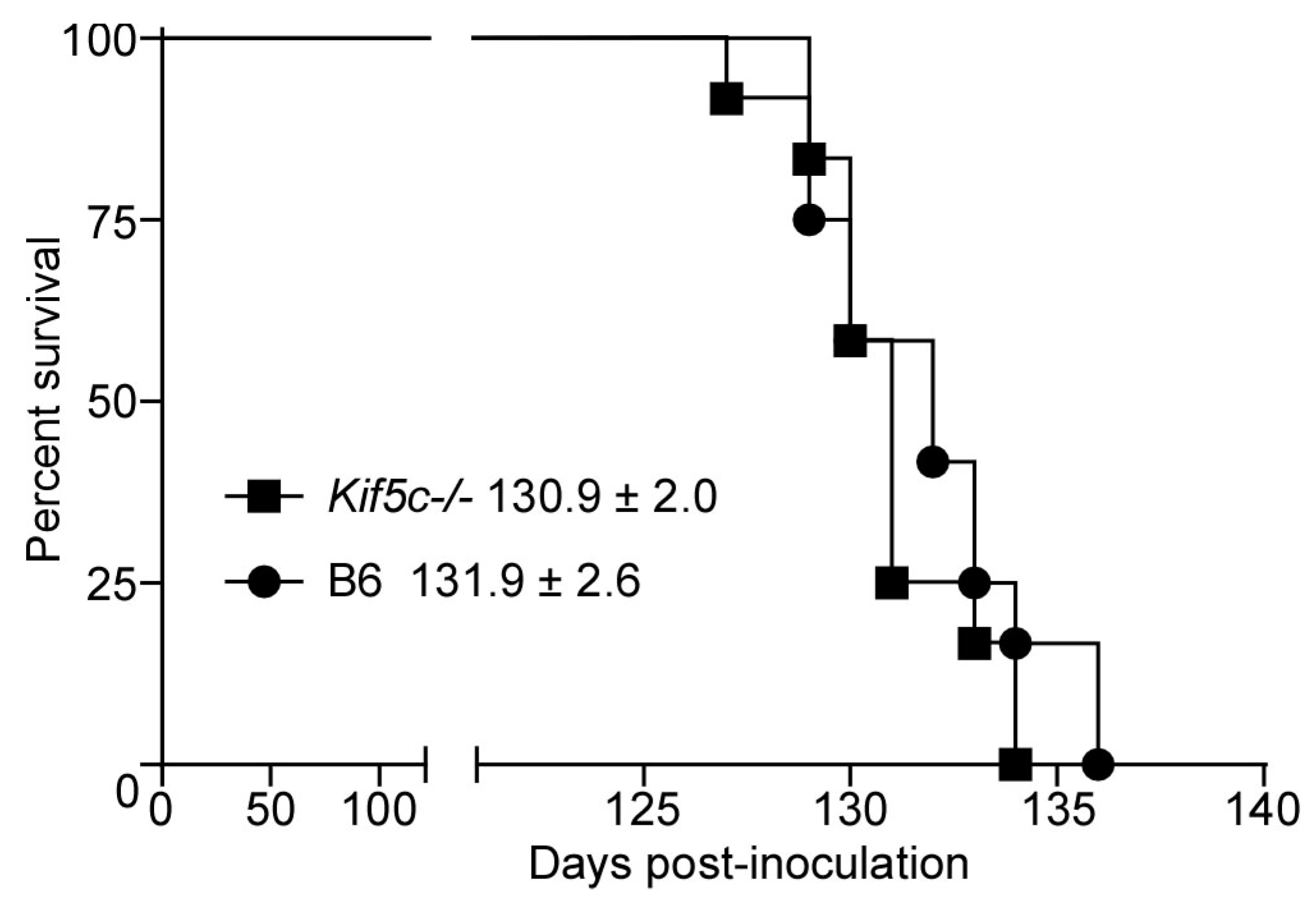
2.4. Survival Curve
2.5. Hematoxylin and Eosin (H&E) and Immunohistochemical (IHC) Staining
2.6. Prion IHC Scoring and Statistical Analysis
3. Results
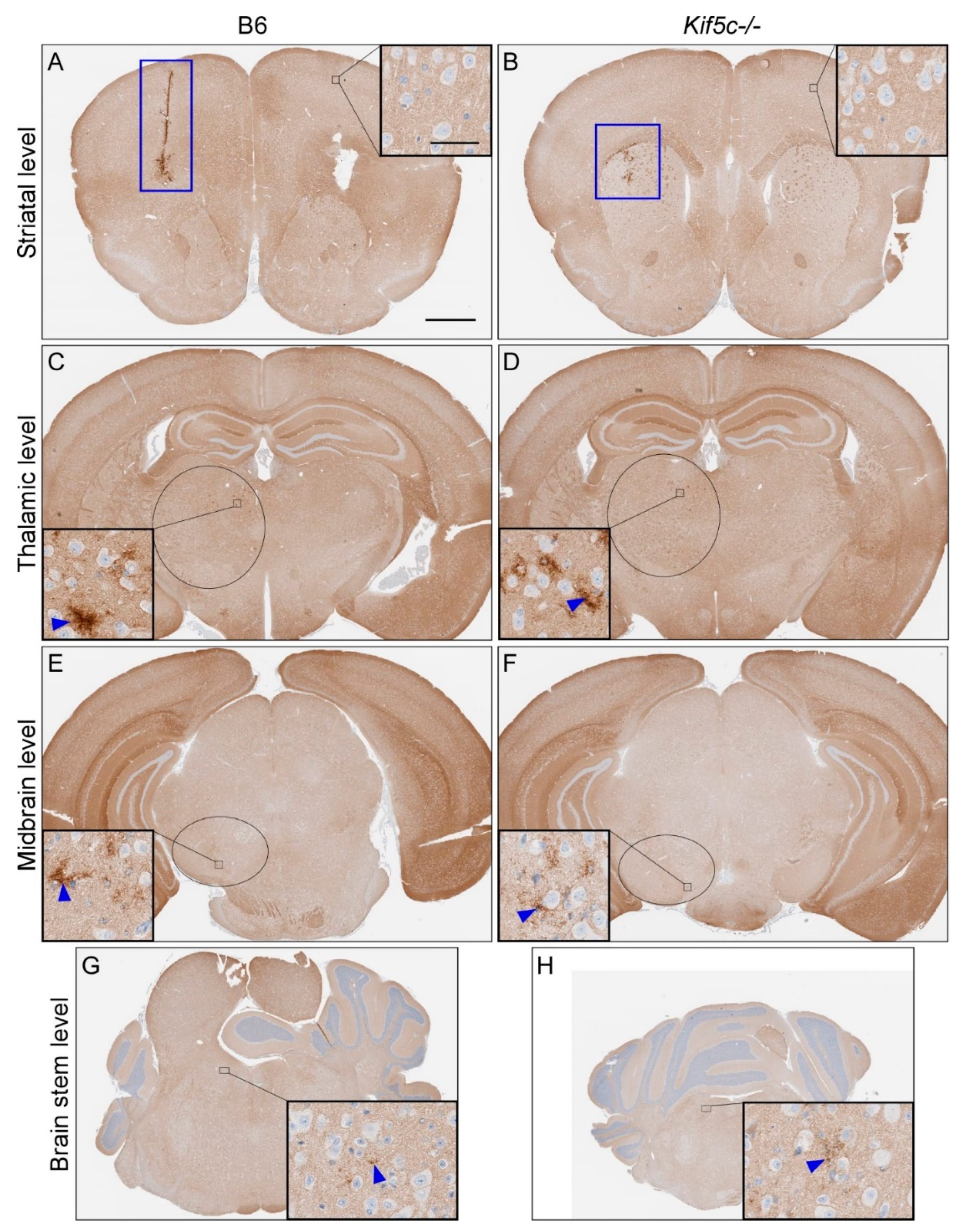
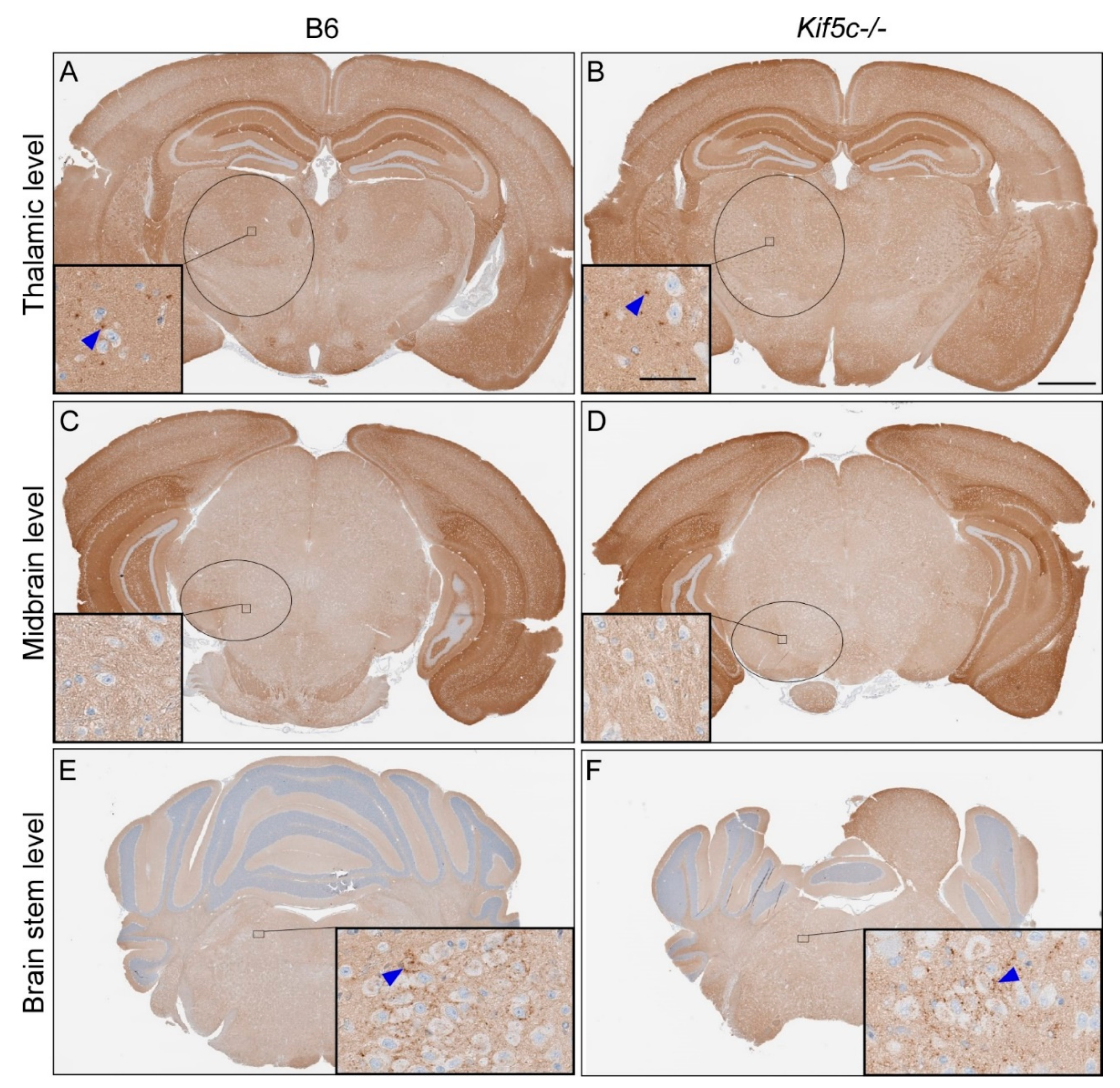
3.1. The 22 L PrPSc Spreading Kinetics in B6 and Kif5c−/− Mice
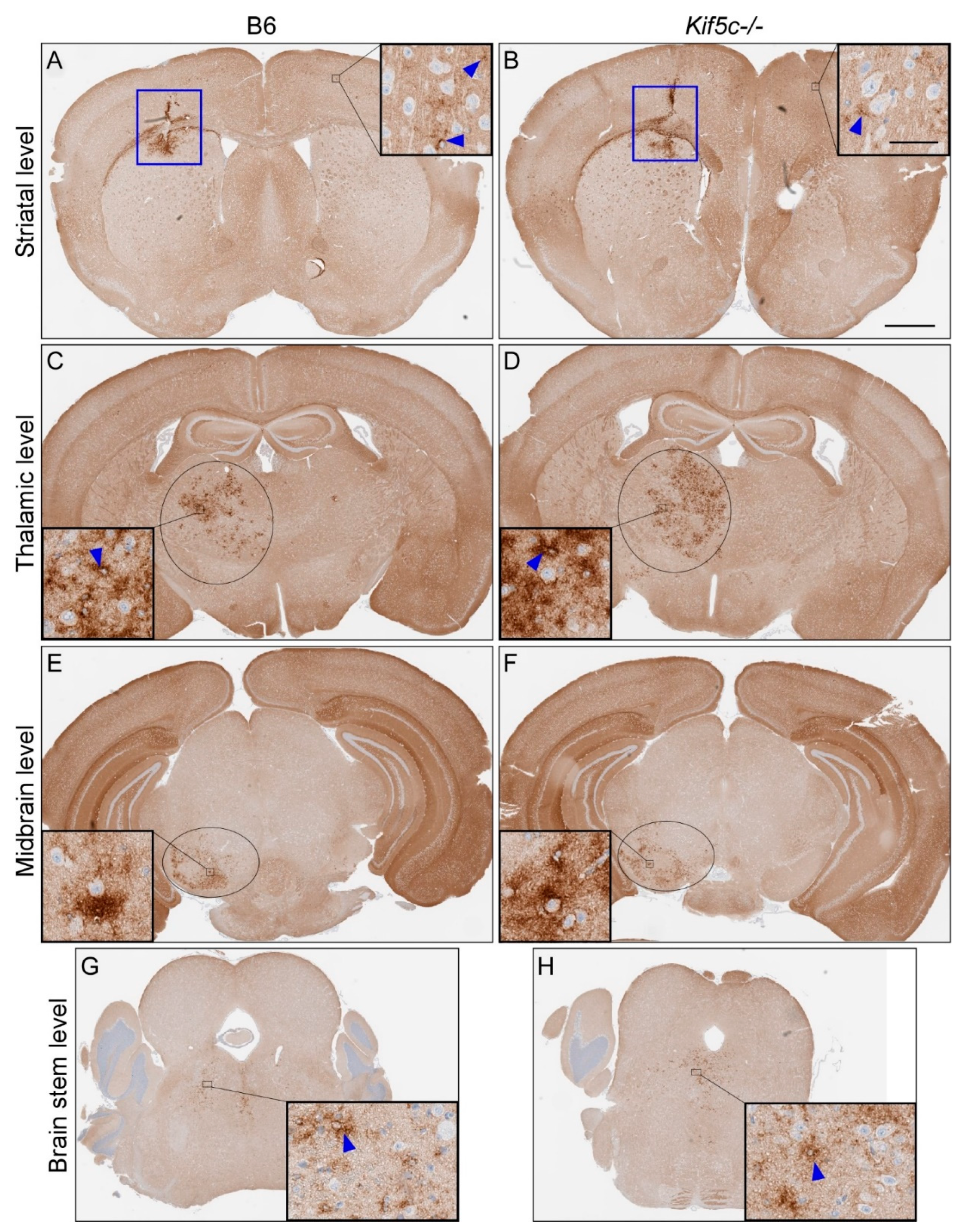
3.2. Stereotactic Kinetics of ME7 Scrapie at 40 Days Post-Inoculation (dpi)
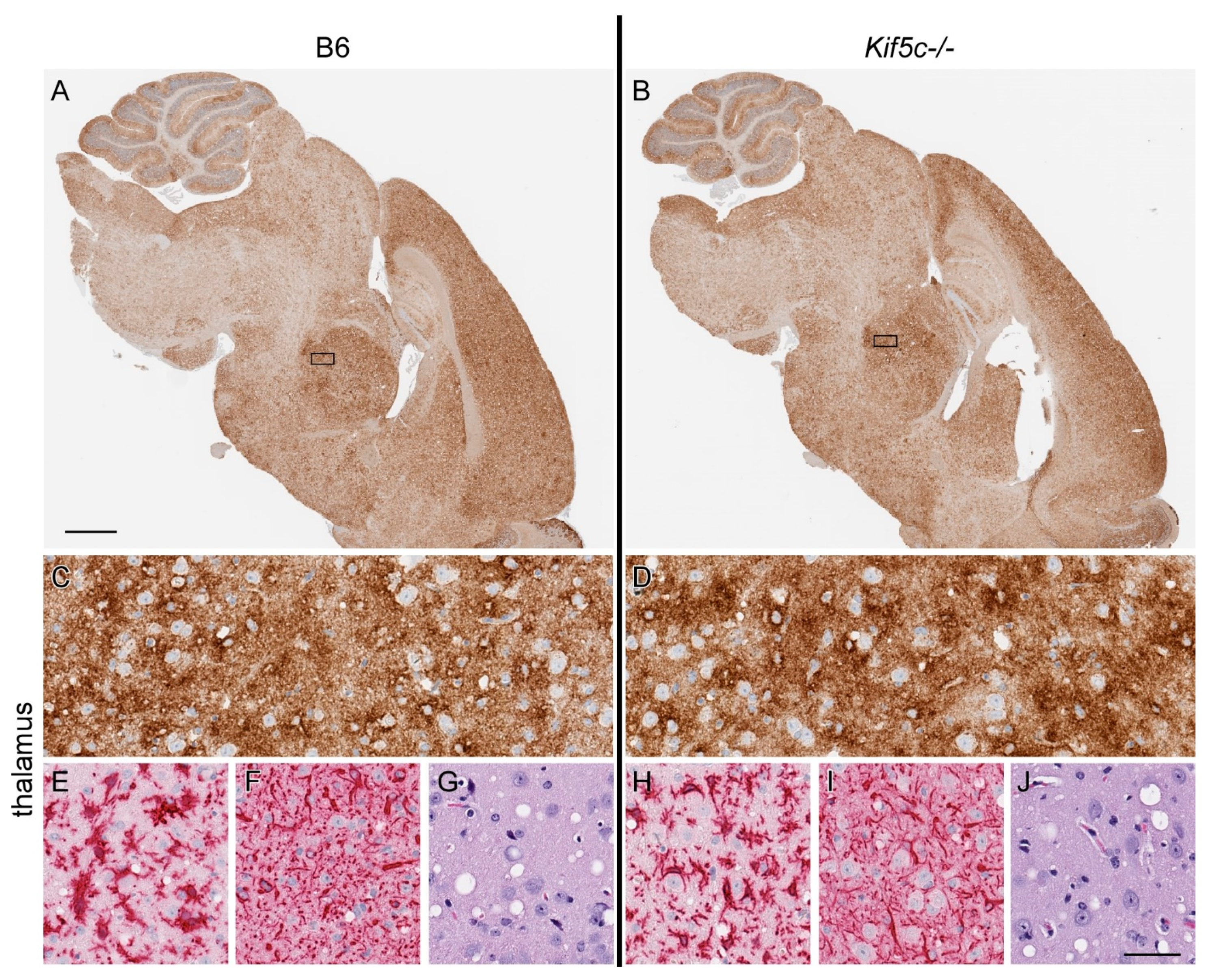
3.3. 22 L Scrapie-Infected Kif5c−/− and B6 Mouse Survival Curve and Neuropathology
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sigurdson, C.J.; Bartz, J.C.; Glatzel, M. Cellular and Molecular Mechanisms of Prion Disease. Annu. Rev. Pathol. Mech. Dis. 2019, 14, 497–516. [Google Scholar] [CrossRef] [PubMed]
- Caughey, B.; Baron, G.S.; Chesebro, B.; Jeffrey, M. Getting a Grip on Prions: Oligomers, Amyloids, and Pathological Membrane Interactions. Annu. Rev. Biochem. 2009, 78, 177–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fraser, H. Neuronal spread of scrapie agent and targeting of lesions within the retino-tectal pathway. Nat. Cell Biol. 1982, 295, 149–150. [Google Scholar] [CrossRef] [PubMed]
