
Oncolytic HSV: Underpinnings of Tumor Susceptibility


Department of Microbiology and Immunology, College of Medicine, University of Illinois, Chicago, IL 60612, USA
*
Author to whom correspondence should be addressed.
Viruses 2021, 13(7), 1408; https://0-doi-org.brum.beds.ac.uk/10.3390/v13071408
Submission received: 15 June 2021
/
Revised: 3 July 2021
/
Accepted: 14 July 2021
/
Published: 20 July 2021
(This article belongs to the Special Issue Oncolytic HSVs)
Abstract
:Oncolytic herpes simplex virus (oHSV) is a therapeutic modality that has seen substantial success for the treatment of cancer, though much remains to be improved. Commonly attenuated through the deletion or alteration of the γ134.5 neurovirulence gene, the basis for the success of oHSV relies in part on the malignant silencing of cellular pathways critical for limiting these viruses in healthy host tissue. However, only recently have the molecular mechanisms underlying the success of these treatments begun to emerge. Further clarification of these mechanisms can strengthen rational design approaches to develop the next generation of oHSV. Herein, we review our current understanding of the molecular basis for tumor susceptibility to γ134.5-attenuated oHSV, with particular focus on the malignant suppression of nucleic acid sensing, along with strategies meant to improve the clinical efficacy of these therapeutic viruses.
Keywords:
herpes simplex virus; oncolytic virus; virotherapy; interferon; STING; T-VEC; immunotherapy1. Introduction
Cancer arises due to genetic or epigenetic changes that activate oncogenes, inactivate tumor-suppressor genes, or suppress innate immune genes. Although advantageous for proliferation or immune escape, such alterations often make cancer cells vulnerable to attack by oncolytic viruses [1]. To date, the most clinically advanced oncolytic virus is talimogene laherparepvec (T-VEC), which is a genetically engineered herpes simplex virus 1 (HSV) that has received international approval for the treatment of advanced melanoma [2,3,4]. T-VEC represents a significant milestone in the translational development of oncolytic immunotherapy and has inspired numerous and diverse oHSV platforms to emerge. Underlying the success of these oncolytic agents are common principles stemming from fundamental virus–host interactions exploited to destroy malignant tissue while leaving healthy tissue unharmed. In this review, we discuss recent progress in intracellular nucleic acid sensing machineries relevant to developing oncolytic viruses based on engineering of the γ134.5 gene.
2. Selectively Engineered HSV as an Oncolytic Virus
HSV-1 is a large, enveloped DNA virus with a 150 kB length genome. The HSV genome is arranged in two regions of unique sequence (UL and US) flanked by two terminal repeated regions (TR) and one internal repeated region (IR) (Figure 1). Upon infection of mucosa, HSV will undergo lytic replication involving the sequential expression of three sets of viral genes: Immediate Early (α), Early (β), and Late (γ), leading to the production of infectious virions [5,6]. Following this, the virus will penetrate peripheral neurons to establish life-long latent infection, where HSV-1 undergoes periodic lytic reactivation in response to stress and other environmental factors [5,7].
The large genome and well-established biology of HSV-1 make it readily amendable to genetic manipulation for therapeutic use. A collection of effective antivirals, such as acyclovir, add an additional layer of safety on top of genetic attenuation. Several genes have been subjected to manipulation as the basis for the attenuation of oHSV. Among the most frequently modified is the γ134.5 gene, which encodes an accessory factor required for neurovirulence [8,9]. HSV γ134.5 is present in two copies per viral genome within the inverted repeat regions [10]. The γ134.5 protein is multifunctional, with roles regulating protein synthesis, autophagy, nucleic acid sensing, and viral egress [11,12,13,14,15,16,17,18,19,20]. Importantly, these roles map to two functionally distinct domains, an amino-terminal domain and a carboxyl-terminal domain, which are connected by a triplet amino acid repeat linker region (Ala-Thr-Pro) (Figure 1). The triplet repeats are a constant feature of the γ134.5 protein in HSV-1, but the number of repeats can vary among different strains [21]. Numerous studies have demonstrated the safety of both HSV deleted of γ134.5 (Δγ134.5) and γ134.5-attenuated viruses at high doses of intracerebral inoculation in HSV-susceptible mice [8,9]. Additionally, to date, completed clinical trials involving γ134.5-attentuated oncolytics ranging from Phase I to III demonstrate significant clinical benefits with limited adverse side effects (Table 1).
