
The Role of Coinfections in the EBV–Host Broken Equilibrium


1
Research Unit in Virology and Cancer, Children’s Hospital of Mexico Federico Gómez, Mexico City 06720, Mexico
2
Doctoral Program in Biomedical Science, National Autonomous University of Mexico, Mexico City 04510, Mexico
*
Author to whom correspondence should be addressed.
Viruses 2021, 13(7), 1399; https://0-doi-org.brum.beds.ac.uk/10.3390/v13071399
Submission received: 15 May 2021
/
Revised: 29 June 2021
/
Accepted: 12 July 2021
/
Published: 19 July 2021
(This article belongs to the Special Issue Epstein Barr Virus)
Abstract
:The Epstein–Barr virus (EBV) is a well-adapted human virus, and its infection is exclusive to our species, generally beginning in the childhood and then persisting throughout the life of most of the affected adults. Although this infection generally remains asymptomatic, EBV can trigger life-threatening conditions under unclear circumstances. The EBV lifecycle is characterized by interactions with other viruses or bacteria, which increases the probability of awakening its pathobiont capacity. For instance, EBV infects B cells with the potential to alter the germinal center reaction (GCR)—an adaptive immune structure wherein mutagenic-driven processes take place. HIV- and Plasmodium falciparum-induced B cell hyperactivation also feeds the GCR. These agents, along with the B cell tropic KSHV, converge in the ontogeny of germinal center (GC) or post-GC lymphomas. EBV oral transmission facilitates interactions with local bacteria and HPV, thereby increasing the risk of periodontal diseases and head and neck carcinomas. It is less clear as to how EBV is localized in the stomach, but together with Helicobacter pylori, they are known to be responsible for gastric cancer. Perhaps this mechanism is reminiscent of the local inflammation that attracts different herpesviruses and enhances graft damage and chances of rejection in transplanted patients. In this review, we discussed the existing evidence suggestive of EBV possessing the potential to synergize or cooperate with these agents to trigger or worsen the disease.
Keywords:
EBV; HIV; beta herpesvirus; HPV; P. falciparum; H. pylori; periodontal bacteria; coinfection; immunosuppression; lymphomagenesis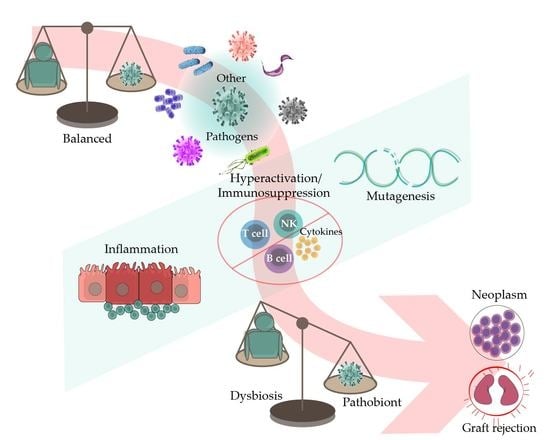
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
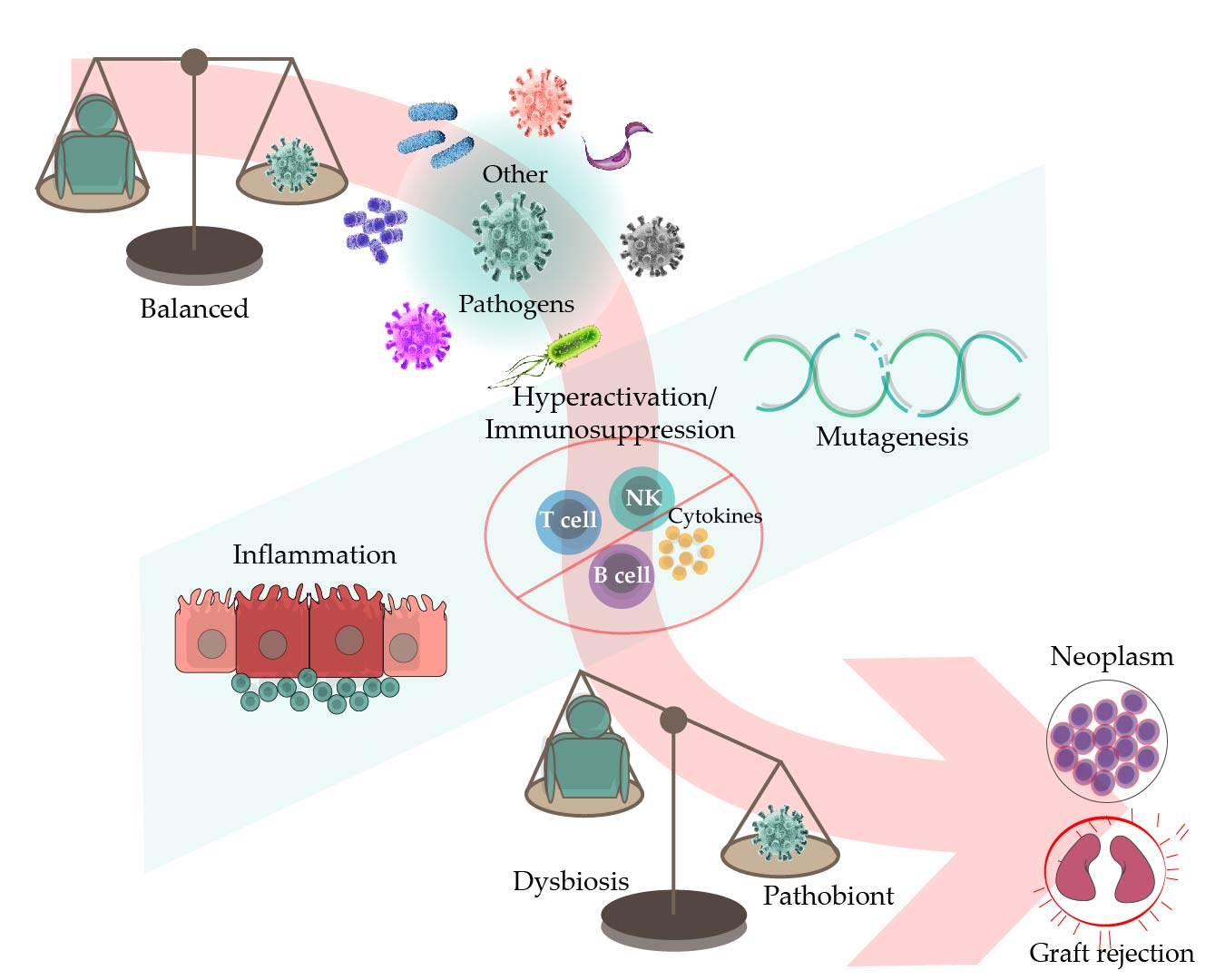
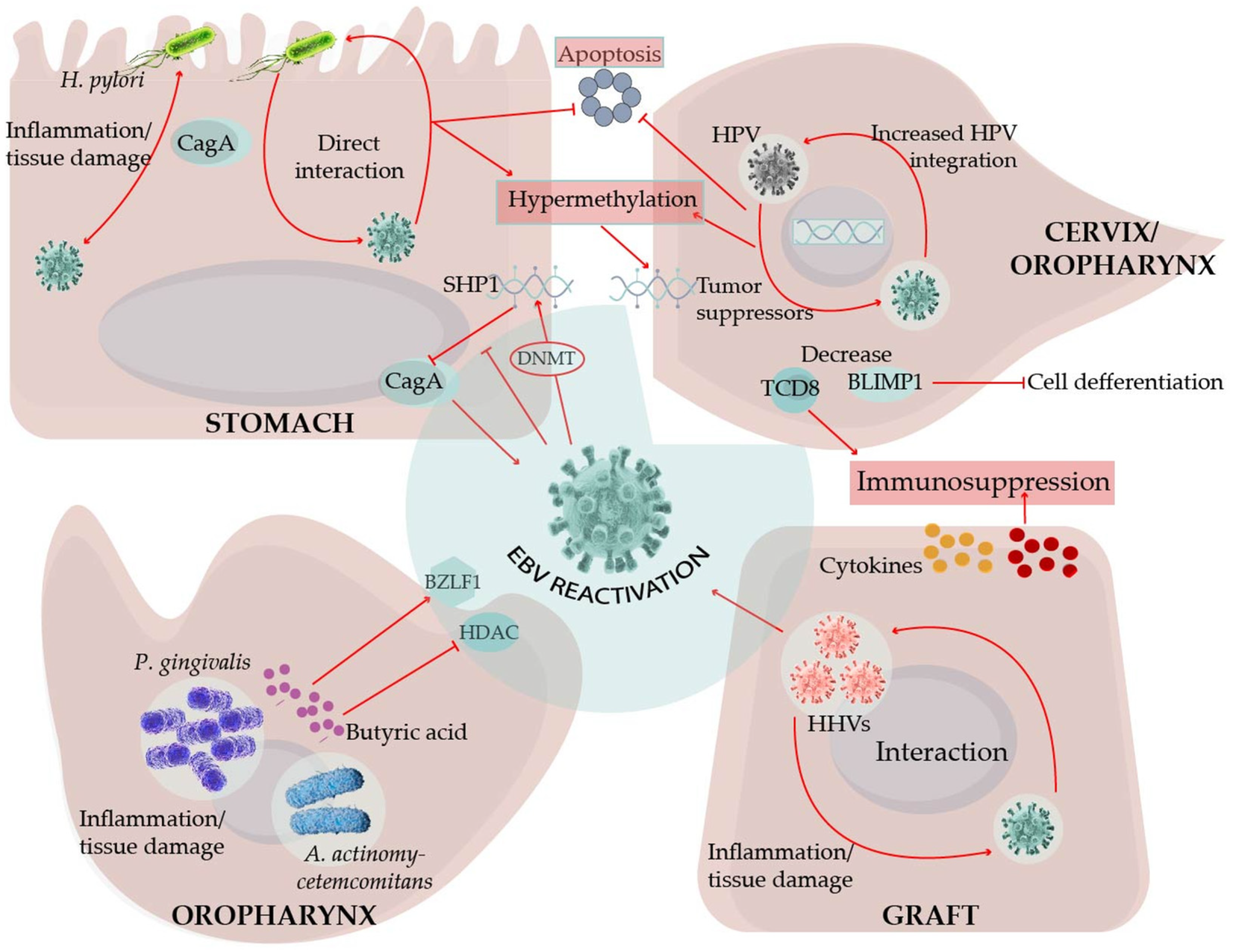
The Epstein–Barr virus (EBV) is a well-adapted human virus, and its infection is exclusive to our species, generally beginning in childhood and persisting throughout the life of most affected adults. This adaptation probably reflects at least a hundred thousand years of viral and human coevolution, helping shape an equilibrium wherein most people live with the virus without showing overt evidence of malaise. Nevertheless, this virus is often associated with severe diseases such as cancer and inflammatory and auto-immune disorders. Thus, EBV seems more of a pathobiont than a true pathogen, that is, an agent with the capacity to unleash its harmful potential in dysbiotic conditions [1]. The circumstances that lead to the disturbance of the viral-host balance are more often unknown. In this review, we focused on the existing evidence indicating that EBV-opportunistic mechanisms of pathogenicity can intersect with those of other infectious agents, resulting in detonation or worsening of the disease (Figure 1).
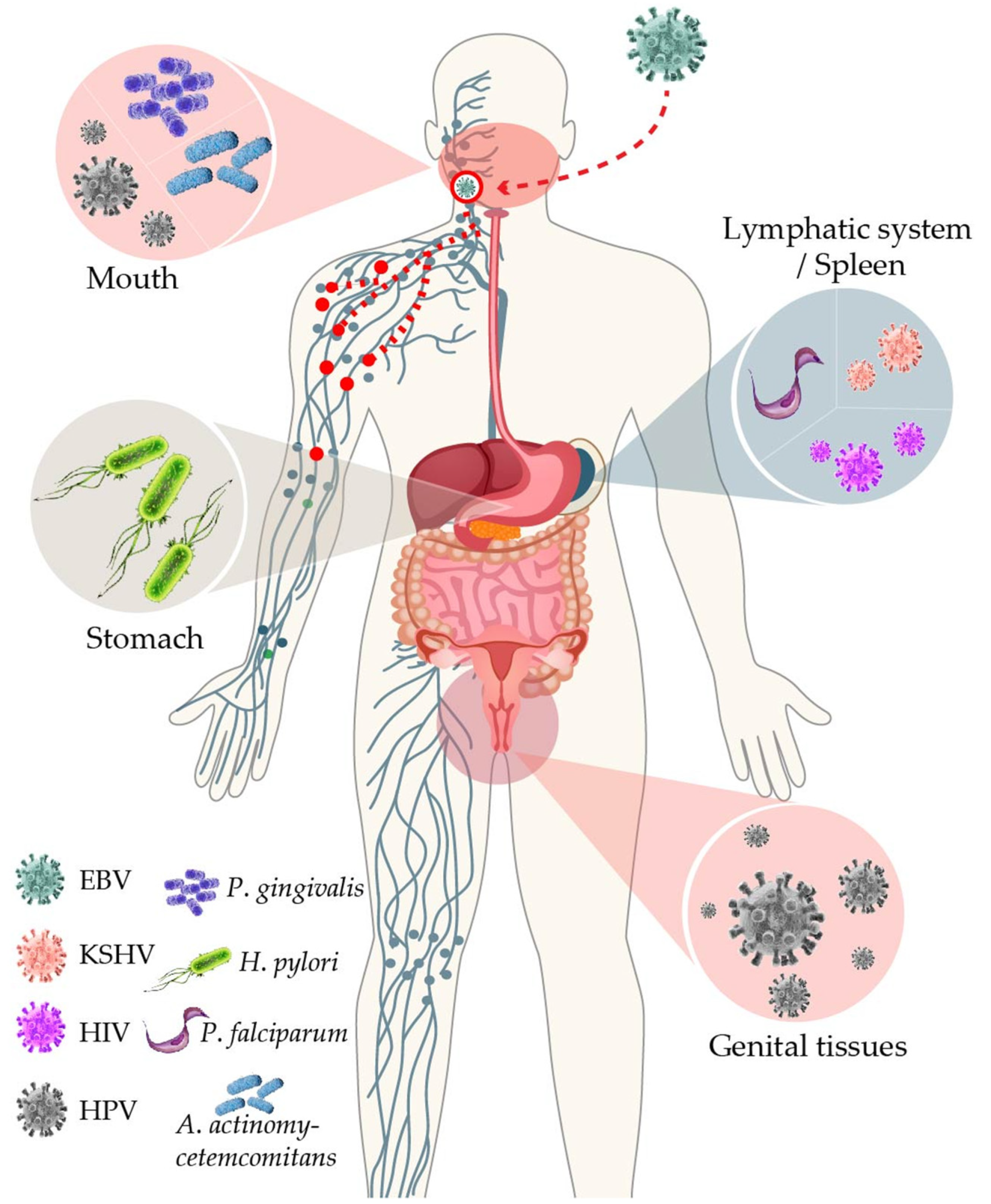
EBV’s main route of transmission is through saliva droplets. EBV first infects the lymphoepithelial tissues of the Waldeyer ring. EBV causes chronic infections oscillating between B cells and epithelial cells, the latter serving to amplify the number of infectious particles released into the saliva for efficient transmission to new hosts and, most likely, B cells providing the niche for persistence, particularly memory B cells [2,3]. Similar to that for the other members of the herpesvirus family, EBV oscillates between lytic and latent infectious cycles, perhaps with the former prevailing more in the epithelial cells and the latter more in the B cells, together assuring successful persistence at the population and individual levels.
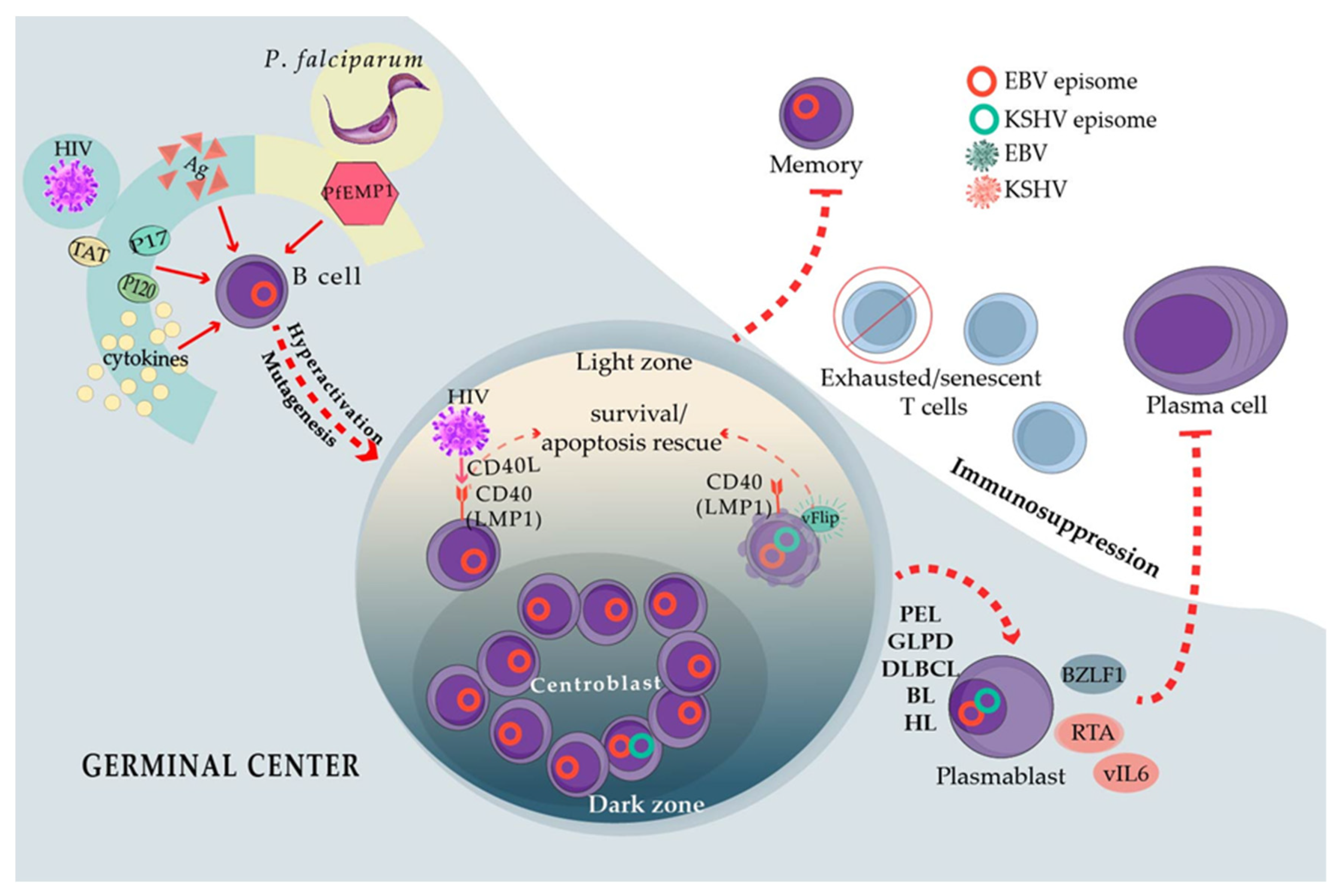
EBV is found in latently infected cells with different active transcriptional programs, which are often referred to as latency programs. For instance, there is a latency III program in which the virus expresses the complete repertoire of latent proteins: the EBV nuclear antigens EBNA1; EBNA2; the family of EBNA 3A, 3B, and 3C; EBNA-LP (leader protein), and three latent membrane proteins LMP1, LMP2A, and LMP2B. The virus progressively switches off the expression of these proteins, beginning with the EBNAs in latency II and then the EBNAs and the LMPs in latency I, except for EBNA1 whose expression is maintained through the three transcriptional programs. EBNA1 serves to replicate the viral genome during the cell cycle and segregates viral genomes to both the daughter cells after mitosis, thus ensuring that the infection is not lost during cell replication. According to the germinal center (GC) model [2], these latency programs allow EBV to transit to the B cell differentiation pathway, changing from an activated to a memory state. Memory B cells are quiescent long-lived cells in which EBV turns off all viral protein expression (latency 0) [3]. Although EBV also expresses several non-coding RNAs throughout all latency programs, latency 0 is most likely the least harmful and the most prevalent one in asymptomatic carriers. The remaining latencies are active in the neoplasms associated with a viral infection and, in the case of EBV-induced B cell lymphomas, these neoplasms usually phenotypically correlate with an intermediate state of the GC reaction (GCR) [3].