- Kimberlin, R.; Hall, S.M.; Walker, C.A. Pathogenesis of mouse scrapie: Evidence for direct neural spread of infection to the CNS after injection of sciatic nerve. J. Neurol. Sci. 1983, 61, 315–325. [Google Scholar] [CrossRef]
- Scott, J.R.; Fraser, H.; Scott, J.R.; Fraser, H. Transport and targeting of scrapie infectivity and pathology in the optic nerve projections following intraocular infection. Prog. Clin. Boil. Res. 1989, 317, 645–652. [Google Scholar] [CrossRef]
- Beekes, M.; Baldauf, E.; McBride, P.A. Cerebral targeting indicates vagal spread of infection in hamsters fed with scrapie. J. Gen. Virol. 1998, 79, 601–607. [Google Scholar] [CrossRef]
- McBride, P.A.; Schulz-Schaeffer, W.J.; Donaldson, M.; Bruce, M.; Diringer, H.; Kretzschmar, H.A.; Beekes, M. Early Spread of Scrapie from the Gastrointestinal Tract to the Central Nervous System Involves Autonomic Fibers of the Splanchnic and Vagus Nerves. J. Virol. 2001, 75, 9320–9327. [Google Scholar] [CrossRef] [Green Version]
- Glatzel, M.; Heppner, F.; Albers, K.; Aguzzi, A. Sympathetic Innervation of Lymphoreticular Organs Is Rate Limiting for Prion Neuroinvasion. Neuron 2001, 31, 25–34. [Google Scholar] [CrossRef] [Green Version]
- Bartz, J.; Kincaid, A.; Bessen, R.A. Rapid Prion Neuroinvasion following Tongue Infection. J. Virol. 2003, 77, 583–591. [Google Scholar] [CrossRef] [Green Version]