3. HSV Grapples with the Host Cytokine Response
HSV infection triggers the activation of host Pathogen Recognition Receptors (PRRs), which are responsible for initiating an antiviral response [22]. Among these PRRs are Toll-like receptors, which are transmembrane proteins on the cell surface and within endosomal compartments that detect the presence of extracellular Pathogen-Associated Molecular Patterns (PAMPs) such as LPS, dsDNA, dsRNA, and viral glycoproteins [23]. Additionally, the host has several intracellular nucleic acid sensors including RIG-I (RNA) [24] and cGAS (dsDNA) [25,26], among others. Activation of these sensing pathways converge on two critical proteins for initiating an anti-pathogen response, TANK Binding Kinase 1 (TBK-1), and Stimulator of Interferon Genes (STING). TBK-1 and STING will form a complex to activate IRF3 and IKK, which initiate Type I Interferon (IFNα/β) production and NF-κB signaling, respectively [22]. IFN and NF-κB will initiate the transcription of hundreds of genes to combat infection, attempting to create a non-permissive environment for pathogen replication while simultaneously secreting extracellular cytokines to direct the immune system toward the site of infection and initiate an adaptive immune response [27].
Wild-type HSV is capable of replication within an IFN-primed environment, and replication is severely restricted upon deletion of γ134.5 [28]. As such, studies over the last decade have drawn light on how γ134.5 overcomes IFN restriction at multiple levels of the immune response. At the level of nucleic acid sensing, γ134.5 has been shown to directly bind and inhibit RIG-I [15]. Concurrently, γ134.5 inhibits downstream adaptors, TBK-1 and STING, which are critical for innate immune signaling and IFN production [14,16]. Should IFN prime cells through upregulation of Interferon Stimulated Genes (ISGs) important for limiting HSV infection, such as dsRNA-dependent Protein Kinase (PKR), γ134.5 and other viral proteins can block PKR and ISG activity to facilitate viral replication [28]. Additionally, γ134.5 inhibits host autophagy, which is capable of degrading HSV capsids [13,29,30]. γ134.5-mediated antagonism of TBK-1 and IKK in dendritic cells (DCs) results in impaired DC maturation, antigen presentation, and subsequent T lymphocyte activation [18]. Ultimately, γ134.5 contributes to the creation of an immunosuppressed environment permissive to HSV replication. However, in the absence of functional γ134.5, as is the case for many oHSV, unperturbed PKR, IFN, and NF-κB signaling can readily restrict replication, creating the need for additional accommodation to facilitate oncolytic replication.
4. Tumor Susceptibility to Δγ134.5 oHSV Replication
Tumor susceptibility to Δγ134.5 oHSV partially stems from the malignant suppression of antiviral pathways needed to limit replication in normal tissues [1]. Malignant antagonism of PKR was the first mechanism identified that could explain Δγ134.5 oHSV replication in cancerous tissue [31]. In addition to PKR restriction, cGAS/STING DNA sensing is critical for limiting HSV infection, and recently, several studies have identified mechanisms in which cancer perturbs cGAS/STING signaling, providing an opportunity for Δγ134.5 oncolytic replication [32,33,34,35].
4.1. PKR Pathway Regulation
PKR is a dsRNA sensing serine threonine kinase constitutively expressed across a number of cell types that is upregulated in response to IFN stimulation [36]. PKR has several antiviral and anti-proliferative functions including shutdown of protein translation, activation of NF-κB signaling, activation of autophagy, and induction of apoptosis, making it a critical target for both virus and pre-malignant tissue [37]. Notably, PKR catalyzes the phosphorylation of eIF2α, which is a cellular protein required for the recycling of eIF2β. eIF2β carries the initial Met-tRNA needed for translation initiation. The phosphorylation of eIF2α at serine 51 greatly enhances eIF2α binding affinity for eIF2β, preventing GDP-GTP exchange factors from recycling eIF2β into the active GTP state needed for translation initiation [38]. Upon the detection of dsRNA, PKR will dimerize, inducing trans-autophosphorylation, which enables PKR to phosphorylate eIF2α at serine 51, resulting in shutdown of protein synthesis [36] (Figure 2).