2. EBV in Immunocompromised Individuals
The capacity of EBV to downregulate the viral protein expression also allows it to hide from antagonistic host immune responses. Presently, the precise mechanisms and immune populations that help maintain the EBV-host equilibrium are mostly unclear. Most likely, this is importantly aided by CD8 T cells, CD4 T cells, and natural killer (NK) cells [4], since one of the most evident mechanisms of a broken equilibrium is made evident by the improper functioning of these immune cells, as it occurs after primary immunodeficiencies (inborn errors of immunity) or because of pharmacological immunosuppression in the organ- or progenitor cell-transplanted patients. Similarly, individuals infected with the human immunodeficiency virus (HIV) often endure uncontrolled bursts of EBV expansion with the capacity to lead to different types of EBV-associated neoplasms. Before the introduction of antiretroviral therapy (ART), the so-called AIDS-defining cancers emerged within a very deteriorated host immune system that was unable to efficiently survey for virus-driven oncogenesis. These neoplasms are often polyclonal cellular expansions, as in Kaposi sarcoma virus (KSHV)-induced sarcomas, Castleman disease, and EBV lymphoproliferative disorders (LPD).
3. EBV and KSHV Coinfect B Cells in Primary Effusion Lymphoma (PEL) and Germinotropic LPD
One of the clearest and best-understood examples of EBV’s cooperation with other pathogens is with KSHV, a gammaherpesvirus similar to EBV. Between 60% and 90% of PEL cases harbor EBV and KSHV genomes coinfecting almost all lymphoma cells [5]. PEL is a neoplasm that almost exclusively surfaces in AIDS patients and is characterized by an accumulation of tumor cells in the serous cavities of the body, such as the pleura, pericardium, and peritoneum, although an extracavitary presentation has also been reported [6]. A different gene expression signature is observed in single-infected PELs when compared with that in coinfected PELs, suggesting distinct PEL subtypes depending on the presence or absence of EBV [6]. EBV-positive PELs exhibit evidence of somatic hypermutation, which is supportive of a post-GC B cell origin. On the contrary, EBV-negative PEL is rarely hypermutated, suggesting a pre-GC origin [7]. Indeed, PEL cells morphologically resemble plasmacytic/plasmablastic cells [8]. KSHV and EBV also coinfect the cells of germinotropic LPD [9]. Germinotropic LPDs (GLPD) morphologically resemble EBV-positive extracavitary PEL, but are mostly found in immunocompetent hosts. Since this LPD is newer and there are only a few reported cases in the literature, most studies addressing EBV and KSHV interaction mechanisms are always considered exclusive of PEL.
It is debatable as to which virus initiates the lymphomagenesis process; since KSHV is present in all PELs, it appears to be the main driving force. However, KSHV cannot transform B cells nor can it persistently infect them in vitro [10]. On the contrary, EBV efficiently immortalizes and activates B cells. Perhaps because of this reason, EBV facilitates the establishment of sustained KSHV infection of B cells, thus allowing KSHV higher loads per cell—an activity credited to EBNA1 [11,12]. Thus, it has been proposed that EBV precedes KSHV, thus rendering EBV-infected cells more susceptible to KSHV infection [7]. However, another study found that persistent infection of peripheral B cells with KSHV was achieved when the cells were exposed to KSHV 24 h before EBV infection [11]. Regardless of which virus infects first, both are believed to be necessary to sustain PEL, as together they provide selective advantages to the coinfected cells [12]. For instance, the coinfected cells present an increased expression of cellular genes involved in the anti-apoptotic processes, mitochondrial functions, and cell cycle, while showing a decreased expression of genes involved in innate immune responses and cytokine signaling [13].
The contribution of EBV and KSHV in the lymphomagenesis process does not strictly follow the canonical patterns of transformation established for each virus individually. It thus seems that EBV and KSHV must establish a delicate balance in coinfected cells, blending and co-regulating their transforming mechanisms. For example, EBV presents a restricted gene expression pattern with a latency I program in coinfected PEL cell lines [14] because the KSHV LANA protein inhibits the master promoter regulator of EBV latency III (the Cp). LANA binds the corepressor mSin3 and together recruits the methylase MeCP2-silencing Cp through methylation-mediated repression [15]. A bidirectional regulatory mechanism that promotes the maintenance of dual latency in coinfected PEL cells through the inhibition of lytic replication has also been proposed [16]. KSHV LANA and RTA proteins transactivate EBV LMP1, which in turn represses lytic replications of both viruses [17,18,19]. RTA and BZLF1 are the main transcription factors controlling the activation of the KSHV and EBV lytic cycles, respectively. Colocalization and coprecipitation experiments suggest that both proteins inhibit each other through direct interactions, which interferes with the formation of their respective multimeric active complexes [18,19,20]. Since EBV is in latency I, it is not clear when the LMP1-mediated lytic cycle suppression is active. Other studies support a loose control of EBV transcription with a sporadic expression of other genes, mainly LMP1 [14,21,22]. Enhanced expression of EBV lytic genes has also been observed in coinfected PEL cells, which can be attributed to an increased expression of the transcription factor BLIMP1 [23]. BLIMP1 is involved not only in the maintenance of PEL [24] but also in the induction of both EBV immediate early genes BZLF1 and BRLF1 [25]. The expression of BLIMP1 marks the end of the GCR, sending GC B cells into antibody-producing plasma cells, which correlates with EBV reactivation [26]. Similarly, the BLIMP1 expression correlates with epithelial cell differentiation and the induction of the EBV lytic program [25].
It remains uncertain how the lymphomagenesis process is enhanced through coinfection. At least for PEL, this is largely aided by the HIV-imposed immunosuppression and HIV potential lymphomagenesis mechanisms (see below). In addition to immunosuppression, it remains reasonable to believe that KSHV, with a limited transforming capacity, is assisted by EBV, whose B cell transforming capacity is notably superior [11]. However, EBV displays a restricted latency, without the expression of its canonical oncogenes. Although KSHV is also responsible for Kaposi sarcoma and multicentric Castleman disease, these are considered to be polyclonal cellular expansions rather than true neoplasms. It has also been proposed that EBV contributes to the lymphomagenesis process through the regulation of the TCL1 oncogene, which also occurs in EBV-positive Burkitt lymphoma (BL), although PEL appears to occur in a TCL1-independent manner [27]. The myc translocation characteristic of BL is also absent in PEL. Nevertheless, the EBERs and EBNA1 are deemed necessary for the proliferation of coinfected PEL cells [21,28]. It is also possible that, at the beginning of infection and before their repression, EBNA2 and other EBV products promote early transcriptional conditions, which modulate the expression of KSHV and cellular genes that together stage the permissive conditions for KSHV-persistent infection and the emergence of PEL [11]. KSHV genes LANA, vFLIP, and vIRF interact with different tumor suppressors and inhibit the apoptotic processes [29,30,31]. In addition, LANA, together with LMP1, can independently induce the promoter of UCH-L1 through the activation of RBP-Jk. UCH-L1 is an oncogenic protein detected in several cancers [32], including some of lymphoid origin [33,34]. In PELs with LANA and LMP1 co-expression, both proteins synergize to activate UCH-L1, which enhances a tumorigenic phenotype with increased proliferation, adhesion, cell migration, and inhibition of apoptosis [35].
Humanized mice models have also aided in the understanding of how infection with both viruses proceeds. These models support that lytic reactivation plays a central role in the development of PEL, as it was observed that a significant proportion of coinfected cells express lytic genes; this observation was also associated with greater efficiency of lymphomagenesis [13,36,37,38]. This event may seem contrary to the idea shaped in cell lines about the negative bidirectional regulation of the KSHV and EBV lytic cycles [16,19]. Lytic genes can mediate the local release of cellular and viral cytokines, such as KSHV vIL-6, vGPCR, K1 and K15, and EBV vIL-10 (BCFR1), v-Bcl-2 (BHRF1), etc., which can aid oncogenesis through the promotion of proliferation, cell survival, immunosuppression, neoangiogenesis, and activation of oncogenic signaling pathways, such as NF-κB [39,40,41,42,43] (Figure 2). Altogether, these studies shed light on the complex interactions between KSHV and EBV, which support a balanced mutual influence that facilitates the persistence of both viruses toward enhancing their tumorigenic potential.
4. EBV and Betaherpesviruses in the Transplanted Patient
The human betaherpesviruses cytomegalovirus (CMV) and the roseoloviruses human herpesvirus 6A (HHV6A), human herpesvirus 6B (HHV6B), and human herpesvirus 7 (HHV7) are also life-persistent common viruses that exert direct cytopathic effects on several organs. These viruses have been proposed as potential cofactors in the progression of different neoplasms [44,45,46], and they are particularly relevant in settings with inadequate immunosurveillance, such as in transplanted patients subjected to pharmacological immunosuppression [47,48]. In these patients, the detection of these viruses has been associated with mutual exacerbation of their infectious cycles, graft rejection, and the development of post-transplant LPD (PTLD) [49,50]. For these reasons, multiple studies have addressed viral loads of EBV and the betaherpesviruses in the peripheral blood of post-transplanted patients as a measure of infection or reactivation. These studies revealed a codetection rate of 2.6–32.7% [51], sometimes without clinical significance [51,52,53,54]. In other studies, however, viral codetection appeared to influence the clinical outcomes. For example, the codetection of EBV and CMV has been associated with graft damage in up to 24.5% of cases [51,55] and with graft rejection in up to 11.1% of cases [51,56]. In children with renal transplants, we observed that EBV and HHV7 codetection was associated with a 5.5-fold-enhanced risk of graft rejection, while that of triple EBV, HHV6, and HHV7 infection was associated with an increased rejection rate by 17.6-fold [57]. Concerning PTLDs, these are a heterogeneous group of lymphoid disorders ranging from indolent polyclonal proliferations to aggressive lymphomas [58]. Although one study did not observe any association between EBV and CMV codetection and PTLD [54], multiple other studies have documented that their codetection increased the risk of PTLD [55,59,60,61].
Unfortunately, all of the above-mentioned reports are associative, and it remains uncertain whether detectable viral loads are indicative of causality or whether a worsened disease influences viral loads. It is therefore important to mention that, although these viruses are present in most of the population, they are generally undetectable or detected in low viral loads in immunocompetent carriers. Moreover, although high viral loads play an important role in the diagnosis, prevention, and treatment of associated diseases, there seems to be a lack of standardization of the methods used to measure them and there is also no consensus on the viral load threshold beyond which they should be considered to be clinically relevant [62]. Different individual or progressing thresholds have been proposed to identify or define groups of transplanted patients with an enhanced risk to develop PTLD, ranging from 500 to 4000 EBV copies per μg or per mL of blood DNA [60,63,64]. The association of viral loads with the clinical outcomes is blurrier when multiple viruses are simultaneously detected. The predictive value of codetection loads is controversial and unclear, with some patients presenting with “high” viral loads and remaining stable for months or years and others presenting with “low” viral loads with significant clinical implications [55]. In the latter study, there was a greater amount of graft dysfunction in patients with CMV and EBV codetection, but with “low” EBV loads (1000–2000 copies/mL of blood) relative to those with a “high” copy number (>5000). It is therefore clear that CMV and EBV individually influence graft damage, graft rejection, and PTLD, since prophylactic treatment with gancyclovir, a nucleoside analog that inhibits the DNA polymerase of herpesviruses, reduces these complications [47]. For this reason, EBV and CMV are routinely followed in transplanted patients. Nonetheless, the sole simultaneous detection of these viruses neither helps elucidate the role played by each virus nor their mechanisms of cooperation.
There are also other scenarios wherein the codetection of multiple herpesviruses has been reported. For instance, EBV and CMV together were associated with more severe forms of infectious mononucleosis [65,66,67], hepatitis, and hemophagocytic lymphohistiocytosis [66,67]. In a study on endemic BL, CMV, and KSHV were detected through immunohistochemistry in the adjacent non-neoplastic tissues. The presence of CMV and KSHV was associated with a shift in the mutational landscape of the neoplasm, with a lower load of classical BL mutations and additional activity of TCF3 and an enhanced expression of EBV lytic cycle genes [68]. In further mechanistic studies, herpesviruses codetection was shown to influence the dynamics of immune cells, such as in the formation and activity of NK cells [69,70,71], as well as the expansion and activity of B cells [72]. These mechanisms may deter immune responses directed against these viruses, facilitating their persistency. Indeed, CMV reactivation has been demonstrated to induce immunosuppression, which predicts EBV reactivation and PTLD risk [60]. CMV and EBV codetection has also been associated with a decreased frequency of CD56dimNKG2AposKIRneg NK cells, thus contributing to suboptimal EBV control in immunosuppressed children with PTLD [59]. HHV6 or EBV single infection of peripheral blood mononuclear cells induces greater levels of IL-6, TNFα, and IL-1β than in coinfection [73]. On the other hand, infection with HHV6 has been shown to influence the in vitro reactivation of EBV-infected B cells [74] (Figure 3). Taken together with the results of the above-mentioned studies, it can be said that a mild inflammatory reaction to the grafted organ can become exacerbated if EBV or other betaherpesviruses-infected immune cells arrive at the site of inflammation.
5. HIV Steady B Cell Stimulation Creates a Lymphomagenesis Permissive Environment
ART introduction shifted the conditions that restrict the life expectancy of people living with HIV (PLWH). Presently, cancer remains one of the most serious threats facing PLHW, and lymphomas are the most common cause of PLWH death in developed nations [75]. EBV is still responsible for more than half of the cases of lymphomas arising in these individuals [76]. For instance, Simard et al. reported that, while the burden of AIDS-defining cancers decreased from 18% to 4.2% with ART, the risk of classical Hodgkin lymphoma (cHL) increased [77], and EBV remains responsible for close to 100% of all cHL cases [78]. Moreover, although DLBCL has significantly decreased with ART, it remains the most common lymphoma post-ART, particularly the immunoblastic subtype that also tends to be EBV positive [79]. A search of The Surveillance, Epidemiology, and End Results tissue repository revealed that EBV is responsible for up to 64% of the BL of PLWH in the USA [80], although other countries have documented lower frequencies. This result indicates that the intersection between EBV and HIV goes beyond the loss of T cells, indicating other levels of interaction between both pathogens. This point is also illustrated by the fact that EBV-associated lymphomas arousing pre-ART tended to be latency III, while in those with controlled HIV viremia and more robust T cells numbers it tended to be latency I or II [81]. Thus, lymphomas arising in PLWH can be divided between those still dependent on strong immunosuppression and those developing within relatively normal CD4 T-cell numbers (e.g., >200/µL), with the EBV-associated cHL, DLBCL, and BL as examples of the latter. There are excellent reviews describing all EBV-positive lymphomas in HIV-infected people [82].
There are four different subtypes of cHL, of which the one that occurs most frequently in PLWH is the mixed-cellularity subtype. Mixed-cellularity cHL in PLWH is extremely similar phenotypically to the cHL of HIV-negative patients, except for a larger numbers of lymphoma cells [83], and EBV expresses a latency II program in both. Likewise, BL in PLWH depends on myc translocations to the immunoglobulin heavy or light chain loci, while EBV presents with a latency I gene expression program [84]. One phenotypic exception is that EBV-positive DLBCL, cHL, and BL more often present with plasmablastic/plasmacytoid features in PLWH. HIV lymphomagenesis mechanisms have been proposed to explain the high frequency of lymphomas in PLWH, despite that HIV has never been detected to infect lymphoma cells [85]. These lymphomagenesis-indirect mechanisms include chronic B cell activation and enhanced mutagenesis resulting from the continuous entry into the GCR, as well as T-cell exhaustion and senescence that may facilitate the immunoescape of lymphoma cells. Although these mechanisms are not EBV-specific, they may directly intersect with EBV-infected B cells transiting the GCR, for instance, cooperating with the capacity of EBV to enhance cell survival and proliferation. GCR elicits both memory B cells and plasma cells, but, contrary to memory cells, plasma cells are short-lived, and EBV-latent genes strongly antagonize the formation of this cell population [86,87] (Figure 2).
HIV-positive individuals harbor B cells with an activated phenotype, which can be explained by direct antigenic or indirect heterologous/polyclonal activation of B cells. Evidence of B cell hyperactivation includes the increased expression of activation markers [88] and hypergammaglobulinemia in in vivo and ex vivo cultures of B cells derived from PLWH [89]. Hyperactivated B cells explain a post-GC cell originating in the EBV-positive lymphomas in PLWH and the plasmablastic/plasmacytoid phenotype. Low levels of HIV replication in the lymph nodes or lymphatic mucosal tissues may directly activate B cells through HIV antigens. In addition, several HIV proteins are secreted, accumulate, and persist in the lymph nodes, in which they may help create a B cell stimulatory microenvironment. Particularly, HIV p17, p120, and Tat induce B cell lymphomas in mice recombinant models [90,91], and p17 and gp120 accumulate in GC even in individuals with undetectable HIV viremia [92]. Tat can be captured by B cells in which it activates oncogenic pathways [93]. This is also true of p17, and CXCR2 acts as a receptor of p17 in B cells triggering the PI3K/Akt oncogenic pathway [94,95]. Moreover, HIV virions carrying the CD40 ligand (CD40L) on their surface have also been reported. CD40L is incorporated into the HIV envelope upon viral budding from infected CD4 T-cells [96]. CD40 ligand-receptor interactions are the most important signals to activate B cells, after antigens, and they are the most important survival signals in the GC [97]. CD40L-positive HIV virions can strongly mimic the activation conferred by the CD40L on follicular CD4 T cells (TFH) [98,99]. In this scenario of specific and heterologous activation, HIV probably sends B cells to the GCR in which highly mutagenic mechanisms occur, such as somatic hypermutation and isotype switching together with extensive proliferation [97]. Enhanced GCR probably amplifies the chance of off-target gene mutagenesis, such as the myc, bcl2, and bcl6 translocations found in BL and DLBCL. Indeed, these lymphomas and cHL often lack the expression of antigen receptors (BCR) because of crippled mutations, which is illustrative of off-target mutagenesis. Receptor-less B cells should die of apoptosis because they lack the signals given by the tonic or antigen-driven BCR activity, but they may be rescued by EBV-latent genes [100,101]. Importantly, the CXCR2 expression is induced by EBV LMP1, a latency II protein expressed in EBV-positive cHL and some DLBCL [102]. EBV LMP1 mimics CD40 coreceptor signals [103], and thus CD40L also enhances the CXCR2 production by B cells, potentially creating a positive loop of B cell activation with HIV and/or secreted p17.
There are other potential mechanisms of HIV heterologous activation of B cells. Despite successful ART, PLWH harbor immune cells that constitutively secrete several different cytokines, chemokines, and other markers of inflammation. The elevated levels of inflammatory molecules contribute to HIV comorbidities, such as cardiovascular disease, neurocognitive impairment, and cancer [104]. The identity of the inflammatory molecules varies among studies, but they often point to IL-6, IL-10, TNFα, IL-1β, IP-10/CXCL10, and C reactive proteins, which may positively loop to further activate B cells. IL-6 has been shown to maintain GCR in murine models of persistent viral infection [105], while IL-10 has been shown to enhance B cell antibody production [106]. IL-6, IL-10, and TNFα are expressed by B cells upon CD40L activation [107], and enhanced expression of IL-6 and IgG has been documented after B cell interaction with CD40L-bearing HIV virions [98]. Indeed, the IL-6 levels do not decline even after years of successful ART [108,109], and the enhanced levels of IL-6 and other B cell stimulatory molecules precede lymphoma development [110,111]. The elevated levels of IL-6, IL-10, TNFα, and IL-1β have also been observed in the heterologous B cell stimulation mediated by p17, gp120, and Tat [90,91] (Figure 2).