- Bessen, R.A.; Martinka, S.; Kelly, J.; Gonzalez, D. Role of the Lymphoreticular System in Prion Neuroinvasion from the Oral and Nasal Mucosa. J. Virol. 2009, 83, 6435–6445. [Google Scholar] [CrossRef] [Green Version]
- Haybaeck, J.; Heikenwalder, M.; Klevenz, B.; Schwarz, P.; Margalith, I.; Bridel, C.; Mertz, K.; Zirdum, E.; Petsch, B.; Fuchs, T.J.; et al. Aerosols Transmit Prions to Immunocompetent and Immunodeficient Mice. PLOS Pathog. 2011, 7, e1001257. [Google Scholar] [CrossRef] [Green Version]
- Shearin, H.; Bessen, R.A. Axonal and Transynaptic Spread of Prions. J. Virol. 2014, 88, 8640–8655. [Google Scholar] [CrossRef] [Green Version]
- Borchelt, D.; Koliatsos, V.; Guarnieri, M.; Pardo, C.; Sisodia, S.; Price, D. Rapid anterograde axonal transport of the cellular prion glycoprotein in the peripheral and central nervous systems. J. Biol. Chem. 1994, 269, 14711–14714. [Google Scholar] [CrossRef]
- Rodolfo, K.; Hässig, R.; Moya, K.L.; Frobert, Y.; Grassi, J.; Di Giamberardino, L. A novel cellular prion protein isoform present in rapid anterograde axonal transport. NeuroReport 1999, 10, 3639–3644. [Google Scholar] [CrossRef] [PubMed]
- Moya, K.L.; Hässig, R.; Créminon, C.; Laffont, I.; Di Giamberardino, L. Enhanced detection and retrograde axonal transport of PrPc in peripheral nerve. J. Neurochem. 2003, 88, 155–160. [Google Scholar] [CrossRef] [PubMed]