The HSV genome contains many complementary transcriptional units thought to serve as a dsRNA source to activate PKR [39]. A major function ascribed to γ134.5 pertains to its ability to maintain protein synthesis during late stages of infection [11]. This involves the C terminal domain of γ134.5, which shares homology with host GADD34. The C terminal domain acts as a mediator to recruit both host Protein Phosphatase 1 α (PP1α) and eIF2α, facilitating the dephosphorylation of eIF2α by PP1α and preventing the shutdown of protein synthesis [12,40]. The importance of inhibition of PKR in HSV infection is evident in several studies showing that Δγ134.5 replication and virulence are restored to wild-type levels in PKR −/− mice [41,42,43]. Although PKR’s status as a tumor suppressor is tenuous, showing both tumor suppressive and oncogenic activity [44,45], subsets of tumors are known to have perturbed PKR, providing an opportunity for Δγ134.5 oncolytic replication in the absence of this restrictive antiviral factor.
Several known oncogenes and tumor suppressors influence PKR activity in the cancerous environment [31,37,46,47]. Approximately 19% of cancers exhibit Ras mutations, predominantly resulting in unregulated cellular proliferation [48]. Within this pathway, MEK, a MAPKK, has been shown to inhibit PKR activity [31]. Ras-transformation itself has been implicated to increase replication of wild-type HSV [49]. However, Smith et al. provide evidence in human cancer cell lines identifying tumor susceptibility to Δγ134.5 oncolytic replication being correlated with MEK activity rather than Ras genotype in addition to being inversely correlated with PKR activation [31]. Pharmacological and genetic inhibition of MEK restores PKR expression and results in reduced Δγ134.5 viral titers [31].
The IFN response is at least partially perturbed in 65–70% of cancers [1], underscoring the selection pressure to inhibit these pathways for tumor development. PKR is a critical ISG for Δγ134.5 virus to overcome for efficient replication [28,41], indicating PKR has an important role in the story of malignant susceptibility to oHSV replication. Although the inhibition of IFN effectors such as PKR provides a precise method to facilitate viral replication or tumor progression, an alternative approach for HSV or pre-malignancy to achieve immune suppression is to target pathways responsible for initiating the IFN response and IFN production.
4.2. STING Pathway Regulation
Intracellular DNA sensing through cGAS/STING is a critical pathway for IFN production in response to both HSV infection and in pre-malignant environments [25,50]. Although the mechanism for HSV viral DNA exposure to the cytosol is unknown, HSV capsids can be degraded in a proteasome-dependent manner yielding cytosolic viral dsDNA for cGAS activation [51,52]. High concentrations of cytosolic dsDNA results in the release of cGAS from phosphatidylinositol 4,5 bisphosphate at the plasma membrane [53]. cGAS will dimerize and bind dsDNA in a sequence-independent manner [54]. Binding to long dsDNA results in optimal conformational changes for the production of cyclic GMP-AMP, a cyclic dinucleotide (CDN) capable of potently activating STING [55,56,57]. CDNs are commonly found as secondary messengers in bacteria and are also capable of activating STING [58]. CDNs bind to STING via its cytoplasmic domain and induce STING oligomerization and translocation from the endoplasmic reticulum to the perinuclear Golgi [59,60]. At the Golgi, STING undergoes post-translational modification, after which STING can recruit TBK-1 [61]. Oligomerized STING brings TBK-1 proteins into proximity, inducing the trans-autophosphorylation and activation of TBK-1. This allows for the recruitment and activation of IRF3, which will then dimerize and translocate to the nucleus to initiate Type I IFN production [62] (Figure 2). DNA sensing and subsequent IFN production places strong selection pressure on HSV to inhibit potent activation of the immune system. Several HSV proteins antagonize various steps of DNA sensing, from the degradation of cGAS itself (vhs) [63] to alteration of STING post-translational modifications (VP1-2) [64] to inhibition of STING translocation and TBK-1 binding (γ134.5) [14,16], among other modes of inhibition [65,66,67,68]. Each of these virus–host interactions contribute to the inhibition of DNA sensing and facilitate successful HSV infection.