Although hefty numbers of CD4 T cells have been observed in the periphery, a different scenario may be occurring in secondary lymph nodes and mucosal tissues. For instance, severe depletion of CCR5pos CD4 T cells in the gastrointestinal (GI) tract has been documented [112,113], which is not restored upon ART [114,115]. This event may compromise the GI epithelial barrier, leading to increased translocation of microbial products into the lamina propria [116] as well as to severe dysfunction of the GI mucosal immune cells. For instance, GI Th17 and Th22 CD4 T cells are impaired, and the non-physiological levels of IL-22 and IL-17 contribute to the disruption of the homeostasis of the epithelial barrier [117,118,119]. Microbial translocation is considered an important contributor to the persistent inflammation and pathological activation of systemic immune cells, which altogether are responsible for the increased mortality of PLWH despite controlled viremia. B cells may also be targeted by this pathological chronic activation, increasing the risk of lymphoma. Among the most important translocated microbial products causing systemic activation of immune cells are TLR and NLRPR agonists and β-d-glucans [120,121].
Some studies suggest that cHL is more common among individuals on ART than in those without it [122]. cHL is also more common in individuals with heftier CD4 T cell numbers (between 200 and 500 cells/µL) [123], perhaps indicating a co-participative role of these cells. CD4 T cells are necessary for the GCR, particularly TFH. HIV and TFH cells’ cooperative mechanisms have been described to affect the GCR function and the B cell terminal differentiation into memory and antibody-producing plasma cells. TFH cells are expanded in PLWH correlating with increased numbers of GC B cells and plasma B cells and with gammaglobulinemia [124]. HIV replication in the lymph node TFH cells has been documented in individuals with controlled viremia [125], and TFH cells have been proposed as the site of HIV long-term persistency [126,127]. HIV-infected TFH cells are phenotypically and functionally different than their HIV-negative counterpart, which supports a dysfunctional GCR [126,127]. Dysfunctional GCR is also supported by a decrease in CD27pos memory B cells [128,129], a remarkable observation considering that memory cells are the site of EBV-unharmful persistent infection [3].
T cells with an exhausted phenotype accumulate in long-term HIV carriers [130] (Figure 2). Exhaustion is executed by a series of immune checkpoint inhibitors (ICI), and the most important ones of these are PD-1, CTLA-4, LAG3, TIGIT, and TIM3 [131]. The expression of ICI should signal for the control of acute antigenic challenges to prevent lingering responses and auto-immunity. However, in chronic antigenic stimulation, such as persistent infections and cancer, T cells expressing ICI markers accumulate, indicating untimely exhaustion [131]. Terminally exhausted T cells do not respond to antigenic stimulation, thus failing to eliminate infected/tumor cells. There are also senescent T cells that are characterized by low to no expression of costimulatory receptors CD28 and CD27 and by the lack of proliferation in response to new antigenic challenges despite an effector memory phenotype. Senescent T cells progressively lose their telomeres and exhibit a metabolic switch that is characterized by enhanced glycolysis and low mitochondrial respiration, but they do not express multiple ICI [132]. Senescent T cells are also characterized by a reduced TCR repertoire, but with enhanced specificity for CMV and EBV antigens. It has also been proposed that senescence is responsible for the lack of response to new antigens in the elderly, for instance, to influenza vaccination [133]. T cells from PLWH also exhibit reduced responses to influenza or tetanus vaccination and attrition of the TCR repertoires [134,135]. EBV viremia correlates with CD4 and CD8 T cell-reduced TCR repertoires, and CD8 T cells isolated from PLWH are not activated by lysates of lymphoblastoid cell lines [136]. Remarkably, exhausted T cells located in the lymph nodes seem to be at least one of the main reservoirs of HIV-persistent infection, probably because of the restraint that ICI expression imposes on T-cell activation [137,138], and CMV and EBV-specific CD4 T cells have been documented as a preferred and even exclusive reservoir for persistent HIV infection [139].
There are multiple levels of evidence supporting an HIV-EBV interaction in addition to the appearance of EBV-positive lymphomas. For instance, Kenyan children on ART exhibited a correlation between EBV primary infection and HIV viremia, which supports that EBV influences HIV reactivation [140]. Similarly, detectable EBV load at the initiation of ART or post-ART was associated with positive markers of inflammation, non-AIDS defined malignancies, and other PLWH morbidities, such as cardiovascular disease [141]. EBV load in the peripheral blood correlates with hypergammaglobulinemia and with the presence of LMP1-transcripts in cells, despite ART-suppressive HIV viremia and increased CD4 numbers [142]. HIV viremia also correlates with other systemic findings, such as enhanced EBV load in saliva, semen, and breast milk. A study revealed that EBV load remains detectable despite successful ART, while the CMV levels became undetectable [143]. EBV load also progressively increases with longer periods since HIV diagnosis and with HIV viremia [144,145]. EBV detection in the semen has also been linked to an increased risk of HIV sexual transmission [146,147], and EBV and HIV have been co-detected in the breast milk of the patients [148]. EBV viremia has been correlated with increased frequencies of CCR5pos CD4 T cells that exhibit an enhanced susceptibility for HIV infection in vitro [149]. HIV infection rendering CD4 and CD8 T cells susceptible for EBV infection in vitro has also been documented. Here, EBV infection of HIV-positive T cells resulted in enhanced HIV replication and enhanced syncytia formation [150]. EBV primary infection and reactivation in children with HIV infection have also been associated with increased apoptosis of CD8 T cells [151]. Altogether, these studies support a close and bidirectional EBV and HIV-interacting scenario, which is beyond the archetypal immunosuppression because of the lack of T cells. They also support that the control of EBV would help control the HIV cellular reservoirs, positively impacting the PLWH general well-being, most likely antagonizing the development of not only EBV-positive but also EBV-negative lymphomas.
6. Plasmodium falciparum Resembles HIV in Its Capacity to Poly-Clonally Activate B Cells and Trigger GC Continuous Re-Entry
BL that develops in P. falciparum holoendemic geographic regions (i.e., sub-Saharan Africa and Papua New Guinea) is known as endemic (eBL), and it is close to 100% associated with EBV. Five different species of Plasmodium infect humans, of which P. falciparum causes the most severe forms of malaria and is the only species associated with BL. The tropical regions in America and Asia wherein malaria is mainly caused by P. vivax have a low incidence of BL [152]. Several mechanisms have been proposed to explain the association between EBV and P. falciparum, some of which resemble the EBV-HIV interactions that enhance the risk of lymphoma. For instance, enhanced B cell activation with hypergammaglobulinemia and the continuous re-entry of B cells to the GCR has also been observed [153,154]. Indeed, BL cells exhibit a phenotype similar to GC centroblasts. Furthermore, P. falciparum infection correlates with the enhanced expression of the activation-induced cytidine deaminase (AID) [155,156], the GCR enzyme in charge of the BCR hypersomatic mutagenesis and isotype switch recombination. The analysis of tonsils derived from P. falciparum-infected individuals has shown enhanced GC formation and enhanced EBV infection of GC cells. Moreover, isolated B cells turned on AID expression upon stimulation with lysates derived from P. falciparum-infected erythrocytes [156]. Infection with murine P. chabaudi also revealed enhanced GCR and AID expression [157]. GCR entry and AID expression may be responsible for the myc translocation that characterizes all types of BL [158]. P. falciparum also encodes the erythrocyte membrane protein 1 (PfEMP1), a super-antigen that poly-clonally activates B cells and protects them from apoptosis [159,160,161]. Thus, PfEMP1 also contributes to B cell hyperactivation and re-entry into GCR, similar to the heterologous B cell activation observed with HIV p17 and gp120 (Figure 2). Interestingly, PfEMP1 is not encoded by other species of Plasmodium [162], which potentially explains the geographical restriction of eBL.
The strict association with P. falciparum but not with other Plasmodium species remains puzzling. The severity of disease presentation does not seem the explanation, since individuals with the hemoglobin variant (HbSS homozygous or HbSA heterozygous) responsible for sickle cell anemia are protected against severe malaria but not against eBL [163]. Because of the similarities with HIV lymphomagenesis, it is unclear why P. falciparum and EBV coinfection only associates with eBL, but not with other types of lymphoma. Moreover, there is a lag between malaria and eBL presentation; while most children are already protected from the former at the age of 5 years, eBL often picks up later, depending on the African country [164,165]. Long-lasting parasitemia subsisting at the sub-clinical levels for several years has been proposed/observed [166]. In this regard, a recent study observed that adult patients with sporadic BL from the United Kingdom also had a history of exposure to P. falciparum [167].
A state of relative immunosuppression has been recorded in patients with acute malaria, with decreased cytotoxic activity and decreased IFNγ secretion in response to latent and lytic EBV antigens. T-cell dysfunction correlates with enhanced EBV reactivation/viremia and is heightened after repeated episodes of P. falciparum infection [168,169,170,171,172,173]. Moreover, enhanced frequencies of exhausted PD-1pos CD8 T cells and regulatory T cells have been recorded in eBL patients who relapsed compared with long-term survivors or healthy controls [174]. Defective NK cell activity has also been observed among Kenyan children with eBL, as characterized by the lack of expression of NKp46, NKp30, CD160, and TNFα markers of NK cell competence [175]. These studies demonstrated unpaired responses to antigens from both infectious agents and increased frequencies of defective NK cells correlated with enhanced EBV viremia. It thus seems to be interesting to compare the phenotypic and functional similarities between the T cell and NK cell defective populations induced by persistent P. falciparum infection with the T-cell HIV-induced exhaustion or senescence phenotypes.
7. EBV Interactions with Oral Bacteria May Facilitate Viral Transmission and Promote Periodontal Diseases
EBV-associated lesions often arise at places that have been the site of residence or colonized by other microbes. Although the presence of EBV together with these agents often promotes or aggravates the disease, it is unclear whether they involve some type of direct or indirect interactions. The oropharynx, as the site of continuous release of infective EBV particles, provides plenty of opportunities for EBV to cross paths with resident or transient pathogens. Particularly, the codetection of EBV and oral bacteria have been associated with periodontal disease. EBV has been detected in deep periodontal pockets in great numbers of up to 2.8 × 109 virus/mL [176], and, in cases of refractory diseases, also in the numbers of almost a million copies/mL [177]. Meta-analyses of 50 studies have revealed that while EBV was detected in only 8% of healthy periodontium samples, in chronic and aggressive periodontitis, it increased to 39% and 52%, respectively [178,179]. Miller et al. treated EBV-positive periodontal patients with valacyclovir and noted a reduction of EBV PCR-positive samples from 29 to 18 after only 3 days of treatment [180], while Sunde et al. observed a 60% reduction in the viral load in the periodontal plaque of a patient treated with the same antiviral agent [177]. The fact that valacyclovir was administered in the absence of antibiotics strongly supports that the sole reduction of EBV helps improve the clinical status of the lesion. EBV has been reported to directly infect epithelial cells from diseased gingival tissues via in situ hybridization (ISH) studies, while healthy tissues were negative for EBV [181]. The viral load in periodontal tissues has been correlated with disease severity, and EBV positivity precedes the onset of the disease [182].
Periodontitis is a poli-microbial disease triggered by dysbiosis of the oral biota. Although the mouth harbors distinct habitats comprising one of the most diverse microbial communities of the human body, Aggregatibacter actinomycetemcomitans and Porphyromonas gingivalis are key low-abundance periodontal bacteria that, when over-grown, can bring these ecosystems to a dysbiotic state, particularly the gingiva [183]. An association between EBV and P. gingivalis in juvenile periodontitis has also been suggested [184]. In addition, the odds ratio (OR) of finding A. actinomycetemcomitans in localized juvenile periodontitis increased nine-fold when EBV was co-detected [184]. Another study on juvenile periodontitis reported ORs of 49 and 10.7 of EBV and A. actinomycetemcomitans or P. gingivalis coinfection, respectively [185]. Multiple studies conducted on patients with adult periodontitis around the world have also observed EBV association with P. gingivalis and A. actinomycetemcomitans [186].
Considering the large numbers of EBV copies reported, and the steady shedding of EBV in the saliva [187], EBV interactions with the oral microbiota may represent a mechanism that evolved to facilitate EBV local reactivation and viral transmission to new hosts. Indeed, P. gingivalis and other resident bacteria produce butyric acid, a short-chain fatty acid that is a metabolic end-product of obligate anaerobes. Butyric acid is a potent class I histone deacetylase (HDAC) inhibitor, and the EBV lytic cycle is importantly controlled by HDACs [188,189]. Butyric acid is widely used to induce the EBV lytic cycle under experimental settings. P. gingivalis, Fusobacterium nucleatum, and Porphyromonas endodontalis reactivate EBV-infected B cells through induction of the BZLF1 expression [190,191]. Moreover, chromatin immunoprecipitation assays have revealed the dissociation of HDAC1, 2, and 7 from the BZLF1 promoter after EBV-infected B cells were treated with conditioned media derived from the cultures of the periodontopathic bacteria [191]. Moreover, high concentrations of butyric acid are present in the periodontal pockets and saliva [192,193]. Butyric acid is also a known potent activator of the HIV lytic cycle [194]. Here, however, contrary to the oral niches, GI mucosal dysfunction has been associated with decreased levels of butyric acid because of the microbiota change from butyrate-producing Firmicutes to Bacteroidetes [195]. In this scenario, intestinal low levels of butyric acid may favor the EBV and HIV latency stages, rather than the reactivation stages, perhaps impacting the synergic lymphomagenic mechanisms of these viruses. It is also worth mentioning that other herpesviruses have also been implicated in the etiology of periodontitis. For instance, mice coinfected with P. gingivalis and CMV exhibited higher mortality rates and lower levels of IFNγ than mice infected only with the bacterium, which supports cooperation in the pathogenic mechanisms [196]. Oral transmission is the most common mechanism for the dissemination of most human herpesviruses. A. actinomycetemcomitans has also been reported to reactivate EBV through the secretion of the bacterial toxin cytolethal distending [197].
8. HPV Is Also a Tumor Virus That Inhabits Oral and Genital Tissues
High-risk HPV16 and HPV18 are the main causes of cervical squamous cell carcinomas (CESC), accounting for nearly 100% of all CESC cases. It has been estimated that only 3.6% of cervical low-grade squamous intra-epithelial lesions (LSIL) progress to a high-grade lesion (HSIL), and approximately half of the progressing lesions eventually regress despite carrying high-risk HPVs [198], indicating additional cooperating factors. EBV has been proposed as one of the cofactors that aggravate HPV infection. The presence of EBV in cervical lesions has been addressed by ISH and immunohistochemistry/immunofluorescence of viral proteins, with some studies supporting a progressive increase in positivity from normal to LSIL to HSIL/CESC [199]. These detection methods have confirmed that EBV positivity arises from the epithelial cells rather than from infiltrating B cells. Some of these data are quite compelling, reporting a positivity of 0–16% in normal or LSIL to 40–80% in advanced lesions [200,201,202,203,204]. Overall, these studies support that EBV increases the risk to develop CESC by almost 4-fold. EBV-HPV coinfection has also been associated with more aggressive tumors [205]. On the contrary, other studies have failed to detect any EBV infection on cervical epithelial cells or it has been detected irrespective of the progressing state of the lesion [206,207,208,209,210,211]. Moreover, arguing that EBV only plays a passenger role in cervical lesions, infection is only present in a small subset of tumor cells, with an estimated load of one viral copy per tumor cell [203]. The reason for these discrepancies is presently unclear. The recent review by Blanco et al. dissects all the studies addressing the EBV and HPV copresence in the cervical tissues and the methods used to determine EBV infection [212].
The aforementioned studies could not conclude the possible EBV and HPV co-influence nor the cooperative mechanisms displayed by both viruses. More mechanistic approaches have demonstrated a decreased EBV-specific cytotoxic immune response in advanced lesions when compared with that in early lesions, relating to EBV-induced immunosuppressive mechanisms [213] (Figure 3). The EBV EBNA2 expression has been detected in advanced coinfected samples, which is suggestive of a viral latency III program and also of an immunosuppressive environment [203,204]. In addition, the more active the cytotoxic activity against EBV, the better the survival chance of the CESC patient, regardless of the status of HPV infection [213]. A different line of evidence associates the presence of EBV with a 5–7-fold increased probability of HPV18 and HPV16 integration, respectively [214,215]. Moreover, increased methylation of tumor suppressor gene promoters has been indicated in EBV-HPV coinfected specimens [216]. Unfortunately, in these studies [214,215,216], the detection of EBV was performed by PCR, which did not distinguish it from epithelial or B cell infection.
HPV has also been associated with approximately 35% of the head and neck squamous cell carcinomas (HNSC) [217]. Nasopharyngeal carcinoma (NPC) is a subtype of HNSC that has been associated with about 100% of EBV infection, particularly the non-keratinizing subtype. HPV has been codetected in 10–47% of NPC from the endemic regions, which are EBV-positive cases [218,219,220,221]. On the contrary, in the study by Lin et al., no HPV-positive NPC was detected in an endemic region in China [222]. In agreement, NPC from the non-endemic regions was either EBV or HPV positive, but not double-positive [222,223,224,225,226]. Another ISH-based study supported EBV-HPV coinfection in 25% and 20% of tonsillar and base of the tongue oral squamous tumors, respectively [227]. High-risk HPV16 and HPV18 have been reported as the most common HPVs, which are more often associated with EBV-coinfection in HNSC.
A common requirement for HPV and EBV persistency in epithelial cells is an undifferentiated state, switching to lytic replication once the cell assumes a differentiation pathway [25,228,229]. EBV LMP2A delays the nasopharyngeal epithelial cell differentiation [230], and HPV oncogenes E6 and E7 do the same in tonsillar organotypic raft cultures, immortalized oral keratinocytes, and primary human foreskin keratinocytes [231,232]. A permissive environment for EBV persistence may be created by HPV-related delayed activity of KLF4 and BLIMP1 [231], two of the central transcription factors required for the EBV lytic cycle in differentiating epithelial cells and B cells [233] (Figure 3). A similar program of EBV gene expression has been reported in HPV-infected CESC and oral carcinomas, with positivity for LMP1, EBNA1, BARF1, and EBNA2 [199,234]. LMP1 and E6 double recombinant expression in murine embryonic fibroblasts associated with oncogenic events, such as enhanced NFκB signaling, increased proliferation and resistance to apoptosis in vitro, and tumorigenesis in nude mice [235] (Figure 3). EBV and HPV coinfected primary oral keratinocytes also demonstrated an increased degree of invasiveness [227].
9. Helicobacter pylori (Hp), Inflammation, and EBV in the Transformation of Gastric Epithelial Cells
Among the organs in which EBV intersects with other pathogens, perhaps one of the most surprising ones is the stomach. Unlike the oropharynx, the stomach lacks secondary lymphoid tissues to support viral latency and may seem like a dead-end for EBV-productive replication. Nevertheless, EBV is responsible for nearly 10% of all gastric cancers (the EBVaGC), and the EBVaGC outnumbers any other EBV-associated cancer. Gastric cancer is a neoplasm with a clear inflammatory etiology. Hp, a stomach resident bacterium, has been considered as the main trigger factor of the inflammatory process.
Direct and indirect EBV-Hp interactions have been documented (see below). However, Hp has been associated mainly with cancers of the distal portions of the stomach region, while EBV has been more associated with cancers of the proximal regions [236], suggesting that both pathogens do not often coexist at the stomach locoregional level. However, an ongoing inflammatory process, most likely induced by Hp, may be responsible for the chemoattraction and reactivation of EBV-infected B cells, which explains the localization of EBV in the gastric mucosa. Evidence of inflammation favoring EBVaGC has been proposed from the tumors arising at the surgical site of partial gastrectomy or gastroenterostomy, the so-called stump or remnant gastric carcinomas, which are up to 35% EBV-positive [236]. Similar to the EBV and HPV interactions, Hp-induced mutagenesis may also shape the cellular genetic background that licenses EBV persistence in the epithelial cells.
Several studies have assessed the presence of both pathogens in gastric lesions [237]. PCR-based studies have observed a significant association between coinfection and more advanced inflammatory lesions [238,239,240,241,242] and between Hp infection and increased EBV load-supporting Hp-induced viral reactivation [240,242]. ISH has often failed to confirm such an association in the pre-neoplastic lesions [243]. Hp-positive EBVaGC exhibits an increased risk for distant metastasis compared with only EBV-infected neoplasms [244]. We reported increased anti-EBV antibody titers correlating with increased anti-Hp titers and with severe inflammation in the pre-neoplastic and neoplastic lesions [245,246]. We made the same observations in pediatric samples, wherein only children seropositive to both agents were associated with severe gastritis [247].