- Butowt, R.; Abdelraheim, S.; Brown, D.R.; Von Bartheld, C.S. Anterograde axonal transport of the exogenous cellular isoform of prion protein in the chick visual system. Mol. Cell. Neurosci. 2006, 31, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Encalada, S.E.; Szpankowski, L.; Xia, C.-H.; Goldstein, L.S. Stable Kinesin and Dynein Assemblies Drive the Axonal Transport of Mammalian Prion Protein Vesicles. Cell 2011, 144, 551–565. [Google Scholar] [CrossRef] [Green Version]
- Heisler, F.F.; Pechmann, Y.; Wieser, I.; Altmeppen, H.; Veenendaal, L.; Muhia, M.; Schweizer, M.; Glatzel, M.; Krasemann, S.; Kneussel, M. Muskelin Coordinates PrPC Lysosome versus Exosome Targeting and Impacts Prion Disease Progression. Neuron 2018, 99, 1155–1169.e9. [Google Scholar] [CrossRef] [Green Version]
- Carroll, J.A.; Striebel, J.F.; Rangel, A.; Woods, T.; Phillips, K.; Peterson, K.E.; Race, B.; Chesebro, B. Prion Strain Differences in Accumulation of PrPSc on Neurons and Glia Are Associated with Similar Expression Profiles of Neuroinflammatory Genes: Comparison of Three Prion Strains. PLOS Pathog. 2016, 12, e1005551. [Google Scholar] [CrossRef]
- Matsunaga, Y.; Peretz, D.; Williamson, A.; Burton, D.; Mehlhorn, I.; Groth, D.; Cohen, F.E.; Prusiner, S.B.; Baldwin, M.A. Cryptic epitopes in N-terminally truncated prion protein are exposed in the full-length molecule: Dependence of conformation on pH. Proteins: Struct. Funct. Bioinform. 2001, 44, 110–118. [Google Scholar] [CrossRef]