Notably, cancer exhibits a heavy reliance on genomic instability for tumor progression [69]. Genomic instability can lead to the formation of micronuclei prone to rupture, reactivation of endogenous retroelements, and cytosolic localization of mitochondrial DNA; each of these dsDNA sources can serve to activate cGAS/STING [70,71,72,73,74]. The activation of cGAS/STING DNA sensing is thought to be antitumorigenic, as the resulting IFN production can alert the immune system to identify and clear pre-malignant cells. Recent evidence has identified several mechanisms malignancy uses to perturb DNA sensing at multiple levels, including epigenetic silencing of cGAS/STING [33], mutant p53 inhibition of TBK-1 recruitment [34], and HER2 inhibition of STING signaling complex formation [35] (Figure 2).
DNA methylation, histone modifications, and non-coding RNAs represent epigenetic mechanisms commonly commandeered by cancer to alter its expression profile. DNA hypermethylation near promoter regions generally silences expression, whereas demethylated DNA is generally activating [75]. The hypermethylation of cGAS/STING promoters was initially implicated in a pair of studies analyzing the role of STING in colorectal cancer and melanoma cell lines [76,77]. Further in silico analysis revealed seven of 32 TCGA tumor types (bladder urothelial carcinoma, breast invasive carcinoma, colon adenocarcinoma, head and neck squamous cell carcinoma, lung adenocarcinoma, lung squamous carcinoma, and prostate adenocarcinoma) have significantly increased promoter methylation in either cGAS or STING as compared to matched normal tissue [33]. Interestingly, increased methylation of RIG-I and MDA5 genes was not seen in any of the 32 TCGA tumor types [33]. This is in line with previous studies indicating a majority of the melanoma and colorectal cancer cell lines analyzed have intact IFN production in response to poly IC dsRNA stimulation [76,77]. Recently, it has been identified that the use of demethylating agents can enhance cGAS/STING expression in silenced melanoma cell lines [78]. This reversal results in improved T lymphocyte recognition and increased IFN-γ production in an in vitro co-culture system [78]. Although much less prevalent (< 1% of tumors within TCGA database), it should be noted that several missense mutants of cGAS and STING were identified in the TCGA database [33]. In vitro reconstitution of mutant cGAS/STING in cGAS −/− or STING −/− MEF cells revealed that mutants had significantly reduced capacity to activate an IFNβ-luciferase promoter and show increased viral replication of Δγ134.5 virus, further demonstrating the opportunity presented by cGAS/STING inhibition for oHSV [33].
DNA methylation contrasts with histone modifications as methyl moieties can be activating or inactivating depending on position and context [75]. Histone modifiers such as Lysine Demethylase 5 (KDM5) have been identified to inhibit STING expression in breast cancer [79]. Interestingly, another histone modifier, SUV39H1, is redirected by the Human Papilloma Virus oncogenic protein E7 to suppress STING expression [80]. Genetic and pharmacological inhibition of KDM5 or SUV39H1 results in increased STING expression and type I IFN production in breast cancer and HPV-transformed cells, respectively, in response to dsDNA treatment [79,80].