Monochloromine is a membrane-permeating oxidant produced by neutrophils in response to Hp gastric colonization. In vitro experiments have demonstrated that monochloramine reactivates EBV from latently infected B cells and epithelial cell lines [248]. In another in vitro model, CagA could increase EBV permissive infection of a gastric epithelial cell line, although the infection seemed lytic rather than latent [249]. CagA is a Hp virulence factor and a bacterial oncogene that is transduced into the epithelial cells, perturbing important oncogenic signaling pathways [250]. The CagA signaling activity is importantly driven by phosphorylation, and, Saju et al. reported that SHP1 phosphatase counteracted CagA activity [251]. EBV epigenetically silenced the SHP1 promoter enhancing the pro-oncogenic activity of CagA. Moreover, SHP1 promoter hypermethylation and SHP1 downregulation have been confirmed in EBVaGC. EBV and Hp coinfected cells have been reported to show an enhanced expression of DNA methyl transferases (DNMT) and hypermethylation of tumor suppressor gene promoters, leading to a decreased expression of key regulatory enzymes participating in the cell cycle, apoptosis, and DNA damage repair [249] (Figure 3). CagA can also be released in bacterial-like exosomes, widening the area of CagA influence [252]. To date, most studies addressing a potential synergy between EBV and Hp have been restricted to EBVaGC, mostly exploring how Hp influences EBV. Whether EBV is also an important trigger of inflammation and gastric mucosa damage has only been indirectly explored [245,246].
10. Conclusions
Although most people live as EBV carriers without showing any clinical manifestation, EBV has the potential to trigger life-threatening conditions. This situation is indicative of how well EBV has adapted to humans as well as illustrative of its pathobiont capacity. Beyond immunosuppression, it remains unclear why healthy people also develop disorders associated with this virus. Increasing lines of evidence support that EBV crosses paths with HIV, P. falciparum, and KSHV pathogenic mechanisms, which enhances the risk of developing lymphomas. These agents directly or indirectly stimulate B cells, which in turn activates the GCR, a highly mutagenic process that increases the risk of lymphomagenesis. Lymphomagenesis may also be aided by mechanisms undermining cancer immunosurveillance, such as T cell exhaustion and senescence. Bacterial products may attract EBV-infected B cells and promote its reactivation in tissues, such as butyric acid by oral bacteria and monochloramine by Hp. This mechanism may have evolved in the oral cavity to facilitate the transmission of EBV to new hosts. HPV and KSHV share the feature of effective modulation of epithelial and B cell terminal differentiation. It is believed that HPV- and KSHV-induced cell arrest in less mature stages may provide more suitable conditions for EBV latent infection. Likewise, Hp-induced mutagenesis may provide the cellular genetic background for EBV persistence in gastric cells.
Several other interactions between EBV and infectious agents have been proposed, although studies that rely only on the copresence of different agents do not necessarily explain the etiopathogenesis of the resultant diseases. Furthermore, these observations are often made through PCR-based techniques, which do not inform about the nature of an EBV-infected cell or the number of infected cells. EBV persists in B cells, and these cells continuously patrol the tissues and organs; hence, positivity does not indubitably mean causality. Even in studies in which the presence of EBV appears to worsen the disease, it is not easy to distinguish whether EBV is causal or an innocent bystander. Gastric cancer exemplifies this ambiguity. Although EBV and Hp cause gastric cancer, it remains unclear whether the emergence of this neoplasm is potentiated after some type of interaction between them or whether both agents transform the gastric epithelial cell through independent mechanisms. Similarly, in transplanted patients, the codetection of EBV and betaherpesviruses has often been correlated with enhanced tissue damage, graft rejection, and PTLD. However, there is still a long way to go before establishing synergies in the infectious cycles of these viruses. The more thoroughly we document the intersections of EBV with other pathogens, the better we would be able to understand the circumstances that lead to its pathogenicity. Unveiling the mechanism(s) responsible for uncontrolled EBV infection is expected to provide the points of prophylactic or therapeutic interventions.
Author Contributions
Conceptualization, E.M.F.-P.; writing—original draft preparation, E.M.F.-P. and Y.S.-P.; visualization, E.M.F.-P. and Y.S.-P.; supervision, E.M.F.-P. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
Y. Sánchez-Ponce is a doctoral student from the Programa de Doctorado en Ciencias Biomédicas, Universidad Nacional Autónoma de México (UNAM) and received fellowship CVU 704288 from the National Council of Science and Technology (CONACyT). We greatly acknowledge to David Sosa Martínez, [email protected], for his support in the digital illustration. We also express our gratitude to the Dirección de Investigación of Hospital Infantil de México for its constant support.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| AIDS | Acquired immunodeficiency syndrome |
| ART | antiretroviral therapy |
| BL | Burkitt lymphoma |
| CESC | Cervical squamous cell carcinomas. |
| cHL | Classical Hodgkin lymphoma |
| CMV | Citomegalovirus |
| DLBCL | Diffuse large B cell lymphomas |
| EBV | Epstein–Barr Virus |
| GCR | Germinal center reaction |
| GI | Gastrointestinal |
| GLPD | Germinotropic lymphoproliferative disorders |
| HHV6A | Human herpesvirus 6A |
| HHV6B | Human herpesvirus 6B |
| HHV7 | Human herpesvirus 7 |
| HIV | Human immunodeficiency virus |
| HNSC | Head and neck squamous cell carcinomas |
| HSIL | High-grade squamous intra-epithelial lesion |
| Hp | Helicobacter pylori |
| HPV | Human papillomavirus |
| IARC | International Agency for Research on Cancer |
| ICGC | International Cancer Genome Consortium |
| ICI | Immune checkpoint inhibitors |
| ISH | In situ hybridization |
| KSHV | Kaposi sarcoma virus |
| LPD | Lymphoproliferative disorders |
| LSIL | Low-grade squamous intra-epithelial lesions |
| NPC | Nasopharyngeal carcinoma |
| OR | Odds ratio |
| PCAWG | Pan-Cancer Analysis of Whole Genomes |
| PEL | Primary effusion lymphoma |
| PLWH | People living with HIV |
| PTLD | Post-transplant lymphoproliferative disease |
| TCGA | The Cancer Genome Atlas |
References
- Cruz-Munoz, M.E.; Fuentes-Panana, E.M. Beta and Gamma Human Herpesviruses: Agonistic and Antagonistic Interactions with the Host Immune System. Front. Microbiol. 2017, 8, 2521. [Google Scholar] [CrossRef]
- Thorley-Lawson, D.A. EBV Persistence—Introducing the Virus. Curr. Top. Microbiol. Immunol. 2015, 390, 151–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Babcock, G.J.; Decker, L.L.; Volk, M.; Thorley-Lawson, D.A. EBV persistence in memory B cells in vivo. Immunity 1998, 9, 395–404. [Google Scholar] [CrossRef] [Green Version]
- Tangye, S.G.; Palendira, U.; Edwards, E.S. Human immunity against EBV-lessons from the clinic. J. Exp. Med. 2017, 214, 269–283. [Google Scholar] [CrossRef] [PubMed]
- Narkhede, M.; Arora, S.; Ujjani, C. Primary effusion lymphoma: Current perspectives. OncoTargets Ther. 2018, 11, 3747–3754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jenner, R.G.; Maillard, K.; Cattini, N.; Weiss, R.A.; Boshoff, C.; Wooster, R.; Kellam, P. Kaposi’s sarcoma-associated herpesvirus-infected primary effusion lymphoma has a plasma cell gene expression profile. Proc. Natl. Acad. Sci. USA 2003, 100, 10399–10404. [Google Scholar] [CrossRef] [Green Version]
- Hamoudi, R.; Diss, T.C.; Oksenhendler, E.; Pan, L.; Carbone, A.; Ascoli, V.; Boshoff, C.; Isaacson, P.; Du, M.Q. Distinct cellular origins of primary effusion lymphoma with and without EBV infection. Leuk. Res. 2004, 28, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Roy, D.; Sin, S.H.; Damania, B.; Dittmer, D.P. Tumor suppressor genes FHIT and WWOX are deleted in primary effusion lymphoma (PEL) cell lines. Blood 2011, 118, e32–e39. [Google Scholar] [CrossRef] [Green Version]
- Chadburn, A.; Said, J.; Gratzinger, D.; Chan, J.K.; de Jong, D.; Jaffe, E.S.; Natkunam, Y.; Goodlad, J.R. HHV8/KSHV-Positive Lymphoproliferative Disorders and the Spectrum of Plasmablastic and Plasma Cell Neoplasms: 2015 SH/EAHP Workshop Report-Part 3. Am. J. Clin. Pathol. 2017, 147, 171–187. [Google Scholar] [CrossRef] [Green Version]
- Mesri, E.A.; Cesarman, E.; Arvanitakis, L.; Rafii, S.; Moore, M.A.; Posnett, D.N.; Knowles, D.M.; Asch, A.S. Human herpesvirus-8/Kaposi’s sarcoma-associated herpesvirus is a new transmissible virus that infects B cells. J. Exp. Med. 1996, 183, 2385–2390. [Google Scholar] [CrossRef]
- Faure, A.; Hayes, M.; Sugden, B. How Kaposi’s sarcoma-associated herpesvirus stably transforms peripheral B cells towards lymphomagenesis. Proc. Natl. Acad. Sci. USA 2019, 116, 16519–16528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bigi, R.; Landis, J.T.; An, H.; Caro-Vegas, C.; Raab-Traub, N.; Dittmer, D.P. Epstein-Barr virus enhances genome maintenance of Kaposi sarcoma-associated herpesvirus. Proc. Natl. Acad. Sci. USA 2018, 115, E11379–E11387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McHugh, D.; Caduff, N.; Barros, M.H.M.; Ramer, P.C.; Raykova, A.; Murer, A.; Landtwing, V.; Quast, I.; Styles, C.T.; Spohn, M.; et al. Persistent KSHV Infection Increases EBV-Associated Tumor Formation In Vivo via Enhanced EBV Lytic Gene Expression. Cell Host Microbe 2017, 22, 61–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szekely, L.; Chen, F.; Teramoto, N.; Ehlin-Henriksson, B.; Pokrovskaja, K.; Szeles, A.; Manneborg-Sandlund, A.; Lowbeer, M.; Lennette, E.T.; Klein, G. Restricted expression of Epstein-Barr virus (EBV)-encoded, growth transformation-associated antigens in an EBV- and human herpesvirus type 8-carrying body cavity lymphoma line. J. Gen. Virol. 1998, 79, 1445–1452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krithivas, A.; Young, D.B.; Liao, G.; Greene, D.; Hayward, S.D. Human herpesvirus 8 LANA interacts with proteins of the mSin3 corepressor complex and negatively regulates Epstein-Barr virus gene expression in dually infected PEL cells. J. Virol. 2000, 74, 9637–9645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, G.; Heston, L.; Grogan, E.; Gradoville, L.; Rigsby, M.; Sun, R.; Shedd, D.; Kushnaryov, V.M.; Grossberg, S.; Chang, Y. Selective switch between latency and lytic replication of Kaposi’s sarcoma herpesvirus and Epstein-Barr virus in dually infected body cavity lymphoma cells. J. Virol. 1997, 71, 314–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groves, A.K.; Cotter, M.A.; Subramanian, C.; Robertson, E.S. The latency-associated nuclear antigen encoded by Kaposi’s sarcoma-associated herpesvirus activates two major essential Epstein-Barr virus latent promoters. J. Virol. 2001, 75, 9446–9457. [Google Scholar] [CrossRef] [Green Version]
- Xu, D.; Coleman, T.; Zhang, J.; Fagot, A.; Kotalik, C.; Zhao, L.; Trivedi, P.; Jones, C.; Zhang, L. Epstein-Barr virus inhibits Kaposi’s sarcoma-associated herpesvirus lytic replication in primary effusion lymphomas. J. Virol. 2007, 81, 6068–6078. [Google Scholar] [CrossRef] [Green Version]
- Spadavecchia, S.; Gonzalez-Lopez, O.; Carroll, K.D.; Palmeri, D.; Lukac, D.M. Convergence of Kaposi’s sarcoma-associated herpesvirus reactivation with Epstein-Barr virus latency and cellular growth mediated by the notch signaling pathway in coinfected cells. J. Virol. 2010, 84, 10488–10500. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.; Xu, D.; Zhao, Y.; Zhang, L. Mutual inhibition between Kaposi’s sarcoma-associated herpesvirus and Epstein-Barr virus lytic replication initiators in dually-infected primary effusion lymphoma. PLoS ONE 2008, 3, e1569. [Google Scholar] [CrossRef]
- Horenstein, M.G.; Nador, R.G.; Chadburn, A.; Hyjek, E.M.; Inghirami, G.; Knowles, D.M.; Cesarman, E. Epstein-Barr virus latent gene expression in primary effusion lymphomas containing Kaposi’s sarcoma-associated herpesvirus/human herpesvirus-8. Blood 1997, 90, 1186–1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Callahan, J.; Pai, S.; Cotter, M.; Robertson, E.S. Distinct patterns of viral antigen expression in Epstein-Barr virus and Kaposi’s sarcoma-associated herpesvirus coinfected body-cavity-based lymphoma cell lines: Potential switches in latent gene expression due to coinfection. Virology 1999, 262, 18–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, U.Y.; Park, A.; Jung, J.U. Double the Trouble When Herpesviruses Join Hands. Cell Host Microbe 2017, 22, 5–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riva, G.; Lagreca, I.; Mattiolo, A.; Belletti, D.; Lignitto, L.; Barozzi, P.; Ruozi, B.; Vallerini, D.; Quadrelli, C.; Corradini, G.; et al. Antineoplastic effects of liposomal short interfering RNA treatment targeting BLIMP1/PRDM1 in primary effusion lymphoma. Haematologica 2015, 100, e467–e470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reusch, J.A.; Nawandar, D.M.; Wright, K.L.; Kenney, S.C.; Mertz, J.E. Cellular differentiation regulator BLIMP1 induces Epstein-Barr virus lytic reactivation in epithelial and B cells by activating transcription from both the R and Z promoters. J. Virol. 2015, 89, 1731–1743. [Google Scholar] [CrossRef] [Green Version]
- Shapiro-Shelef, M.; Lin, K.I.; McHeyzer-Williams, L.J.; Liao, J.; McHeyzer-Williams, M.G.; Calame, K. Blimp-1 is required for the formation of immunoglobulin secreting plasma cells and pre-plasma memory B cells. Immunity 2003, 19, 607–620. [Google Scholar] [CrossRef] [Green Version]
- Trivedi, P.; Takazawa, K.; Zompetta, C.; Cuomo, L.; Anastasiadou, E.; Carbone, A.; Uccini, S.; Belardelli, F.; Takada, K.; Frati, L.; et al. Infection of HHV-8+ primary effusion lymphoma cells with a recombinant Epstein-Barr virus leads to restricted EBV latency, altered phenotype, and increased tumorigenicity without affecting TCL1 expression. Blood 2004, 103, 313–316. [Google Scholar] [CrossRef] [Green Version]
- Mack, A.A.; Sugden, B. EBV is necessary for proliferation of dually infected primary effusion lymphoma cells. Cancer Res. 2008, 68, 6963–6968. [Google Scholar] [CrossRef] [Green Version]
- Dittmer, D.P.; Damania, B. Kaposi sarcoma-associated herpesvirus: Immunobiology, oncogenesis, and therapy. J. Clin. Investig. 2016, 126, 3165–3175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, Y.C.; Nakamura, H.; Liang, X.; Feng, P.; Chang, H.; Kowalik, T.F.; Jung, J.U. Inhibition of the ATM/p53 signal transduction pathway by Kaposi’s sarcoma-associated herpesvirus interferon regulatory factor 1. J. Virol. 2006, 80, 2257–2266. [Google Scholar] [CrossRef] [Green Version]
- Seo, T.; Park, J.; Choe, J. Kaposi’s sarcoma-associated herpesvirus viral IFN regulatory factor 1 inhibits transforming growth factor-beta signaling. Cancer Res. 2005, 65, 1738–1747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.J.; Kim, Y.M.; Lim, S.; Nam, Y.K.; Jeong, J.; Kim, H.J.; Lee, K.J. Ubiquitin C-terminal hydrolase-L1 is a key regulator of tumor cell invasion and metastasis. Oncogene 2009, 28, 117–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otsuki, T.; Yata, K.; Takata-Tomokuni, A.; Hyodoh, F.; Miura, Y.; Sakaguchi, H.; Hatayama, T.; Hatada, S.; Tsujioka, T.; Sato, Y.; et al. Expression of protein gene product 9.5 (PGP9.5)/ubiquitin-C-terminal hydrolase 1 (UCHL-1) in human myeloma cells. Br. J. Haematol. 2004, 127, 292–298. [Google Scholar] [CrossRef]
- Ovaa, H.; Kessler, B.M.; Rolen, U.; Galardy, P.J.; Ploegh, H.L.; Masucci, M.G. Activity-based ubiquitin-specific protease (USP) profiling of virus-infected and malignant human cells. Proc. Natl. Acad. Sci. USA 2004, 101, 2253–2258. [Google Scholar] [CrossRef] [Green Version]
- Bentz, G.L.; Bheda-Malge, A.; Wang, L.; Shackelford, J.; Damania, B.; Pagano, J.S. KSHV LANA and EBV LMP1 induce the expression of UCH-L1 following viral transformation. Virology 2014, 448, 293–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gruffat, H.; Manet, E. EBV/KSHV co-infection: An effective partnership. Med. Sci. 2018, 34, 79–82. [Google Scholar] [CrossRef]
- Ma, S.D.; Hegde, S.; Young, K.H.; Sullivan, R.; Rajesh, D.; Zhou, Y.; Jankowska-Gan, E.; Burlingham, W.J.; Sun, X.; Gulley, M.L.; et al. A new model of Epstein-Barr virus infection reveals an important role for early lytic viral protein expression in the development of lymphomas. J. Virol. 2011, 85, 165–177. [Google Scholar] [CrossRef] [Green Version]