- Striebel, J.F.; Race, B.; Williams, K.; Carroll, J.A.; Klingeborn, M.; Chesebro, B. Microglia are not required for prion-induced retinal photoreceptor degeneration. Acta Neuropathol. Commun. 2019, 7, 48. [Google Scholar] [CrossRef] [PubMed]
- Klingeborn, M.; Race, B.; Meade-White, K.D.; Rosenke, R.; Striebel, J.F.; Chesebro, B. Crucial Role for Prion Protein Membrane Anchoring in the Neuroinvasion and Neural Spread of Prion Infection. J. Virol. 2010, 85, 1484–1494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imai, Y.; Ibata, I.; Ito, D.; Ohsawa, K.; Kohsaka, S. A Novel Geneiba1in the Major Histocompatibility Complex Class III Region Encoding an EF Hand Protein Expressed in a Monocytic Lineage. Biochem. Biophys. Res. Commun. 1996, 224, 855–862. [Google Scholar] [CrossRef]
- Chesebro, B.; Striebel, J.; Rangel, A.; Phillips, K.; Hughson, A.; Caughey, B.; Race, B. Early Generation of New PrPSc on Blood Vessels after Brain Microinjection of Scrapie in Mice. mBio 2015, 6, e01419-15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mabbott, N.A. How do PrPSc Prions Spread between Host Species, and within Hosts? Pathogenes 2017, 6, 60. [Google Scholar] [CrossRef] [Green Version]