Widely known for its roles in apoptosis and tumor suppression, the transcription factor p53 is one of the most commonly mutated genes across cancer types: 47% ovarian, 43% colorectal, 40% head and neck, 38% lung, 35% melanoma, and 32% pancreatic [81,82]. Several in silico analyses have correlated p53 inactivation with immunosuppressed tumor phenotypes and decreased immune cell infiltration [83,84,85], but only recently has the role for mutant p53 in the suppression of cGAS–STING–TBK–IRF3 signaling been demonstrated [34]. Ghosh et al. found that nine forms of p53, which were mutated in either the DNA binding region or its structural domain region, were capable of binding TBK-1 in a cell transfection system. All nine p53 mutants reduced IRF3 activation by at least 60%, but three mutants show near complete abrogation of IRF3 activation. One mutant displaying near complete inhibition of IRF3 activation, p53R249S, was further characterized. The interaction between p53R249S and TBK-1 is sufficient to suppress IRF3-mediated apoptosis and IFN production in response to dsDNA stimulation. IFN reduction resulted in decreased CD4+ and CD8+ T lymphocyte infiltration in syngeneic mouse models of 4T1 breast cancer engineered with mutated p53R249S—as compared to wild-type p53 in non-modified 4T1. The extrinsic production of IFNβ was critical for the infiltration of tumors by CD4+ and CD8+ T lymphocytes. A lack of IFNβ, resultant from mutated p53R249S binding to TBK-1, leads to increased infiltration of immunosuppressive, M2 polarized macrophage in tumor tissues. Interestingly, mutant p53 interaction also blocks IFN production in response to poly IC and LPS stimulation, showing that the blockade of TBK-1 activity extends to other innate immune signaling pathways reliant on TBK-1 [34].
HER2 is a ligand-independent receptor tyrosine kinase. Upon activation, HER2 will homo/heterodimerize with other HER-family proteins at the cell surface, where it will then recruit and activate downstream effectors to influence cell proliferation and survival [86]. HER2 is mutated into a constitutively active form in ≈3.5% of cancers [87]. Recently, Wu et al. have identified HER2 as a potent inhibitor of STING signaling [35]. The intracellular domain of HER2 binds with the TBK-1-interacting C-terminal tail of STING. While this interaction is partially inhibitory itself, HER2 functions to recruit AKT1 to the STING signaling complex. AKT1 phosphorylates TBK-1 at serine 510, inhibiting TBK-1′s ability to bind in the STING complex, resulting in a significant decrease in IRF3 activation and subsequent production of ISGs. Importantly, constitutive STING signaling reduces tumor size in a mouse xenograft model using B16 melanoma, and tumor reduction is emulated when HER2 inhibitors are administered. Additionally, constitutively active STING increased CD4+ and CD8+ T lymphocyte infiltration, and the expression of HER2 significantly reduces infiltration in the B16 xenograft model [35].
5. Improving Tumor Selective Replication
While inhibition of cGAS/STING and PKR can provide an opportunity for Δγ134.5 oncolytic replication, early evidence regarding first-generation oHSV such as HSV1716, R3616, and G207 show a wide range of replication efficiencies across tumor types [88,89,90,91]. The dichotomous role of PKR in tumor progression suggests that PKR is a barrier that must be overcome for efficient Δγ134.5 oHSV replication in many tumor types [44,45], and studies characterizing cGAS/STING in melanoma, colorectal, and ovarian cancer cell lines have identified that epigenetic silencing, while prevalent and measurable, is not ubiquitous [76,77,92]. These studies have also indicated that activity of dsRNA sensing for the most part remains intact within these malignant cell lines [76,77,92]. Active PKR, TBK-1, cGAS/STING, and RIG-I all have the potential to restrict Δγ134.5 replication [14,15,16,41], and the heterogenous inhibition of these pathways across cancer types provides a possible explanation for the replication variability observed with first-generation Δγ134.5 oncolytics. Thus, several strategies have evolved from first-generation oHSV to enhance oncolytic replication and ultimately improve therapeutic efficacy (Table 1). Examined in further detail here are examples of oHSV that attempt to restore select functions of γ134.5 to enhance oncolytic replication. The selective restoration of γ134.5 has been the basis for using α expression of Us11, chimeric oncolytic HSV/HCMV hybrids, partial deletion of γ134.5, and tumor-selective expression of γ134.5 or γ134.5 homologs to enhance oncolytic replication without restoring wild-type pathogenicity.

{kind=link}
{kind=link}
Table 1.