- Gloghini, A.; Volpi, C.C.; Gualeni, A.V.; Dolcetti, R.; Bongarzone, I.; De Paoli, P.; Carbone, A. Multiple viral infections in primary effusion lymphoma: A model of viral cooperation in lymphomagenesis. Expert Rev. Hematol. 2017, 10, 505–514. [Google Scholar] [CrossRef]
- Schulz, T.F.; Cesarman, E. Kaposi Sarcoma-associated Herpesvirus: Mechanisms of oncogenesis. Curr. Opin. Virol. 2015, 14, 116–128. [Google Scholar] [CrossRef]
- Pantanowitz, L.; Carbone, A.; Dolcetti, R. Microenvironment and HIV-related lymphomagenesis. Semin. Cancer Biol. 2015, 34, 52–57. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, M.; Osborne, J.; Bestetti, G.; Chang, Y.; Moore, P.S. Viral IL-6-induced cell proliferation and immune evasion of interferon activity. Science 2002, 298, 1432–1435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, K.D.; Aoki, Y.; Chang, Y.; Moore, P.S.; Yarchoan, R.; Tosato, G. Involvement of interleukin-10 (IL-10) and viral IL-6 in the spontaneous growth of Kaposi’s sarcoma herpesvirus-associated infected primary effusion lymphoma cells. Blood 1999, 94, 2871–2879. [Google Scholar] [CrossRef] [PubMed]
- Punj, V.; Matta, H.; Schamus, S.; Yang, T.; Chang, Y.; Chaudhary, P.M. Induction of CCL20 production by Kaposi sarcoma-associated herpesvirus: Role of viral FLICE inhibitory protein K13-induced NF-kappaB activation. Blood 2009, 113, 5660–5668. [Google Scholar] [CrossRef] [Green Version]
- Cinatl, J., Jr.; Vogel, J.U.; Kotchetkov, R.; Wilhelm Doerr, H. Oncomodulatory signals by regulatory proteins encoded by human cytomegalovirus: A novel role for viral infection in tumor progression. FEMS Microbiol. Rev. 2004, 28, 59–77. [Google Scholar] [CrossRef] [Green Version]
- Flamand, L.; Gosselin, J.; Stefanescu, I.; Ablashi, D.; Menezes, J. Immunosuppressive effect of human herpesvirus 6 on T-cell functions: Suppression of interleukin-2 synthesis and cell proliferation. Blood 1995, 85, 1263–1271. [Google Scholar] [CrossRef]
- Ogata, M. Human herpesvirus 6 in hematological malignancies. J. Clin. Exp. Hematop. 2009, 49, 57–67. [Google Scholar] [CrossRef] [Green Version]
- Jenkins, F.J.; Rowe, D.T.; Rinaldo, C.R., Jr. Herpesvirus infections in organ transplant recipients. Clin. Diagn. Lab. Immunol. 2003, 10, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Cohen, J.I. Chapter 142—Human Herpesvirus Types 6 and 7 (Exanthem Subitum). In Mandell, Douglas, and Bennett’s Principles and Practice of Infectious Diseases, 8th ed.; Bennett, J.E., Dolin, R., Blaser, M.J., Eds.; W.B. Saunders: Philadelphia, PA, USA, 2015; pp. 1772–1776.e1. [Google Scholar]
- Mendez, J.C.; Dockrell, D.H.; Espy, M.J.; Smith, T.F.; Wilson, J.A.; Harmsen, W.S.; Ilstrup, D.; Paya, C.V. Human beta-herpesvirus interactions in solid organ transplant recipients. J. Infect. Dis. 2001, 183, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Razonable, R.R.; Humar, A.; Practice, A.S.T.I.D.C.o. Cytomegalovirus in solid organ transplantation. Am. J. Transplant. 2013, 13, 93–106. [Google Scholar] [CrossRef] [PubMed]
- Anderson-Smits, C.; Baker, E.R.; Hirji, I. Coinfection rates and clinical outcome data for cytomegalovirus and Epstein-Barr virus in post-transplant patients: A systematic review of the literature. Transpl. Infect. Dis. 2020, 22, e13396. [Google Scholar] [CrossRef]
- Indolfi, G.; Heaton, N.; Smith, M.; Mieli-Vergani, G.; Zuckerman, M. Effect of early EBV and/or CMV viremia on graft function and acute cellular rejection in pediatric liver transplantation. Clin. Transplant. 2012, 26, E55–E61. [Google Scholar] [CrossRef]
- Ono, Y.; Ito, Y.; Kaneko, K.; Shibata-Watanabe, Y.; Tainaka, T.; Sumida, W.; Nakamura, T.; Kamei, H.; Kiuchi, T.; Ando, H.; et al. Simultaneous monitoring by real-time polymerase chain reaction of epstein-barr virus, human cytomegalovirus, and human herpesvirus-6 in juvenile and adult liver transplant recipients. Transplant. Proc. 2008, 40, 3578–3582. [Google Scholar] [CrossRef]
- Garcia-Cadenas, I.; Castillo, N.; Martino, R.; Barba, P.; Esquirol, A.; Novelli, S.; Orti, G.; Garrido, A.; Saavedra, S.; Moreno, C.; et al. Impact of Epstein Barr virus-related complications after high-risk allo-SCT in the era of pre-emptive rituximab. Bone Marrow Transplant. 2015, 50, 579–584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barani, R.; Ravi, Y.; Seshan, V.; Reju, S.; Soundararajan, P.; Palani, G.; Srikanth, P. Epstein-Barr Virus DNAemia and co-occurrence with cytomegalovirus DNAemia in postrenal transplant recipients from a tertiary care center. Indian J. Transplant. 2018, 12, 95–102. [Google Scholar] [CrossRef]
- Shivanesan, P.; Minz, M.; Minz, R.W.; Kumar, Y.; Sharma, A.; Kanwar, D.B.; Singh, S.; Kohli, H.S.; Anand, S.; Nada, R. Utility of quantitative real time PCR in detection and monitoring of viral infections in post renal transplant recipients. Indian J. Transplant. 2016, 10, 9–14. [Google Scholar] [CrossRef]
- Sanchez-Ponce, Y.; Varela-Fascinetto, G.; Romo-Vazquez, J.C.; Lopez-Martinez, B.; Sanchez-Huerta, J.L.; Parra-Ortega, I.; Fuentes-Panana, E.M.; Morales-Sanchez, A. Simultaneous Detection of Beta and Gamma Human Herpesviruses by Multiplex qPCR Reveals Simple Infection and Coinfection Episodes Increasing Risk for Graft Rejection in Solid Organ Transplantation. Viruses 2018, 10, 730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Mansour, Z.; Nelson, B.P.; Evens, A.M. Post-transplant lymphoproliferative disease (PTLD): Risk factors, diagnosis, and current treatment strategies. Curr. Hematol. Malig. Rep. 2013, 8, 173–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lam, J.K.P.; Azzi, T.; Hui, K.F.; Wong, A.M.G.; McHugh, D.; Caduff, N.; Chan, K.H.; Munz, C.; Chiang, A.K.S. Co-infection of Cytomegalovirus and Epstein-Barr Virus Diminishes the Frequency of CD56(dim)NKG2A(+)KIR(-) NK Cells and Contributes to Suboptimal Control of EBV in Immunosuppressed Children With Post-transplant Lymphoproliferative Disorder. Front. Immunol. 2020, 11, 1231. [Google Scholar] [CrossRef]
- Zallio, F.; Primon, V.; Tamiazzo, S.; Pini, M.; Baraldi, A.; Corsetti, M.T.; Gotta, F.; Bertassello, C.; Salvi, F.; Rocchetti, A.; et al. Epstein-Barr virus reactivation in allogeneic stem cell transplantation is highly related to cytomegalovirus reactivation. Clin. Transplant. 2013, 27, E491–E497. [Google Scholar] [CrossRef]
- Allen, U.D.; Preiksaitis, J.K.; Practice, A.S.T.I.D.C.o. Post-transplant lymphoproliferative disorders, Epstein-Barr virus infection, and disease in solid organ transplantation: Guidelines from the American Society of Transplantation Infectious Diseases Community of Practice. Clin. Transplant. 2019, 33, e13652. [Google Scholar] [CrossRef] [PubMed]
- Kimura, H.; Kwong, Y.L. EBV Viral Loads in Diagnosis, Monitoring, and Response Assessment. Front. Oncol. 2019, 9, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimura, H.; Ito, Y.; Suzuki, R.; Nishiyama, Y. Measuring Epstein-Barr virus (EBV) load: The significance and application for each EBV-associated disease. Rev. Med. Virol. 2008, 18, 305–319. [Google Scholar] [CrossRef]
- Wadowsky, R.M.; Laus, S.; Green, M.; Webber, S.A.; Rowe, D. Measurement of Epstein-Barr virus DNA loads in whole blood and plasma by TaqMan PCR and in peripheral blood lymphocytes by competitive PCR. J. Clin. Microbiol. 2003, 41, 5245–5249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Yang, K.; Wei, C.; Huang, Y.; Zhao, D. Coinfection with EBV/CMV and other respiratory agents in children with suspected infectious mononucleosis. Virol. J. 2010, 7, 247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, Y.; Shibata-Watanabe, Y.; Kawada, J.; Maruyama, K.; Yagasaki, H.; Kojima, S.; Kimura, H. Cytomegalovirus and Epstein-Barr virus coinfection in three toddlers with prolonged illnesses. J. Med. Virol. 2009, 81, 1399–1402. [Google Scholar] [CrossRef]
- Olson, D.; Huntington, M.K. Co-infection with cytomegalovirus and Epstein-Barr virus in mononucleosis: Case report and review of literature. S D Med. 2009, 62, 349, 351–353. [Google Scholar]
- Abate, F.; Ambrosio, M.R.; Mundo, L.; Laginestra, M.A.; Fuligni, F.; Rossi, M.; Zairis, S.; Gazaneo, S.; De Falco, G.; Lazzi, S.; et al. Distinct Viral and Mutational Spectrum of Endemic Burkitt Lymphoma. PLoS Pathog. 2015, 11, e1005158. [Google Scholar] [CrossRef] [Green Version]
- Saghafian-Hedengren, S.; Sundstrom, Y.; Sohlberg, E.; Nilsson, C.; Linde, A.; Troye-Blomberg, M.; Berg, L.; Sverremark-Ekstrom, E. Herpesvirus seropositivity in childhood associates with decreased monocyte-induced NK cell IFN-gamma production. J. Immunol. 2009, 182, 2511–2517. [Google Scholar] [CrossRef]
- Muller-Durovic, B.; Grahlert, J.; Devine, O.P.; Akbar, A.N.; Hess, C. CD56-negative NK cells with impaired effector function expand in CMV and EBV co-infected healthy donors with age. Aging 2019, 11, 724–740. [Google Scholar] [CrossRef]
- Saghafian-Hedengren, S.; Sohlberg, E.; Theorell, J.; Carvalho-Queiroz, C.; Nagy, N.; Persson, J.O.; Nilsson, C.; Bryceson, Y.T.; Sverremark-Ekstrom, E. Epstein-Barr virus coinfection in children boosts cytomegalovirus-induced differentiation of natural killer cells. J. Virol. 2013, 87, 13446–13455. [Google Scholar] [CrossRef] [Green Version]
- Sohlberg, E.; Saghafian-Hedengren, S.; Rasul, E.; Marchini, G.; Nilsson, C.; Klein, E.; Nagy, N.; Sverremark-Ekstrom, E. Cytomegalovirus-seropositive children show inhibition of in vitro EBV infection that is associated with CD8+CD57+ T cell enrichment and IFN-gamma. J. Immunol. 2013, 191, 5669–5676. [Google Scholar] [CrossRef] [Green Version]
- Gosselin, J.; Flamand, L.; D’Addario, M.; Hiscott, J.; Stefanescu, I.; Ablashi, D.V.; Gallo, R.C.; Menezes, J. Modulatory effects of Epstein-Barr, herpes simplex, and human herpes-6 viral infections and coinfections on cytokine synthesis. A comparative study. J. Immunol. 1992, 149, 181–187. [Google Scholar]
- Flamand, L.; Stefanescu, I.; Ablashi, D.V.; Menezes, J. Activation of the Epstein-Barr virus replicative cycle by human herpesvirus 6. J. Virol. 1993, 67, 6768–6777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simard, E.P.; Engels, E.A. Cancer as a cause of death among people with AIDS in the United States. Clin. Infect. Dis. 2010, 51, 957–962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shindiapina, P.; Ahmed, E.H.; Mozhenkova, A.; Abebe, T.; Baiocchi, R.A. Immunology of EBV-Related Lymphoproliferative Disease in HIV-Positive Individuals. Front. Oncol. 2020, 10, 1723. [Google Scholar] [CrossRef]
- Simard, E.P.; Pfeiffer, R.M.; Engels, E.A. Cumulative incidence of cancer among individuals with acquired immunodeficiency syndrome in the United States. Cancer 2011, 117, 1089–1096. [Google Scholar] [CrossRef]
- Carbone, A.; Gloghini, A.; Larocca, L.M.; Antinori, A.; Falini, B.; Tirelli, U.; Dalla-Favera, R.; Gaidano, G. Human immunodeficiency virus-associated Hodgkin’s disease derives from post-germinal center B cells. Blood 1999, 93, 2319–2326. [Google Scholar] [PubMed]
- Gibson, T.M.; Morton, L.M.; Shiels, M.S.; Clarke, C.A.; Engels, E.A. Risk of non-Hodgkin lymphoma subtypes in HIV-infected people during the HAART era: A population-based study. AIDS 2014, 28, 2313–2318. [Google Scholar] [CrossRef] [Green Version]
- Mbulaiteye, S.M.; Pullarkat, S.T.; Nathwani, B.N.; Weiss, L.M.; Rao, N.; Emmanuel, B.; Lynch, C.F.; Hernandez, B.; Neppalli, V.; Hawes, D.; et al. Epstein-Barr virus patterns in US Burkitt lymphoma tumors from the SEER residual tissue repository during 1979-2009. APMIS 2014, 122, 5–15. [Google Scholar] [CrossRef] [Green Version]
- Kersten, M.J.; Van Gorp, J.; Pals, S.T.; Boon, F.; Van Oers, M.H. Expression of Epstein-Barr virus latent genes and adhesion molecules in AIDS-related non-Hodgkin’s lymphomas: Correlation with histology and CD4-cell number. Leuk. Lymphoma 1998, 30, 515–524. [Google Scholar] [CrossRef]
- Linke-Serinsoz, E.; Fend, F.; Quintanilla-Martinez, L. Human immunodeficiency virus (HIV) and Epstein-Barr virus (EBV) related lymphomas, pathology view point. Semin. Diagn. Pathol. 2017, 34, 352–363. [Google Scholar] [CrossRef]
- Carbone, A.; Gloghini, A.; Caruso, A.; De Paoli, P.; Dolcetti, R. The impact of EBV and HIV infection on the microenvironmental niche underlying Hodgkin lymphoma pathogenesis. Int. J. Cancer 2017, 140, 1233–1245. [Google Scholar] [CrossRef]
- Kelly, G.L.; Stylianou, J.; Rasaiyaah, J.; Wei, W.; Thomas, W.; Croom-Carter, D.; Kohler, C.; Spang, R.; Woodman, C.; Kellam, P.; et al. Different patterns of Epstein-Barr virus latency in endemic Burkitt lymphoma (BL) lead to distinct variants within the BL-associated gene expression signature. J. Virol. 2013, 87, 2882–2894. [Google Scholar] [CrossRef] [Green Version]
- Dolcetti, R.; Gloghini, A.; Caruso, A.; Carbone, A. A lymphomagenic role for HIV beyond immune suppression? Blood 2016, 127, 1403–1409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vrzalikova, K.; Woodman, C.B.; Murray, P.G. BLIMP1alpha, the master regulator of plasma cell differentiation is a tumor supressor gene in B cell lymphomas. Biomed. Pap. Med. Fac. Univ. Palacky Olomouc Czech Repub. 2012, 156, 1–6. [Google Scholar] [CrossRef]
- Vrzalikova, K.; Vockerodt, M.; Leonard, S.; Bell, A.; Wei, W.; Schrader, A.; Wright, K.L.; Kube, D.; Rowe, M.; Woodman, C.B.; et al. Down-regulation of BLIMP1alpha by the EBV oncogene, LMP-1, disrupts the plasma cell differentiation program and prevents viral replication in B cells: Implications for the pathogenesis of EBV-associated B-cell lymphomas. Blood 2011, 117, 5907–5917. [Google Scholar] [CrossRef] [PubMed]
- Malaspina, A.; Moir, S.; Kottilil, S.; Hallahan, C.W.; Ehler, L.A.; Liu, S.; Planta, M.A.; Chun, T.W.; Fauci, A.S. Deleterious effect of HIV-1 plasma viremia on B cell costimulatory function. J. Immunol. 2003, 170, 5965–5972. [Google Scholar] [CrossRef] [Green Version]
- Nagase, H.; Agematsu, K.; Kitano, K.; Takamoto, M.; Okubo, Y.; Komiyama, A.; Sugane, K. Mechanism of hypergammaglobulinemia by HIV infection: Circulating memory B-cell reduction with plasmacytosis. Clin. Immunol. 2001, 100, 250–259. [Google Scholar] [CrossRef] [PubMed]
- Curreli, S.; Krishnan, S.; Reitz, M.; Lunardi-Iskandar, Y.; Lafferty, M.K.; Garzino-Demo, A.; Zella, D.; Gallo, R.C.; Bryant, J. B cell lymphoma in HIV transgenic mice. Retrovirology 2013, 10, 92. [Google Scholar] [CrossRef] [Green Version]
- Kundu, R.K.; Sangiorgi, F.; Wu, L.Y.; Pattengale, P.K.; Hinton, D.R.; Gill, P.S.; Maxson, R. Expression of the human immunodeficiency virus-Tat gene in lymphoid tissues of transgenic mice is associated with B-cell lymphoma. Blood 1999, 94, 275–282. [Google Scholar] [CrossRef]
- Popovic, M.; Tenner-Racz, K.; Pelser, C.; Stellbrink, H.J.; van Lunzen, J.; Lewis, G.; Kalyanaraman, V.S.; Gallo, R.C.; Racz, P. Persistence of HIV-1 structural proteins and glycoproteins in lymph nodes of patients under highly active antiretroviral therapy. Proc. Natl. Acad. Sci. USA 2005, 102, 14807–14812. [Google Scholar] [CrossRef] [Green Version]
- Lazzi, S.; Bellan, C.; De Falco, G.; Cinti, C.; Ferrari, F.; Nyongo, A.; Claudio, P.P.; Tosi, G.M.; Vatti, R.; Gloghini, A.; et al. Expression of RB2/p130 tumor-suppressor gene in AIDS-related non-Hodgkin’s lymphomas: Implications for disease pathogenesis. Hum. Pathol. 2002, 33, 723–731. [Google Scholar] [CrossRef] [Green Version]
- Giagulli, C.; Marsico, S.; Magiera, A.K.; Bruno, R.; Caccuri, F.; Barone, I.; Fiorentini, S.; Ando, S.; Caruso, A. Opposite effects of HIV-1 p17 variants on PTEN activation and cell growth in B cells. PLoS ONE 2011, 6, e17831. [Google Scholar] [CrossRef] [PubMed]
- Caccuri, F.; Giagulli, C.; Reichelt, J.; Martorelli, D.; Marsico, S.; Bugatti, A.; Barone, I.; Rusnati, M.; Guzman, C.A.; Dolcetti, R.; et al. Simian immunodeficiency virus and human immunodeficiency virus type 1 matrix proteins specify different capabilities to modulate B cell growth. J. Virol. 2014, 88, 5706–5717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, G.; Tremblay, M.J. HLA-DR, ICAM-1, CD40, CD40L, and CD86 are incorporated to a similar degree into clinical human immunodeficiency virus type 1 variants expanded in natural reservoirs such as peripheral blood mononuclear cells and human lymphoid tissue cultured ex vivo. Clin. Immunol. 2004, 111, 275–285. [Google Scholar] [CrossRef]
- Klein, U.; Dalla-Favera, R. Germinal centres: Role in B-cell physiology and malignancy. Nat. Rev. Immunol. 2008, 8, 22–33. [Google Scholar] [CrossRef] [PubMed]
- Martin, G.; Roy, J.; Barat, C.; Ouellet, M.; Gilbert, C.; Tremblay, M.J. Human immunodeficiency virus type 1-associated CD40 ligand transactivates B lymphocytes and promotes infection of CD4+ T cells. J. Virol. 2007, 81, 5872–5881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imbeault, M.; Ouellet, M.; Giguere, K.; Bertin, J.; Belanger, D.; Martin, G.; Tremblay, M.J. Acquisition of host-derived CD40L by HIV-1 in vivo and its functional consequences in the B-cell compartment. J. Virol. 2011, 85, 2189–2200. [Google Scholar] [CrossRef] [Green Version]