- Kanai, Y.; Okada, Y.; Tanaka, Y.; Harada, A.; Terada, S.; Hirokawa, N. KIF5C, A Novel Neuronal Kinesin Enriched in Motor Neurons. J. Neurosci. 2000, 20, 6374–6384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandner, S.; Raeber, A.; Sailer, A.; Blattler, T.; Fischer, M.; Weissmann, C.; Aguzzi, A. Normal host prion protein (PrPC) is required for scrapie spread within the central nervous system. Proc. Natl. Acad. Sci. USA 1996, 93, 13148–13151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glatzel, M.; Aguzzi, A. PrPC expression in the peripheral nervous system is a determinant of prion neuroinvasion. J. Gen. Virol. 2000, 81, 2813–2821. [Google Scholar] [CrossRef]
- Saborio, G.P.; Permanne, B.; Soto, C. Sensitive detection of pathological prion protein by cyclic amplification of protein misfolding. Nat. Cell Biol. 2001, 411, 810–813. [Google Scholar] [CrossRef] [PubMed]
- Atarashi, R.; Moore, R.A.; Sim, V.L.; Hughson, A.G.; Dorward, D.W.; Onwubiko, H.A.; Priola, S.A.; Caughey, B. Ultrasensitive detection of scrapie prion protein using seeded conversion of recombinant prion protein. Nat. Methods 2007, 4, 645–650. [Google Scholar] [CrossRef]