Clinical stage oncolytic HSV based on γ134.5 engineering.
| Virus | Modifications | Clinical Trials | PMID | Reference |
|---|---|---|---|---|
| Seprehvir (HSV1716) | Deletion of two copies of γ134.5, HSV-1 strain 17 | NCT00931931, NCT01721018, NCT02031965 | 1848598 | [9] |
| NV1020 (R7020) | Deletion of the HSV IR region (deletion of UL56, one copy of ICP0, ICP4, and γ134.5), insertion of US2-2, US2-3, US2-4, and US2-5 derived from HSV-2 | NCT00149396, NCT00012155 | 2842408 | [93] |
| G207 | Deletion of two copies of γ134.5, insertion of lacZ into ICP6 | NCT04482933, NCT03911388, NCT02457845, NCT00028158, NCT00157703 | 7585221 | [94] |
| RP1 | Deletion two copies of γ134.5, deletion of ICP47 (α expression of Us11), insertion of GALV fusogenic protein; insertion of GM-CSF | NCT04050436, NCT03767348, NCT04349436 | 31399043 | [95] |
| RP2 | Deletion of two copies of γ134.5, deletion of ICP47 (α expression of Us11), insertion of GALV fusogenic protein, insertion of anti-CTLA4 antibody | NCT04336241 | 31399043 | [95] |
| RP3 | Deletion of two copies of γ134.5, deletion of ICP47, insertion of GALV fusogenic protein, insertion of proprietary stimulatory agents | NCT04735978 | 33072862 | [96] |
| M032 | Deletion of two copies of γ134.5, expression of IL-12 | NCT02062827 | 27314913 | [97] |
| VG161 | Deletion of two copies of γ134.5; insertion of IL-12, IL-15, IL-15RA, and TF-Fc peptide capable of disrupting PD-1/PD-L1 interactions | NCT04758897, NCT04806464 | 33182232 | [98] |
| ONCR-177 | Deletion of the IR region, null ICP47 mutation; mutations in gB and UL37; insertion of miR-124-3p; insertion of miR-T cassettes targeting HSV-1 genes ICP4, ICP27, UL8, and γ134.5; insertion of anti-CTLA4 (ipilimumab), FLT3LG, CCL4, IL12, IL12B, anti-PD-1 | NCT04348916 | 33355229 | [99] |
| talimogene laherparepvec (T-VEC) | Deletion of two copies of γ134.5, deletion of ICP47 (α expression of Us11), insertion of GM-CSF | >50 Trials in clinicaltrials.gov, Select Trials: NCT00402025, NCT00289016, NCT00769704, NCT02263508, NCT02366195 | 12595888 | [100] |
| OH2 | HSV-2 backbone: Deletion of two copies of γ134.5, ICP47 deletion (α expression of Us11), insertion of GM-CSF | NCT04637698, NCT04616443, NCT04386967, NCT03866525, NCT04755543 | 24671154 | [101] |
| G47Δ | Deletion of two copies of γ134.5, deletion of ICP6, deletion of ICP47 (α expression of Us11) | UMIN000002661, UMIN000010463, UMIN000011636, UMIN000034063 | 11353831 | [102] |
| C134 | Deletion of two copies of γ134.5, insertion of HCMV IRS1 | NCT03657576 | 15994764 | [103] |
| rQNestin34.5v.2 | Single copy of γ134.5 under the nestin promoter | NCT03152318 | 32373649 | [104] |
5.1. α Expression of Us11
Us11 is a 21 kDa late-stage viral protein whose expression typically coincides with the onset of viral DNA replication [105]. Since their discovery, several functional overlaps between γ134.5 and Us11 have been identified. Both exhibit inhibitory activity toward PKR [12,106], autophagy [13,107], RIG-I [15,108], and both contribute to full IFN resistance in late stages of HSV infection [109]. In hindsight, these overlaps make it clear why the serial evolution of Δγ134.5 HSV in non-permissive glioma can select for α expression of Us11 [110,111]. α expression of Us11 can partially yet substantially restore viral replication without enhancing pathogenicity. As such, several oHSV use α expression of Us11 as a strategy to enhance oncolysis (Table 1).
T-VEC contains several modifications, including α expression of Us11 [100]. T-VEC has undergone clinical trials for the treatment of a myriad of solid tumors (Table 1). Approved as a standalone therapeutic for melanoma, many ongoing clinical trials use T-VEC in combination with either traditional cancer therapies (surgery and radiation) or with immune checkpoint blockade inhibitors [2]. G47Δ is an oHSV built upon the G207 backbone, containing deletions in both copies of γ134.5 as well as the insertion of lacZ into the ICP6 gene along with the deletion of ICP47 yielding α expression of Us11 [102]. G47Δ shows enhanced in vitro replication and therapeutic efficacy in preclinical models as compared to G207, and this inspired several clinical trials in Japan for the treatment of glioblastoma, prostate cancer, olfactory neuroblastoma, and malignant mesothelioma (Table 1).