- Fish, K.; Comoglio, F.; Shaffer, A.L., 3rd; Ji, Y.; Pan, K.T.; Scheich, S.; Oellerich, A.; Doebele, C.; Ikeda, M.; Schaller, S.J.; et al. Rewiring of B cell receptor signaling by Epstein-Barr virus LMP2A. Proc. Natl. Acad. Sci. USA 2020, 117, 26318–26327. [Google Scholar] [CrossRef]
- Mancao, C.; Altmann, M.; Jungnickel, B.; Hammerschmidt, W. Rescue of “crippled” germinal center B cells from apoptosis by Epstein-Barr virus. Blood 2005, 106, 4339–4344. [Google Scholar] [CrossRef] [PubMed]
- Martorelli, D.; Muraro, E.; Mastorci, K.; Dal Col, J.; Fae, D.A.; Furlan, C.; Giagulli, C.; Caccuri, F.; Rusnati, M.; Fiorentini, S.; et al. A natural HIV p17 protein variant up-regulates the LMP-1 EBV oncoprotein and promotes the growth of EBV-infected B-lymphocytes: Implications for EBV-driven lymphomagenesis in the HIV setting. Int. J. Cancer 2015, 137, 1374–1385. [Google Scholar] [CrossRef]
- Lam, N.; Sugden, B. CD40 and its viral mimic, LMP1: Similar means to different ends. Cell Signal. 2003, 15, 9–16. [Google Scholar] [CrossRef]
- Wada, N.I.; Jacobson, L.P.; Margolick, J.B.; Breen, E.C.; Macatangay, B.; Penugonda, S.; Martinez-Maza, O.; Bream, J.H. The effect of HAART-induced HIV suppression on circulating markers of inflammation and immune activation. AIDS 2015, 29, 463–471. [Google Scholar] [CrossRef] [Green Version]
- Harker, J.A.; Lewis, G.M.; Mack, L.; Zuniga, E.I. Late interleukin-6 escalates T follicular helper cell responses and controls a chronic viral infection. Science 2011, 334, 825–829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rousset, F.; Garcia, E.; Defrance, T.; Peronne, C.; Vezzio, N.; Hsu, D.H.; Kastelein, R.; Moore, K.W.; Banchereau, J. Interleukin 10 is a potent growth and differentiation factor for activated human B lymphocytes. Proc. Natl. Acad. Sci. USA 1992, 89, 1890–1893. [Google Scholar] [CrossRef] [Green Version]
- Bishop, G.A.; Hostager, B.S. The CD40-CD154 interaction in B cell-T cell liaisons. Cytokine Growth Factor Rev. 2003, 14, 297–309. [Google Scholar] [CrossRef]
- Regidor, D.L.; Detels, R.; Breen, E.C.; Widney, D.P.; Jacobson, L.P.; Palella, F.; Rinaldo, C.R.; Bream, J.H.; Martinez-Maza, O. Effect of highly active antiretroviral therapy on biomarkers of B-lymphocyte activation and inflammation. AIDS 2011, 25, 303–314. [Google Scholar] [CrossRef] [Green Version]
- Borges, A.H.; Silverberg, M.J.; Wentworth, D.; Grulich, A.E.; Fatkenheuer, G.; Mitsuyasu, R.; Tambussi, G.; Sabin, C.A.; Neaton, J.D.; Lundgren, J.D.; et al. Predicting risk of cancer during HIV infection: The role of inflammatory and coagulation biomarkers. AIDS 2013, 27, 1433–1441. [Google Scholar] [CrossRef] [Green Version]
- Vendrame, E.; Hussain, S.K.; Breen, E.C.; Magpantay, L.I.; Widney, D.P.; Jacobson, L.P.; Variakojis, D.; Knowlton, E.R.; Bream, J.H.; Ambinder, R.F.; et al. Serum levels of cytokines and biomarkers for inflammation and immune activation, and HIV-associated non-Hodgkin B-cell lymphoma risk. Cancer Epidemiol. Biomark. Prev. 2014, 23, 343–349. [Google Scholar] [CrossRef] [Green Version]
- Breen, E.C.; Hussain, S.K.; Magpantay, L.; Jacobson, L.P.; Detels, R.; Rabkin, C.S.; Kaslow, R.A.; Variakojis, D.; Bream, J.H.; Rinaldo, C.R.; et al. B-cell stimulatory cytokines and markers of immune activation are elevated several years prior to the diagnosis of systemic AIDS-associated non-Hodgkin B-cell lymphoma. Cancer Epidemiol. Biomark. Prev. 2011, 20, 1303–1314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brenchley, J.M.; Schacker, T.W.; Ruff, L.E.; Price, D.A.; Taylor, J.H.; Beilman, G.J.; Nguyen, P.L.; Khoruts, A.; Larson, M.; Haase, A.T.; et al. CD4+ T cell depletion during all stages of HIV disease occurs predominantly in the gastrointestinal tract. J. Exp. Med. 2004, 200, 749–759. [Google Scholar] [CrossRef] [Green Version]
- Veazey, R.S.; DeMaria, M.; Chalifoux, L.V.; Shvetz, D.E.; Pauley, D.R.; Knight, H.L.; Rosenzweig, M.; Johnson, R.P.; Desrosiers, R.C.; Lackner, A.A. Gastrointestinal tract as a major site of CD4+ T cell depletion and viral replication in SIV infection. Science 1998, 280, 427–431. [Google Scholar] [CrossRef]
- Mehandru, S.; Poles, M.A.; Tenner-Racz, K.; Jean-Pierre, P.; Manuelli, V.; Lopez, P.; Shet, A.; Low, A.; Mohri, H.; Boden, D.; et al. Lack of mucosal immune reconstitution during prolonged treatment of acute and early HIV-1 infection. PLoS Med. 2006, 3, e484. [Google Scholar] [CrossRef]
- Tincati, C.; Biasin, M.; Bandera, A.; Violin, M.; Marchetti, G.; Piacentini, L.; Vago, G.L.; Balotta, C.; Moroni, M.; Franzetti, F.; et al. Early initiation of highly active antiretroviral therapy fails to reverse immunovirological abnormalities in gut-associated lymphoid tissue induced by acute HIV infection. Antivir. Ther. 2009, 14, 321–330. [Google Scholar]
- Nazli, A.; Chan, O.; Dobson-Belaire, W.N.; Ouellet, M.; Tremblay, M.J.; Gray-Owen, S.D.; Arsenault, A.L.; Kaushic, C. Exposure to HIV-1 directly impairs mucosal epithelial barrier integrity allowing microbial translocation. PLoS Pathog. 2010, 6, e1000852. [Google Scholar] [CrossRef]
- Klatt, N.R.; Estes, J.D.; Sun, X.; Ortiz, A.M.; Barber, J.S.; Harris, L.D.; Cervasi, B.; Yokomizo, L.K.; Pan, L.; Vinton, C.L.; et al. Loss of mucosal CD103+ DCs and IL-17+ and IL-22+ lymphocytes is associated with mucosal damage in SIV infection. Mucosal Immunol. 2012, 5, 646–657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryan, E.S.; Micci, L.; Fromentin, R.; Paganini, S.; McGary, C.S.; Easley, K.; Chomont, N.; Paiardini, M. Loss of Function of Intestinal IL-17 and IL-22 Producing Cells Contributes to Inflammation and Viral Persistence in SIV-Infected Rhesus Macaques. PLoS Pathog. 2016, 12, e1005412. [Google Scholar] [CrossRef]
- Kim, C.J.; Nazli, A.; Rojas, O.L.; Chege, D.; Alidina, Z.; Huibner, S.; Mujib, S.; Benko, E.; Kovacs, C.; Shin, L.Y.; et al. A role for mucosal IL-22 production and Th22 cells in HIV-associated mucosal immunopathogenesis. Mucosal Immunol. 2012, 5, 670–680. [Google Scholar] [CrossRef]
- Sandler, N.G.; Douek, D.C. Microbial translocation in HIV infection: Causes, consequences and treatment opportunities. Nat. Rev. Microbiol. 2012, 10, 655–666. [Google Scholar] [CrossRef]
- Mehraj, V.; Ramendra, R.; Isnard, S.; Dupuy, F.P.; Ponte, R.; Chen, J.; Kema, I.; Jenabian, M.A.; Costinuik, C.T.; Lebouche, B.; et al. Circulating (1-->3)-beta-D-glucan Is Associated with Immune Activation During Human Immunodeficiency Virus Infection. Clin. Infect. Dis. 2020, 70, 232–241. [Google Scholar] [CrossRef]
- Clifford, G.M.; Polesel, J.; Rickenbach, M.; Dal Maso, L.; Keiser, O.; Kofler, A.; Rapiti, E.; Levi, F.; Jundt, G.; Fisch, T.; et al. Cancer risk in the Swiss HIV Cohort Study: Associations with immunodeficiency, smoking, and highly active antiretroviral therapy. J. Natl. Cancer Inst. 2005, 97, 425–432. [Google Scholar] [CrossRef] [Green Version]
- Biggar, R.J.; Jaffe, E.S.; Goedert, J.J.; Chaturvedi, A.; Pfeiffer, R.; Engels, E.A. Hodgkin lymphoma and immunodeficiency in persons with HIV/AIDS. Blood 2006, 108, 3786–3791. [Google Scholar] [CrossRef] [PubMed]
- Lindqvist, M.; van Lunzen, J.; Soghoian, D.Z.; Kuhl, B.D.; Ranasinghe, S.; Kranias, G.; Flanders, M.D.; Cutler, S.; Yudanin, N.; Muller, M.I.; et al. Expansion of HIV-specific T follicular helper cells in chronic HIV infection. J. Clin. Investig. 2012, 122, 3271–3280. [Google Scholar] [CrossRef] [PubMed]
- Lorenzo-Redondo, R.; Fryer, H.R.; Bedford, T.; Kim, E.Y.; Archer, J.; Pond, S.L.K.; Chung, Y.S.; Penugonda, S.; Chipman, J.; Fletcher, C.V.; et al. Persistent HIV-1 replication maintains the tissue reservoir during therapy. Nature 2016, 530, 51–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perreau, M.; Savoye, A.L.; De Crignis, E.; Corpataux, J.M.; Cubas, R.; Haddad, E.K.; De Leval, L.; Graziosi, C.; Pantaleo, G. Follicular helper T cells serve as the major CD4 T cell compartment for HIV-1 infection, replication, and production. J. Exp. Med. 2013, 210, 143–156. [Google Scholar] [CrossRef] [PubMed]
- Cubas, R.A.; Mudd, J.C.; Savoye, A.L.; Perreau, M.; van Grevenynghe, J.; Metcalf, T.; Connick, E.; Meditz, A.; Freeman, G.J.; Abesada-Terk, G., Jr.; et al. Inadequate T follicular cell help impairs B cell immunity during HIV infection. Nat. Med. 2013, 19, 494–499. [Google Scholar] [CrossRef] [Green Version]
- De Milito, A.; Morch, C.; Sonnerborg, A.; Chiodi, F. Loss of memory (CD27) B lymphocytes in HIV-1 infection. AIDS 2001, 15, 957–964. [Google Scholar] [CrossRef]
- Van Grevenynghe, J.; Cubas, R.A.; Noto, A.; DaFonseca, S.; He, Z.; Peretz, Y.; Filali-Mouhim, A.; Dupuy, F.P.; Procopio, F.A.; Chomont, N.; et al. Loss of memory B cells during chronic HIV infection is driven by Foxo3a- and TRAIL-mediated apoptosis. J. Clin. Investig. 2011, 121, 3877–3888. [Google Scholar] [CrossRef] [PubMed]
- Fenwick, C.; Joo, V.; Jacquier, P.; Noto, A.; Banga, R.; Perreau, M.; Pantaleo, G. T-cell exhaustion in HIV infection. Immunol. Rev. 2019, 292, 149–163. [Google Scholar] [CrossRef]
- Wherry, E.J.; Kurachi, M. Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 2015, 15, 486–499. [Google Scholar] [CrossRef]
- Deeks, S.G. HIV infection, inflammation, immunosenescence, and aging. Annu. Rev. Med. 2011, 62, 141–155. [Google Scholar] [CrossRef] [Green Version]
- Allen, J.C.; Toapanta, F.R.; Chen, W.; Tennant, S.M. Understanding immunosenescence and its impact on vaccination of older adults. Vaccine 2020, 38, 8264–8272. [Google Scholar] [CrossRef]
- Clerici, M.; Stocks, N.I.; Zajac, R.A.; Boswell, R.N.; Lucey, D.R.; Via, C.S.; Shearer, G.M. Detection of three distinct patterns of T helper cell dysfunction in asymptomatic, human immunodeficiency virus-seropositive patients. Independence of CD4+ cell numbers and clinical staging. J. Clin. Investig. 1989, 84, 1892–1899. [Google Scholar] [CrossRef]
- Heather, J.M.; Best, K.; Oakes, T.; Gray, E.R.; Roe, J.K.; Thomas, N.; Friedman, N.; Noursadeghi, M.; Chain, B. Dynamic Perturbations of the T-Cell Receptor Repertoire in Chronic HIV Infection and following Antiretroviral Therapy. Front. Immunol. 2015, 6, 644. [Google Scholar] [CrossRef] [Green Version]
- Hernandez, D.M.; Valderrama, S.; Gualtero, S.; Hernandez, C.; Lopez, M.; Herrera, M.V.; Solano, J.; Fiorentino, S.; Quijano, S. Loss of T-Cell Multifunctionality and TCR-Vbeta Repertoire Against Epstein-Barr Virus Is Associated With Worse Prognosis and Clinical Parameters in HIV(+) Patients. Front. Immunol. 2018, 9, 2291. [Google Scholar] [CrossRef] [Green Version]
- Fromentin, R.; Bakeman, W.; Lawani, M.B.; Khoury, G.; Hartogensis, W.; DaFonseca, S.; Killian, M.; Epling, L.; Hoh, R.; Sinclair, E.; et al. CD4+ T Cells Expressing PD-1, TIGIT and LAG-3 Contribute to HIV Persistence during ART. PLoS Pathog. 2016, 12, e1005761. [Google Scholar] [CrossRef]
- Evans, V.A.; van der Sluis, R.M.; Solomon, A.; Dantanarayana, A.; McNeil, C.; Garsia, R.; Palmer, S.; Fromentin, R.; Chomont, N.; Sekaly, R.P.; et al. Programmed cell death-1 contributes to the establishment and maintenance of HIV-1 latency. AIDS 2018, 32, 1491–1497. [Google Scholar] [CrossRef]
- Henrich, T.J.; Hobbs, K.S.; Hanhauser, E.; Scully, E.; Hogan, L.E.; Robles, Y.P.; Leadabrand, K.S.; Marty, F.M.; Palmer, C.D.; Jost, S.; et al. Human Immunodeficiency Virus Type 1 Persistence Following Systemic Chemotherapy for Malignancy. J. Infect. Dis. 2017, 216, 254–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slyker, J.A.; Guthrie, B.; Pankau, M.; Tapia, K.; Wamalwa, D.; Benki-Nugent, S.; Ngugi, E.; Huang, M.L.; Njuguna, I.; Langat, A.; et al. Cytomegalovirus and Epstein-Barr virus viremia are associated with HIV DNA levels in the reservoir of Kenyan infants on antiretroviral therapy. J. Infect. Dis. 2020. [Google Scholar] [CrossRef] [PubMed]
- Gianella, S.; Moser, C.; Vitomirov, A.; McKhann, A.; Layman, L.; Scott, B.; Caballero, G.; Lada, S.; Bosch, R.J.; Hoenigl, M.; et al. Presence of asymptomatic cytomegalovirus and Epstein-Barr virus DNA in blood of persons with HIV starting antiretroviral therapy is associated with non-AIDS clinical events. AIDS 2020, 34, 849–857. [Google Scholar] [CrossRef] [PubMed]
- Righetti, E.; Ballon, G.; Ometto, L.; Cattelan, A.M.; Menin, C.; Zanchetta, M.; Chieco-Bianchi, L.; De Rossi, A. Dynamics of Epstein-Barr virus in HIV-1-infected subjects on highly active antiretroviral therapy. AIDS 2002, 16, 63–73. [Google Scholar] [CrossRef]
- Basso, M.; Andreis, S.; Scaggiante, R.; Franchin, E.; Zago, D.; Biasolo, M.A.; Del Vecchio, C.; Mengoli, C.; Sarmati, L.; Andreoni, M.; et al. Cytomegalovirus, Epstein-Barr virus and human herpesvirus 8 salivary shedding in HIV positive men who have sex with men with controlled and uncontrolled plasma HIV viremia: A 24-month longitudinal study. BMC Infect. Dis. 2018, 18, 683. [Google Scholar] [CrossRef] [PubMed]
- Giron, L.B.; Ramos da Silva, S.; Barbosa, A.N.; Monteiro de Barros Almeida, R.A.; Rosario de Souza, L.; Elgui de Oliveira, D. Impact of Epstein-Barr virus load, virus genotype, and frequency of the 30 bp deletion in the viral BNLF-1 gene in patients harboring the human immunodeficiency virus. J. Med. Virol. 2013, 85, 2110–2118. [Google Scholar] [CrossRef] [PubMed]
- De Franca, T.R.; de Albuquerque Tavares Carvalho, A.; Gomes, V.B.; Gueiros, L.A.; Porter, S.R.; Leao, J.C. Salivary shedding of Epstein-Barr virus and cytomegalovirus in people infected or not by human immunodeficiency virus 1. Clin. Oral Investig. 2012, 16, 659–664. [Google Scholar] [CrossRef]
- Lisco, A.; Munawwar, A.; Introini, A.; Vanpouille, C.; Saba, E.; Feng, X.; Grivel, J.C.; Singh, S.; Margolis, L. Semen of HIV-1-infected individuals: Local shedding of herpesviruses and reprogrammed cytokine network. J. Infect. Dis. 2012, 205, 97–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gianella, S.; Morris, S.R.; Vargas, M.V.; Young, J.A.; Callahan, B.; Richman, D.D.; Little, S.J.; Smith, D.M. Role of seminal shedding of herpesviruses in HIV Type 1 Transmission. J. Infect. Dis. 2013, 207, 257–261. [Google Scholar] [CrossRef]
- Gantt, S.; Carlsson, J.; Shetty, A.K.; Seidel, K.D.; Qin, X.; Mutsvangwa, J.; Musingwini, G.; Woelk, G.; Zijenah, L.S.; Katzenstein, D.A.; et al. Cytomegalovirus and Epstein-Barr virus in breast milk are associated with HIV-1 shedding but not with mastitis. AIDS 2008, 22, 1453–1460. [Google Scholar] [CrossRef] [Green Version]
- Moriuchi, M.; Moriuchi, H. Increased susceptibility to HIV-1 of peripheral blood lymphocytes in acute infection with Epstein-Barr virus. J. Med. Virol. 2003, 71, 343–346. [Google Scholar] [CrossRef] [PubMed]
- Guan, M.; Zhang, R.D.; Wu, B.; Henderson, E.E. Infection of primary CD4+ and CD8+ T lymphocytes by Epstein-Barr virus enhances human immunodeficiency virus expression. J. Virol. 1996, 70, 7341–7346. [Google Scholar] [CrossRef] [Green Version]
- Lauener, R.P.; Huttner, S.; Buisson, M.; Hossle, J.P.; Albisetti, M.; Seigneurin, J.M.; Seger, R.A.; Nadal, D. T-cell death by apoptosis in vertically human immunodeficiency virus-infected children coincides with expansion of CD8+/interleukin-2 receptor-/HLA-DR+ T cells: Sign of a possible role for herpes viruses as cofactors? Blood 1995, 86, 1400–1407. [Google Scholar] [CrossRef]
- Quintana, M.D.P.; Smith-Togobo, C.; Moormann, A.; Hviid, L. Endemic Burkitt lymphoma—An aggressive childhood cancer linked to Plasmodium falciparum exposure, but not to exposure to other malaria parasites. APMIS 2020, 128, 129–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sebina, I.; Fogg, L.G.; James, K.R.; Soon, M.S.F.; Akter, J.; Thomas, B.S.; Hill, G.R.; Engwerda, C.R.; Haque, A. IL-6 promotes CD4(+) T-cell and B-cell activation during Plasmodium infection. Parasite Immunol. 2017, 39. [Google Scholar] [CrossRef]
- Alves, F.A.; Pelajo-Machado, M.; Totino, P.R.; Souza, M.T.; Goncalves, E.C.; Schneider, M.P.; Muniz, J.A.; Krieger, M.A.; Andrade, M.C.; Daniel-Ribeiro, C.T.; et al. Splenic architecture disruption and parasite-induced splenocyte activation and anergy in Plasmodium falciparum-infected Saimiri sciureus monkeys. Malar. J. 2015, 14, 128. [Google Scholar] [CrossRef] [Green Version]
- Wilmore, J.R.; Asito, A.S.; Wei, C.; Piriou, E.; Sumba, P.O.; Sanz, I.; Rochford, R. AID expression in peripheral blood of children living in a malaria holoendemic region is associated with changes in B cell subsets and Epstein-Barr virus. Int. J. Cancer 2015, 136, 1371–1380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torgbor, C.; Awuah, P.; Deitsch, K.; Kalantari, P.; Duca, K.A.; Thorley-Lawson, D.A. A multifactorial role for P. falciparum malaria in endemic Burkitt’s lymphoma pathogenesis. PLoS Pathog. 2014, 10, e1004170. [Google Scholar] [CrossRef] [Green Version]
- Robbiani, D.F.; Deroubaix, S.; Feldhahn, N.; Oliveira, T.Y.; Callen, E.; Wang, Q.; Jankovic, M.; Silva, I.T.; Rommel, P.C.; Bosque, D.; et al. Plasmodium Infection Promotes Genomic Instability and AID-Dependent B Cell Lymphoma. Cell 2015, 162, 727–737. [Google Scholar] [CrossRef] [Green Version]