- Baron, G.S.; Caughey, B.; Starke, D.W.; Chock, P.B.; Mieyal, J.J. Effect of Glycosylphosphatidylinositol Anchor-dependent and -independent Prion Protein Association with Model Raft Membranes on Conversion to the Protease-resistant Isoform. J. Biol. Chem. 2003, 278, 14883–14892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hafezparast, M.; Brandner, S.; Linehan, J.; Martin, J.E.; Collinge, J.; Fisher, E. Prion disease incubation time is not affected in mice heterozygous for a dynein mutation. Biochem. Biophys. Res. Commun. 2004, 326, 18–22. [Google Scholar] [CrossRef] [PubMed]
- Künzi, V.; Glatzel, M.; Nakano, M.Y.; Greber, U.F.; Van Leuven, F.; Aguzzi, A. Unhampered Prion Neuroinvasion despite Impaired Fast Axonal Transport in Transgenic Mice Overexpressing Four-Repeat Tau. J. Neurosci. 2002, 22, 7471–7477. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Scrapie Strain | Mouse Strain | Time Point (dpi 2) | Brain Region | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Striatum (0) 1 | Thalamus (3.2) 1 | Midbrain (5) 1 | Rostral Pons (7) 1 | |||||||||
| PrPSc Present 3 | PrPSc Score 4 | PrPSc Present 3 | PrPSc Score 4 | PrPSc Present 3 | PrPSc Score 4 | p-Value 5 | PrPSc Present 3 | PrPSc Score 4 | p-Value 5 | |||
| 22 L | B6 | 7 | 2/2 | 1 | 0/2 | 0 | 0/2 | 0 | 0/2 | 0 | ||
| Kif5c−/− | 7 | 2/2 | 1 | 0/2 | 0 | 0/2 | 0 | 0/2 | 0 | |||
| B6 | 25 | 5/5 | 2 | 5/5 | 1 | 3/5 | 0–1 | 0.46 | 4/5 | 0–1 | >0.99 | |