- Ramiro, A.R.; Jankovic, M.; Eisenreich, T.; Difilippantonio, S.; Chen-Kiang, S.; Muramatsu, M.; Honjo, T.; Nussenzweig, A.; Nussenzweig, M.C. AID is required for c-myc/IgH chromosome translocations in vivo. Cell 2004, 118, 431–438. [Google Scholar] [CrossRef] [Green Version]
- Donati, D.; Zhang, L.P.; Chene, A.; Chen, Q.; Flick, K.; Nystrom, M.; Wahlgren, M.; Bejarano, M.T. Identification of a polyclonal B-cell activator in Plasmodium falciparum. Infect. Immun. 2004, 72, 5412–5418. [Google Scholar] [CrossRef] [Green Version]
- Donati, D.; Mok, B.; Chene, A.; Xu, H.; Thangarajh, M.; Glas, R.; Chen, Q.; Wahlgren, M.; Bejarano, M.T. Increased B cell survival and preferential activation of the memory compartment by a malaria polyclonal B cell activator. J. Immunol. 2006, 177, 3035–3044. [Google Scholar] [CrossRef]
- Chene, A.; Donati, D.; Guerreiro-Cacais, A.O.; Levitsky, V.; Chen, Q.; Falk, K.I.; Orem, J.; Kironde, F.; Wahlgren, M.; Bejarano, M.T. A molecular link between malaria and Epstein-Barr virus reactivation. PLoS Pathog. 2007, 3, e80. [Google Scholar] [CrossRef] [PubMed]
- Frech, C.; Chen, N. Genome comparison of human and non-human malaria parasites reveals species subset-specific genes potentially linked to human disease. PLoS Comput. Biol. 2011, 7, e1002320. [Google Scholar] [CrossRef]
- Mulama, D.H.; Bailey, J.A.; Foley, J.; Chelimo, K.; Ouma, C.; Jura, W.G.; Otieno, J.; Vulule, J.; Moormann, A.M. Sickle cell trait is not associated with endemic Burkitt lymphoma: An ethnicity and malaria endemicity-matched case-control study suggests factors controlling EBV may serve as a predictive biomarker for this pediatric cancer. Int. J. Cancer 2014, 134, 645–653. [Google Scholar] [CrossRef] [Green Version]
- Hviid, L. Naturally acquired immunity to Plasmodium falciparum malaria in Africa. Acta Trop. 2005, 95, 270–275. [Google Scholar] [CrossRef] [PubMed]
- Burkitt, D.; Wright, D. Geographical and tribal distribution of the African lymphoma in Uganda. Br. Med. J. 1966, 1, 569–573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, I.; Clarke, S.E.; Gosling, R.; Hamainza, B.; Killeen, G.; Magill, A.; O’Meara, W.; Price, R.N.; Riley, E.M. “Asymptomatic” Malaria: A Chronic and Debilitating Infection That Should Be Treated. PLoS Med. 2016, 13, e1001942. [Google Scholar] [CrossRef]
- Karimi, P.; Birmann, B.M.; Anderson, L.A.; McShane, C.M.; Gadalla, S.M.; Sampson, J.N.; Mbulaiteye, S.M. Risk factors for Burkitt lymphoma: A nested case-control study in the UK Clinical Practice Research Datalink. Br. J. Haematol. 2018, 181, 505–514. [Google Scholar] [CrossRef] [PubMed]
- Moormann, A.M.; Chelimo, K.; Sumba, O.P.; Lutzke, M.L.; Ploutz-Snyder, R.; Newton, D.; Kazura, J.; Rochford, R. Exposure to holoendemic malaria results in elevated Epstein-Barr virus loads in children. J. Infect. Dis. 2005, 191, 1233–1238. [Google Scholar] [CrossRef] [Green Version]
- Njie, R.; Bell, A.I.; Jia, H.; Croom-Carter, D.; Chaganti, S.; Hislop, A.D.; Whittle, H.; Rickinson, A.B. The effects of acute malaria on Epstein-Barr virus (EBV) load and EBV-specific T cell immunity in Gambian children. J. Infect. Dis. 2009, 199, 31–38. [Google Scholar] [CrossRef] [Green Version]
- Reynaldi, A.; Schlub, T.E.; Chelimo, K.; Sumba, P.O.; Piriou, E.; Ogolla, S.; Moormann, A.M.; Rochford, R.; Davenport, M.P. Impact of Plasmodium falciparum Coinfection on Longitudinal Epstein-Barr Virus Kinetics in Kenyan Children. J. Infect. Dis. 2016, 213, 985–991. [Google Scholar] [CrossRef] [Green Version]
- Chattopadhyay, P.K.; Chelimo, K.; Embury, P.B.; Mulama, D.H.; Sumba, P.O.; Gostick, E.; Ladell, K.; Brodie, T.M.; Vulule, J.; Roederer, M.; et al. Holoendemic malaria exposure is associated with altered Epstein-Barr virus-specific CD8(+) T-cell differentiation. J. Virol. 2013, 87, 1779–1788. [Google Scholar] [CrossRef] [Green Version]
- Snider, C.J.; Cole, S.R.; Chelimo, K.; Sumba, P.O.; Macdonald, P.D.; John, C.C.; Meshnick, S.R.; Moormann, A.M. Recurrent Plasmodium falciparum malaria infections in Kenyan children diminish T-cell immunity to Epstein Barr virus lytic but not latent antigens. PLoS ONE 2012, 7, e31753. [Google Scholar] [CrossRef]
- Moormann, A.M.; Heller, K.N.; Chelimo, K.; Embury, P.; Ploutz-Snyder, R.; Otieno, J.A.; Oduor, M.; Munz, C.; Rochford, R. Children with endemic Burkitt lymphoma are deficient in EBNA1-specific IFN-gamma T cell responses. Int. J. Cancer 2009, 124, 1721–1726. [Google Scholar] [CrossRef] [Green Version]
- Parsons, E.; Otieno, J.A.; Ong’echa, J.M.; Nixon, C.E.; Vulule, J.; Munz, C.; Stewart, V.A.; Moormann, A.M. Regulatory T Cells in Endemic Burkitt Lymphoma Patients Are Associated with Poor Outcomes: A Prospective, Longitudinal Study. PLoS ONE 2016, 11, e0167841. [Google Scholar] [CrossRef] [Green Version]
- Forconi, C.S.; Cosgrove, C.P.; Saikumar-Lakshmi, P.; Nixon, C.E.; Foley, J.; Ong’echa, J.M.; Otieno, J.A.; Alter, G.; Munz, C.; Moormann, A.M. Poorly cytotoxic terminally differentiated CD56(neg)CD16(pos) NK cells accumulate in Kenyan children with Burkitt lymphomas. Blood Adv. 2018, 2, 1101–1114. [Google Scholar] [CrossRef] [Green Version]
- Kato, A.; Imai, K.; Ochiai, K.; Ogata, Y. Prevalence and quantitative analysis of Epstein-Barr virus DNA and Porphyromonas gingivalis associated with Japanese chronic periodontitis patients. Clin. Oral Investig. 2015, 19, 1605–1610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sunde, P.T.; Olsen, I.; Enersen, M.; Grinde, B. Patient with severe periodontitis and subgingival Epstein-Barr virus treated with antiviral therapy. J. Clin. Virol. 2008, 42, 176–178. [Google Scholar] [CrossRef]
- Slots, J. Periodontal herpesviruses: Prevalence, pathogenicity, systemic risk. Periodontol. 2000 2015, 69, 28–45. [Google Scholar] [CrossRef]
- Slots, J.; Slots, H. Periodontal herpesvirus morbidity and treatment. Periodontol. 2000 2019, 79, 210–220. [Google Scholar] [CrossRef]
- Miller, C.S.; Avdiushko, S.A.; Kryscio, R.J.; Danaher, R.J.; Jacob, R.J. Effect of prophylactic valacyclovir on the presence of human herpesvirus DNA in saliva of healthy individuals after dental treatment. J. Clin. Microbiol. 2005, 43, 2173–2180. [Google Scholar] [CrossRef] [Green Version]
- Kikuchi, K.; Noguchi, Y.; de Rivera, M.W.; Hoshino, M.; Sakashita, H.; Yamada, T.; Inoue, H.; Miyazaki, Y.; Nozaki, T.; Gonzalez-Lopez, B.S.; et al. Detection of Epstein-Barr virus genome and latent infection gene expression in normal epithelia, epithelial dysplasia, and squamous cell carcinoma of the oral cavity. Tumour Biol. 2016, 37, 3389–3404. [Google Scholar] [CrossRef]
- Vincent-Bugnas, S.; Vitale, S.; Mouline, C.C.; Khaali, W.; Charbit, Y.; Mahler, P.; Precheur, I.; Hofman, P.; Maryanski, J.L.; Doglio, A. EBV infection is common in gingival epithelial cells of the periodontium and worsens during chronic periodontitis. PLoS ONE 2013, 8, e80336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imai, K.; Ogata, Y. How Does Epstein-Barr Virus Contribute to Chronic Periodontitis? Int. J. Mol. Sci. 2020, 21, 1940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michalowicz, B.S.; Ronderos, M.; Camara-Silva, R.; Contreras, A.; Slots, J. Human herpesviruses and Porphyromonas gingivalis are associated with juvenile periodontitis. J. Periodontol. 2000, 71, 981–988. [Google Scholar] [CrossRef]
- Elamin, A.; Ali, R.W.; Bakken, V. Putative periodontopathic bacteria and herpes viruses interactions in the subgingival plaque of patients with aggressive periodontitis and healthy controls. Clin. Exp. Dent. Res. 2017, 3, 183–190. [Google Scholar] [CrossRef]
- Chen, C.; Feng, P.; Slots, J. Herpesvirus-bacteria synergistic interaction in periodontitis. Periodontol. 2000 2020, 82, 42–64. [Google Scholar] [CrossRef]
- Hadinoto, V.; Shapiro, M.; Sun, C.C.; Thorley-Lawson, D.A. The dynamics of EBV shedding implicate a central role for epithelial cells in amplifying viral output. PLoS Pathog. 2009, 5, e1000496. [Google Scholar] [CrossRef] [Green Version]
- Countryman, J.K.; Gradoville, L.; Miller, G. Histone hyperacetylation occurs on promoters of lytic cycle regulatory genes in Epstein-Barr virus-infected cell lines which are refractory to disruption of latency by histone deacetylase inhibitors. J. Virol. 2008, 82, 4706–4719. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, S.K.; Perrine, S.P.; Williams, R.M.; Faller, D.V. Histone deacetylase inhibitors are potent inducers of gene expression in latent EBV and sensitize lymphoma cells to nucleoside antiviral agents. Blood 2012, 119, 1008–1017. [Google Scholar] [CrossRef] [Green Version]
- Makino, K.; Takeichi, O.; Imai, K.; Inoue, H.; Hatori, K.; Himi, K.; Saito, I.; Ochiai, K.; Ogiso, B. Porphyromonas endodontalis reactivates latent Epstein-Barr virus. Int. Endod. J. 2018, 51, 1410–1419. [Google Scholar] [CrossRef] [PubMed]
- Imai, K.; Inoue, H.; Tamura, M.; Cueno, M.E.; Inoue, H.; Takeichi, O.; Kusama, K.; Saito, I.; Ochiai, K. The periodontal pathogen Porphyromonas gingivalis induces the Epstein-Barr virus lytic switch transactivator ZEBRA by histone modification. Biochimie 2012, 94, 839–846. [Google Scholar] [CrossRef] [PubMed]
- Niederman, R.; Buyle-Bodin, Y.; Lu, B.Y.; Robinson, P.; Naleway, C. Short-chain carboxylic acid concentration in human gingival crevicular fluid. J. Dent. Res. 1997, 76, 575–579. [Google Scholar] [CrossRef] [PubMed]
- Koike, R.; Nodomi, K.; Watanabe, N.; Ogata, Y.; Takeichi, O.; Takei, M.; Kaneko, T.; Tonogi, M.; Kotani, A.I.; Imai, K. Butyric Acid in Saliva of Chronic Periodontitis Patients Induces Transcription of the EBV Lytic Switch Activator BZLF1: A Pilot Study. In Vivo 2020, 34, 587–594. [Google Scholar] [CrossRef] [Green Version]
- Imai, K.; Yamada, K.; Tamura, M.; Ochiai, K.; Okamoto, T. Reactivation of latent HIV-1 by a wide variety of butyric acid-producing bacteria. Cell. Mol. Life Sci. 2012, 69, 2583–2592. [Google Scholar] [CrossRef] [PubMed]
- Desai, S.N.; Landay, A.L. HIV and aging: Role of the microbiome. Curr. Opin. HIV AIDS 2018, 13, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Stern, J.; Shai, E.; Zaks, B.; Halabi, A.; Houri-Haddad, Y.; Shapira, L.; Palmon, A. Reduced expression of gamma interferon in serum and marked lymphoid depletion induced by Porphyromonas gingivalis increase murine morbidity and mortality due to cytomegalovirus infection. Infect. Immun. 2004, 72, 5791–5798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rush, M.C.; Simon, M.W. Occurrence of Epstein-Barr virus illness in children diagnosed with group A streptococcal pharyngitis. Clin. Pediatr. 2003, 42, 417–420. [Google Scholar] [CrossRef]
- Schlecht, N.F.; Platt, R.W.; Duarte-Franco, E.; Costa, M.C.; Sobrinho, J.P.; Prado, J.C.; Ferenczy, A.; Rohan, T.E.; Villa, L.L.; Franco, E.L. Human papillomavirus infection and time to progression and regression of cervical intraepithelial neoplasia. J. Natl. Cancer Inst. 2003, 95, 1336–1343. [Google Scholar] [CrossRef] [Green Version]
- De Lima, M.A.P.; Neto, P.J.N.; Lima, L.P.M.; Goncalves Junior, J.; Teixeira Junior, A.G.; Teodoro, I.P.P.; Facundo, H.T.; da Silva, C.G.L.; Lima, M.V.A. Association between Epstein-Barr virus (EBV) and cervical carcinoma: A meta-analysis. Gynecol. Oncol. 2018, 148, 317–328. [Google Scholar] [CrossRef]
- Khenchouche, A.; Sadouki, N.; Boudriche, A.; Houali, K.; Graba, A.; Ooka, T.; Bouguermouh, A. Human papillomavirus and Epstein-Barr virus co-infection in cervical carcinoma in Algerian women. Virol. J. 2013, 10, 340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Se Thoe, S.Y.; Wong, K.K.; Pathmanathan, R.; Sam, C.K.; Cheng, H.M.; Prasad, U. Elevated secretory IgA antibodies to Epstein-Barr virus (EBV) and presence of EBV DNA and EBV receptors in patients with cervical carcinoma. Gynecol. Oncol. 1993, 50, 168–172. [Google Scholar] [CrossRef]
- Landers, R.J.; O’Leary, J.J.; Crowley, M.; Healy, I.; Annis, P.; Burke, L.; O’Brien, D.; Hogan, J.; Kealy, W.F.; Lewis, F.A.; et al. Epstein-Barr virus in normal, pre-malignant, and malignant lesions of the uterine cervix. J. Clin. Pathol. 1993, 46, 931–935. [Google Scholar] [CrossRef] [Green Version]
- Sasagawa, T.; Shimakage, M.; Nakamura, M.; Sakaike, J.; Ishikawa, H.; Inoue, M. Epstein-Barr virus (EBV) genes expression in cervical intraepithelial neoplasia and invasive cervical cancer: A comparative study with human papillomavirus (HPV) infection. Hum. Pathol. 2000, 31, 318–326. [Google Scholar] [CrossRef]
- Shimakage, M.; Sasagawa, T. Detection of Epstein-Barr virus-determined nuclear antigen-2 mRNA by in situ hybridization. J. Virol. Methods 2001, 93, 23–32. [Google Scholar] [CrossRef]
- Al-Thawadi, H.; Ghabreau, L.; Aboulkassim, T.; Yasmeen, A.; Vranic, S.; Batist, G.; Al Moustafa, A.E. Co-Incidence of Epstein-Barr Virus and High-Risk Human Papillomaviruses in Cervical Cancer of Syrian Women. Front. Oncol. 2018, 8, 250. [Google Scholar] [CrossRef] [Green Version]
- Aromseree, S.; Pientong, C.; Swangphon, P.; Chaiwongkot, A.; Patarapadungkit, N.; Kleebkaow, P.; Tungsiriwattana, T.; Kongyingyoes, B.; Vendrig, T.; Middeldorp, J.M.; et al. Possible contributing role of Epstein-Barr virus (EBV) as a cofactor in human papillomavirus (HPV)-associated cervical carcinogenesis. J. Clin. Virol. 2015, 73, 70–76. [Google Scholar] [CrossRef]
- Hilton, D.A.; Brown, L.J.; Pringle, J.H.; Nandha, H. Absence of Epstein-Barr virus in carcinoma of the cervix. Cancer 1993, 72, 1946–1948. [Google Scholar] [CrossRef]
- Shoji, Y.; Saegusa, M.; Takano, Y.; Hashimura, M.; Okayasu, I. Detection of the Epstein-Barr virus genome in cervical neoplasia is closely related to the degree of infiltrating lymphoid cells: A polymerase chain reaction and in situ hybridization approach. Pathol. Int. 1997, 47, 507–511. [Google Scholar] [CrossRef]
- Elgui de Oliveira, D.; Furtado Monteiro, T.A.; Alencar de Melo, W.; Amaral Reboucas Moreira, M.; Alvarenga, M.; Bacchi, C.E. Lack of Epstein-Barr virus infection in cervical carcinomas. Arch. Pathol. Lab. Med. 1999, 123, 1098–1100. [Google Scholar] [CrossRef]
- Yang, Y.Y.; Koh, L.W.; Tsai, J.H.; Tsai, C.H.; Wong, E.F.; Lin, S.J.; Yang, C.C. Correlation of viral factors with cervical cancer in Taiwan. J. Microbiol. Immunol. Infect. 2004, 37, 282–287. [Google Scholar]
- Seo, S.S.; Kim, W.H.; Song, Y.S.; Kim, S.H.; Kim, J.W.; Park, N.H.; Kang, S.B.; Lee, H.P. Epstein-Barr virus plays little role in cervical carcinogenesis in Korean women. Int. J. Gynecol. Cancer. 2005, 15, 312–318. [Google Scholar] [CrossRef]
- Blanco, R.; Carrillo-Beltran, D.; Osorio, J.C.; Calaf, G.M.; Aguayo, F. Role of Epstein-Barr Virus and Human Papillomavirus Coinfection in Cervical Cancer: Epidemiology, Mechanisms and Perspectives. Pathogens 2020, 9, 685. [Google Scholar] [CrossRef]
- Kitano, Y.; Fujisaki, S.; Nakamura, N.; Miyazaki, K.; Katsuki, T.; Okamura, H. Immunological disorder against the Epstein-Barr virus infection and prognosis in patients with cervical carcinoma. Gynecol. Oncol. 1995, 57, 150–157. [Google Scholar] [CrossRef]
- Kahla, S.; Oueslati, S.; Achour, M.; Kochbati, L.; Chanoufi, M.B.; Maalej, M.; Oueslati, R. Correlation between ebv co-infection and HPV16 genome integrity in Tunisian cervical cancer patients. Braz. J. Microbiol. 2012, 43, 744–753. [Google Scholar] [CrossRef] [Green Version]
- Szostek, S.; Zawilinska, B.; Kopec, J.; Kosz-Vnenchak, M. Herpesviruses as possible cofactors in HPV-16-related oncogenesis. Acta Biochim. Pol. 2009, 56, 337–342. [Google Scholar] [CrossRef] [Green Version]
- McCormick, T.M.; Canedo, N.H.; Furtado, Y.L.; Silveira, F.A.; de Lima, R.J.; Rosman, A.D.; Almeida Filho, G.L.; Carvalho Mda, G. Association between human papillomavirus and Epstein—Barr virus DNA and gene promoter methylation of RB1 and CDH1 in the cervical lesions: A transversal study. Diagn. Pathol. 2015, 10, 59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Termine, N.; Panzarella, V.; Falaschini, S.; Russo, A.; Matranga, D.; Lo Muzio, L.; Campisi, G. HPV in oral squamous cell carcinoma vs head and neck squamous cell carcinoma biopsies: A meta-analysis (1988–2007). Ann. Oncol. 2008, 19, 1681–1690. [Google Scholar] [CrossRef]
- Staff, P.O. Correction: Epstein-Barr virus and human papillomavirus infections and genotype distribution in head and neck cancers. PLoS ONE 2015, 10, e0118439. [Google Scholar] [CrossRef] [Green Version]
- Laantri, N.; Attaleb, M.; Kandil, M.; Naji, F.; Mouttaki, T.; Dardari, R.; Belghmi, K.; Benchakroun, N.; El Mzibri, M.; Khyatti, M. Human papillomavirus detection in moroccan patients with nasopharyngeal carcinoma. Infect. Agent Cancer 2011, 6, 3. [Google Scholar] [CrossRef] [Green Version]
- Mirzamani, N.; Salehian, P.; Farhadi, M.; Tehran, E.A. Detection of EBV and HPV in nasopharyngeal carcinoma by in situ hybridization. Exp. Mol. Pathol. 2006, 81, 231–234. [Google Scholar] [CrossRef] [PubMed]
- Tung, Y.C.; Lin, K.H.; Chu, P.Y.; Hsu, C.C.; Kuo, W.R. Detection of human papilloma virus and Epstein-Barr virus DNA in nasopharyngeal carcinoma by polymerase chain reaction. Kaohsiung J. Med. Sci. 1999, 15, 256–262. [Google Scholar] [PubMed]
- Lin, Z.; Khong, B.; Kwok, S.; Cao, H.; West, R.B.; Le, Q.T.; Kong, C.S. Human papillomavirus 16 detected in nasopharyngeal carcinomas in white Americans but not in endemic Southern Chinese patients. Head Neck 2014, 36, 709–714. [Google Scholar] [CrossRef]
- Stenmark, M.H.; McHugh, J.B.; Schipper, M.; Walline, H.M.; Komarck, C.; Feng, F.Y.; Worden, F.P.; Wolf, G.T.; Chepeha, D.B.; Prince, M.E.; et al. Nonendemic HPV-positive nasopharyngeal carcinoma: Association with poor prognosis. Int. J. Radiat. Oncol. Biol. Phys. 2014, 88, 580–588. [Google Scholar] [CrossRef] [Green Version]
- Dogan, S.; Hedberg, M.L.; Ferris, R.L.; Rath, T.J.; Assaad, A.M.; Chiosea, S.I. Human papillomavirus and Epstein-Barr virus in nasopharyngeal carcinoma in a low-incidence population. Head Neck 2014, 36, 511–516. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, J.H.; Kumar, B.; Feng, F.Y.; McHugh, J.B.; Cordell, K.G.; Eisbruch, A.; Worden, F.P.; Wolf, G.T.; Prince, M.E.; Moyer, J.S.; et al. HPV-positive/p16-positive/EBV-negative nasopharyngeal carcinoma in white North Americans. Head Neck 2010, 32, 562–567. [Google Scholar] [CrossRef]
- Robinson, M.; Suh, Y.E.; Paleri, V.; Devlin, D.; Ayaz, B.; Pertl, L.; Thavaraj, S. Oncogenic human papillomavirus-associated nasopharyngeal carcinoma: An observational study of correlation with ethnicity, histological subtype and outcome in a UK population. Infect Agent Cancer 2013, 8, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, R.; Ekshyyan, O.; Moore-Medlin, T.; Rong, X.; Nathan, S.; Gu, X.; Abreo, F.; Rosenthal, E.L.; Shi, M.; Guidry, J.T.; et al. Association between human papilloma virus/Epstein-Barr virus coinfection and oral carcinogenesis. J. Oral Pathol. Med. 2015, 44, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Nawandar, D.M.; Ohashi, M.; Djavadian, R.; Barlow, E.; Makielski, K.; Ali, A.; Lee, D.; Lambert, P.F.; Johannsen, E.; Kenney, S.C. Differentiation-Dependent LMP1 Expression Is Required for Efficient Lytic Epstein-Barr Virus Reactivation in Epithelial Cells. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, E.A. Manipulation of Epithelial Differentiation by HPV Oncoproteins. Viruses 2019, 11, 369. [Google Scholar] [CrossRef] [Green Version]
- Scholle, F.; Bendt, K.M.; Raab-Traub, N. Epstein-Barr virus LMP2A transforms epithelial cells, inhibits cell differentiation, and activates Akt. J. Virol. 2000, 74, 10681–10689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guidry, J.T.; Myers, J.E.; Bienkowska-Haba, M.; Songock, W.K.; Ma, X.; Shi, M.; Nathan, C.O.; Bodily, J.M.; Sapp, M.J.; Scott, R.S. Inhibition of Epstein-Barr Virus Replication in Human Papillomavirus-Immortalized Keratinocytes. J. Virol. 2019, 93. [Google Scholar] [CrossRef] [Green Version]
- Makielski, K.R.; Lee, D.; Lorenz, L.D.; Nawandar, D.M.; Chiu, Y.F.; Kenney, S.C.; Lambert, P.F. Human papillomavirus promotes Epstein-Barr virus maintenance and lytic reactivation in immortalized oral keratinocytes. Virology 2016, 495, 52–62. [Google Scholar] [CrossRef] [PubMed]
- Nawandar, D.M.; Wang, A.; Makielski, K.; Lee, D.; Ma, S.; Barlow, E.; Reusch, J.; Jiang, R.; Wille, C.K.; Greenspan, D.; et al. Differentiation-Dependent KLF4 Expression Promotes Lytic Epstein-Barr Virus Infection in Epithelial Cells. PLoS Pathog. 2015, 11, e1005195. [Google Scholar] [CrossRef]
- De Lima, M.A.P.; Teodoro, I.P.P.; Galiza, L.E.; Filho, P.; Marques, F.M.; Pinheiro Junior, R.F.F.; Macedo, G.E.C.; Facundo, H.T.; da Silva, C.G.L.; Lima, M.V.A. Association between Epstein-Barr Virus and Oral Carcinoma: A Systematic Review with Meta-Analysis. Crit. Rev. Oncog. 2019, 24, 349–368. [Google Scholar] [CrossRef] [PubMed]
- Shimabuku, T.; Tamanaha, A.; Kitamura, B.; Tanabe, Y.; Tawata, N.; Ikehara, F.; Arakaki, K.; Kinjo, T. Dual expression of Epstein-Barr virus, latent membrane protein-1 and human papillomavirus-16 E6 transform primary mouse embryonic fibroblasts through NF-kappaB signaling. Int. J. Clin. Exp. Pathol. 2014, 7, 1920–1934. [Google Scholar] [PubMed]
- Murphy, G.; Pfeiffer, R.; Camargo, M.C.; Rabkin, C.S. Meta-analysis shows that prevalence of Epstein-Barr virus-positive gastric cancer differs based on sex and anatomic location. Gastroenterology 2009, 137, 824–833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davila-Collado, R.; Jarquin-Duran, O.; Dong, L.T.; Espinoza, J.L. Epstein-Barr Virus and Helicobacter pylori Co-Infection in Non-Malignant Gastroduodenal Disorders. Pathogens 2020, 9, 104. [Google Scholar] [CrossRef] [Green Version]
- Saxena, A.; Nath Prasad, K.; Chand Ghoshal, U.; Krishnani, N.; Roshan Bhagat, M.; Husain, N. Association of Helicobacter pylori and Epstein-Barr virus with gastric cancer and peptic ulcer disease. Scand. J. Gastroenterol. 2008, 43, 669–674. [Google Scholar] [CrossRef]
- Castaneda, C.A.; Castillo, M.; Chavez, I.; Barreda, F.; Suarez, N.; Nieves, J.; Bernabe, L.A.; Valdivia, D.; Ruiz, E.; Dias-Neto, E.; et al. Prevalence of Helicobacter pylori Infection, Its Virulent Genotypes, and Epstein-Barr Virus in Peruvian Patients with Chronic Gastritis and Gastric Cancer. J. Glob. Oncol. 2019, 5, 1–9. [Google Scholar] [CrossRef]
- Shukla, S.K.; Prasad, K.N.; Tripathi, A.; Ghoshal, U.C.; Krishnani, N.; Husain, N. Expression profile of latent and lytic transcripts of epstein-barr virus in patients with gastroduodenal diseases: A study from northern India. J. Med. Virol. 2012, 84, 1289–1297. [Google Scholar] [CrossRef]
- Del Moral-Hernandez, O.; Castanon-Sanchez, C.A.; Reyes-Navarrete, S.; Martinez-Carrillo, D.N.; Betancourt-Linares, R.; Jimenez-Wences, H.; de la Pena, S.; Roman-Roman, A.; Hernandez-Sotelo, D.; Fernandez-Tilapa, G. Multiple infections by EBV, HCMV and Helicobacter pylori are highly frequent in patients with chronic gastritis and gastric cancer from Southwest Mexico: An observational study. Medicine 2019, 98, e14124. [Google Scholar] [CrossRef]
- Shukla, S.K.; Prasad, K.N.; Tripathi, A.; Singh, A.; Saxena, A.; Ghoshal, U.C.; Krishnani, N.; Husain, N. Epstein-Barr virus DNA load and its association with Helicobacter pylori infection in gastroduodenal diseases. Braz. J. Infect. Dis. 2011, 15, 583–590. [Google Scholar] [CrossRef] [Green Version]
- De Souza, C.R.; de Oliveira, K.S.; Ferraz, J.J.; Leal, M.F.; Calcagno, D.Q.; Seabra, A.D.; Khayat, A.S.; Montenegro, R.C.; Alves, A.P.; Assumpcao, P.P.; et al. Occurrence of Helicobacter pylori and Epstein-Barr virus infection in endoscopic and gastric cancer patients from Northern Brazil. BMC Gastroenterol. 2014, 14, 179. [Google Scholar] [CrossRef] [Green Version]
- De Souza, C.R.T.; Almeida, M.C.A.; Khayat, A.S.; da Silva, E.L.; Soares, P.C.; Chaves, L.C.; Burbano, R.M.R. Association between Helicobacter pylori, Epstein-Barr virus, human papillomavirus and gastric adenocarcinomas. World J. Gastroenterol. 2018, 24, 4928–4938. [Google Scholar] [CrossRef]
- Cardenas-Mondragon, M.G.; Torres, J.; Sanchez-Zauco, N.; Gomez-Delgado, A.; Camorlinga-Ponce, M.; Maldonado-Bernal, C.; Fuentes-Panana, E.M. Elevated Levels of Interferon-gamma Are Associated with High Levels of Epstein-Barr Virus Reactivation in Patients with the Intestinal Type of Gastric Cancer. J. Immunol. Res. 2017, 2017, 7069242. [Google Scholar] [CrossRef] [Green Version]
- Cardenas-Mondragon, M.G.; Torres, J.; Flores-Luna, L.; Camorlinga-Ponce, M.; Carreon-Talavera, R.; Gomez-Delgado, A.; Kasamatsu, E.; Fuentes-Panana, E.M. Case-control study of Epstein-Barr virus and Helicobacter pylori serology in Latin American patients with gastric disease. Br. J. Cancer 2015, 112, 1866–1873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cardenas-Mondragon, M.G.; Carreon-Talavera, R.; Camorlinga-Ponce, M.; Gomez-Delgado, A.; Torres, J.; Fuentes-Panana, E.M. Epstein Barr virus and Helicobacter pylori co-infection are positively associated with severe gastritis in pediatric patients. PLoS ONE 2013, 8, e62850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minoura-Etoh, J.; Gotoh, K.; Sato, R.; Ogata, M.; Kaku, N.; Fujioka, T.; Nishizono, A. Helicobacter pylori-associated oxidant monochloramine induces reactivation of Epstein-Barr virus (EBV) in gastric epithelial cells latently infected with EBV. J. Med. Microbiol. 2006, 55, 905–911. [Google Scholar] [CrossRef] [Green Version]
- Pandey, S.; Jha, H.C.; Shukla, S.K.; Shirley, M.K.; Robertson, E.S. Epigenetic Regulation of Tumor Suppressors by Helicobacter pylori Enhances EBV-Induced Proliferation of Gastric Epithelial Cells. mBio 2018, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ansari, S.; Yamaoka, Y. Helicobacter pylori Virulence Factor Cytotoxin-Associated Gene A (CagA)-Mediated Gastric Pathogenicity. Int. J. Mol. Sci. 2020, 21, 7430. [Google Scholar] [CrossRef] [PubMed]
- Saju, P.; Murata-Kamiya, N.; Hayashi, T.; Senda, Y.; Nagase, L.; Noda, S.; Matsusaka, K.; Funata, S.; Kunita, A.; Urabe, M.; et al. Host SHP1 phosphatase antagonizes Helicobacter pylori CagA and can be downregulated by Epstein-Barr virus. Nat. Microbiol. 2016, 1, 16026. [Google Scholar] [CrossRef]
- Olofsson, A.; Vallstrom, A.; Petzold, K.; Tegtmeyer, N.; Schleucher, J.; Carlsson, S.; Haas, R.; Backert, S.; Wai, S.N.; Grobner, G.; et al. Biochemical and functional characterization of Helicobacter pylori vesicles. Mol. Microbiol. 2010, 77, 1539–1555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
Shared microenvironments between EBV and other infectious agents. EBV infection’s main portal of entry and exit is the oral mucosa, where it coexists with the periodontal bacteria P. gingivalis and A. actinomycetemcomitans, and HPV. EBV persists in B cells residing in the lymphatic system, where it can cross paths with other infectious agents such as HIV, P. falciparum, or KSHV (here represented in the spleen as an example of a secondary lymph node). From the lymphatic system, EBV-infected cells can infiltrate certain tissues, such as the stomach or cervix, where EBV can intersect with H. pylori and HPV, respectively.
Figure 1.
Shared microenvironments between EBV and other infectious agents. EBV infection’s main portal of entry and exit is the oral mucosa, where it coexists with the periodontal bacteria P. gingivalis and A. actinomycetemcomitans, and HPV. EBV persists in B cells residing in the lymphatic system, where it can cross paths with other infectious agents such as HIV, P. falciparum, or KSHV (here represented in the spleen as an example of a secondary lymph node). From the lymphatic system, EBV-infected cells can infiltrate certain tissues, such as the stomach or cervix, where EBV can intersect with H. pylori and HPV, respectively.

Figure 2.
EBV-infected B cells transiting through the germinal center reaction (GCR) cross paths with HIV, P. falciparum, and KSHV. The key convergent mechanisms that potentiate lymphomagenesis are antigen (Ag)-specific and heterologous hyperactivation of B cells, which, upon enhancing the passage through the GCR, increase the likelihood of off-target mutagenesis. Mutated B cells normally die of apoptosis. However, if EBV alone or in conjunction with KSHV resides in these lymphocytes, viral proteins can rescue the cells from dying (illustrated here with LMP1 and vFLIP). In addition, the transformed/mutated B cells should be eliminated by the immune system, but infection-induced exhaustion or senescence causes unpaired immunosurveillance and immunoescape of these cells. The viral products also activate the oncogenic pathways and oncogenic processes and block B cell differentiation into memory cells. Immunosuppression by viral orthologs of cellular cytokines may also contribute to lymphomagenesis. Altogether, these events result in the development of conditions that favor the emergence of lymphomas with a plasmablastic morphology (see text for a more detailed explanation): Burkitt’s lymphoma (BL), Hodgkin’s lymphoma (HL), diffuse large B cell lymphoma (DLBCL), primary effusion lymphoma (PEL), and germinotropic lymphoproliferative disorder (GLPD).
Figure 2.
EBV-infected B cells transiting through the germinal center reaction (GCR) cross paths with HIV, P. falciparum, and KSHV. The key convergent mechanisms that potentiate lymphomagenesis are antigen (Ag)-specific and heterologous hyperactivation of B cells, which, upon enhancing the passage through the GCR, increase the likelihood of off-target mutagenesis. Mutated B cells normally die of apoptosis. However, if EBV alone or in conjunction with KSHV resides in these lymphocytes, viral proteins can rescue the cells from dying (illustrated here with LMP1 and vFLIP). In addition, the transformed/mutated B cells should be eliminated by the immune system, but infection-induced exhaustion or senescence causes unpaired immunosurveillance and immunoescape of these cells. The viral products also activate the oncogenic pathways and oncogenic processes and block B cell differentiation into memory cells. Immunosuppression by viral orthologs of cellular cytokines may also contribute to lymphomagenesis. Altogether, these events result in the development of conditions that favor the emergence of lymphomas with a plasmablastic morphology (see text for a more detailed explanation): Burkitt’s lymphoma (BL), Hodgkin’s lymphoma (HL), diffuse large B cell lymphoma (DLBCL), primary effusion lymphoma (PEL), and germinotropic lymphoproliferative disorder (GLPD).

Figure 3.
Interactions between EBV and other infectious agents in non-lymphatic organs. The arrival and reactivation of EBV-infected B cells in different tissues may be influenced by ongoing inflammation. Reactivation is better understood by the secretion of butyric acid by oral bacteria. Released viral particles can infect epithelial cells aggravating inflammation and worsening tissue damage. EBV together with H. pylori or HPV alter the expression of methyltransferases (DNMT), which silences the tumor suppressor genes, such as the phosphatase SHP1, rendering the bacterium oncoprotein CagA more active. H. pylori, HPV, and EBV products also enhance the survival of cells. EBV and HPV also turned off the expression of differentiation genes, such as BLIMP1, thus halting cells in the proliferative states to become more permissive for viral persistency, and EBV may favor HPV integration into the host DNA. EBV immunomodulatory mechanisms may help create an immunosuppressive microenvironment that may cooperate with the immunomodulatory mechanisms of other herpesviruses (HHV). Altogether, these interactions promote the appearance of tissue lesions that may be partially responsible for graft rejection and carcinogenesis (see text for details).
Figure 3.
Interactions between EBV and other infectious agents in non-lymphatic organs. The arrival and reactivation of EBV-infected B cells in different tissues may be influenced by ongoing inflammation. Reactivation is better understood by the secretion of butyric acid by oral bacteria. Released viral particles can infect epithelial cells aggravating inflammation and worsening tissue damage. EBV together with H. pylori or HPV alter the expression of methyltransferases (DNMT), which silences the tumor suppressor genes, such as the phosphatase SHP1, rendering the bacterium oncoprotein CagA more active. H. pylori, HPV, and EBV products also enhance the survival of cells. EBV and HPV also turned off the expression of differentiation genes, such as BLIMP1, thus halting cells in the proliferative states to become more permissive for viral persistency, and EBV may favor HPV integration into the host DNA. EBV immunomodulatory mechanisms may help create an immunosuppressive microenvironment that may cooperate with the immunomodulatory mechanisms of other herpesviruses (HHV). Altogether, these interactions promote the appearance of tissue lesions that may be partially responsible for graft rejection and carcinogenesis (see text for details).

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Sánchez-Ponce, Y.; Fuentes-Pananá, E.M. The Role of Coinfections in the EBV–Host Broken Equilibrium. Viruses 2021, 13, 1399. https://0-doi-org.brum.beds.ac.uk/10.3390/v13071399
AMA Style
Sánchez-Ponce Y, Fuentes-Pananá EM. The Role of Coinfections in the EBV–Host Broken Equilibrium. Viruses. 2021; 13(7):1399. https://0-doi-org.brum.beds.ac.uk/10.3390/v13071399
Chicago/Turabian StyleSánchez-Ponce, Yessica, and Ezequiel M. Fuentes-Pananá. 2021. "The Role of Coinfections in the EBV–Host Broken Equilibrium" Viruses 13, no. 7: 1399. https://0-doi-org.brum.beds.ac.uk/10.3390/v13071399
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.