| Kif5c−/− | 25 | 3/3 | 2 | 3/3 | 1 | 3/3 | 1 | 2/3 | 0–1 | |||
| B6 | 40 | 4/4 | 3 | 4/4 | 2 | 4/4 | 2 | 4/4 | 1 | >0.99 | ||
| Kif5c−/− | 40 | 4/4 | 3 | 4/4 | 2 | 4/4 | 2 | 3/4 | 1 | |||
| B6 | 60 | 3/3 | 2–3 | 3/3 | 3 | 3/3 | 1–2 | 3/3 | 1–2 | |||
| Kif5c−/− | 60 | 3/3 | 3 | 3/3 | 3 | 3/3 | 2 | 3/3 | 1–2 | |||
| ME7 | B6 | 40 | Not tested | 4/4 | 1 | 0/4 | 0 | 0.17 | 2/4 | 0–1 | >0.99 | |
| Kif5c−/− | 40 | Not tested | 5/5 | 1 | 3/5 | 0–1 | 3/5 | 0–1 | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Race, B.; Williams, K.; Baune, C.; Striebel, J.F.; Winkler, C.W.; Carroll, J.A.; Encalada, S.E.; Chesebro, B. Deletion of Kif5c Does Not Alter Prion Disease Tempo or Spread in Mouse Brain. Viruses 2021, 13, 1391. https://0-doi-org.brum.beds.ac.uk/10.3390/v13071391
Race B, Williams K, Baune C, Striebel JF, Winkler CW, Carroll JA, Encalada SE, Chesebro B. Deletion of Kif5c Does Not Alter Prion Disease Tempo or Spread in Mouse Brain. Viruses. 2021; 13(7):1391. https://0-doi-org.brum.beds.ac.uk/10.3390/v13071391
Chicago/Turabian StyleRace, Brent, Katie Williams, Chase Baune, James F. Striebel, Clayton W. Winkler, James A. Carroll, Sandra E. Encalada, and Bruce Chesebro. 2021. "Deletion of Kif5c Does Not Alter Prion Disease Tempo or Spread in Mouse Brain" Viruses 13, no. 7: 1391. https://0-doi-org.brum.beds.ac.uk/10.3390/v13071391