5.2. Chimeric oHSV
Another approach to improve oncolytic replication is to utilize non-native viral proteins functionally similar to γ134.5 or proteins to perturb angiogenesis [112]. One such virus, C134, expresses IRS1, derived from Human Cytomegalovirus (HCMV). IRS1 is a dsRNA binding protein capable of binding to and blocking PKR-mediated shutdown of protein synthesis in the context of Δγ134.5 infection [103,113] but is unable to prevent IRF3 signaling when expressed in the Δγ134.5 backbone [114]. These functions serve to enhance the replication of C134 in several glioma models [115]. Preclinical testing of C134 in Aotus nancymaae demonstrated safety in this model [116], and in 2019, C134 entered Phase I clinical trials enrolling 24 patients for the treatment of glioblastoma multiforme set to complete in late 2024 (Table 1).
5.3. Tumor-Selective Expression of γ134.5
Several oHSVs utilize tumor-selective promoters to express γ134.5. The selective expression of γ134.5 allows for increased viral spread and replication within transcriptionally targeted tissues. To date, these viruses have been developed using an oncolytic backbone deficient in both copies of γ134.5; then, a single copy of γ134.5 is reintroduced under tumor-selective promoters for proteins such as b-myb and nestin [117,118]. Nestin is an intermediate filament expressed specifically in glioma. Several oHSV use the nestin promoter, including rQNestinv.2, which is set to complete a Phase 1 clinical trial in July 2021 for the treatment of recurrent glioma. NG34 is a recent experimental oncolytic virus using the nestin promoter to selectively express host GADD34 [119]. GADD34 is homologous to the γ134.5 C-terminal domain and functions in the cell to negatively regulate eIF2α kinases such as PKR. Interestingly, chemotherapy and radiation have been shown to upregulate GADD34, which specifically enhances therapeutic effect of other single deletion γ134.5 oHSVs [120,121]. ONCR-177 is another oHSV that employs miRNA to selectively inhibit the expression of γ134.5 in normal tissues and thereby enables tumor-selective viral replication [122]. In addition to tumor-selective expression of γ134.5 itself, tumor selectivity can be achieved through an alteration of HSV tropism. Retargeting HSV to tumor biomarkers has an advantage through the selective expression of wild-type viral proteins such as γ134.5, which can facilitate replication in targeted tissue. One example involves the alteration of oHSV entry proteins to target cancer cells expressing HER2 [123,124].
5.4. Partial Deletion of γ134.5
Three decades of research have mapped numerous functions within the γ134.5 protein. The TBK-1, STING, IKK, and Beclin 1 interaction regions all lie within the N-terminal domain of γ134.5 [13,14,17,18]. ΔN146 is an experimental oncolytic virus in which the N-terminal domain of γ134.5 has been deleted but retains two copies of the γ134.5 C-terminal domain. ΔN146 has been shown to inhibit translational shutdown, overall contributing to enhanced replication in a number of tumor cells lines and enhanced therapeutic efficacy in 4T1 syngeneic tumor models as compared to first-generation oHSV [125]. The oncolytic virus Δ68H-6 contains a deletion of amino acids 68–87 of γ134.5 along with a deletion of ICP6 [126]. This region binds to Beclin 1 to prevent autophagy [13] but also overlaps with regions on γ134.5 needed for viral egress and TBK-1 binding [16,127]. Δ68H-6 was shown to extend the survival time in a U87 glioma mouse xenograft model as compared to HSV1716 [126].
6. Improving Antitumor Immunity
It is now well recognized that in addition to tumor-selective replication, robust immune activation is critical for therapeutic efficacy of oHSV. In addition to ISGs to directly combat infection, IFN also stimulates the production of cytokines to recruit Dendritic Cells (DCs), Macrophages, and T lymphocytes to a site of infection. DCs and Macrophages are Antigen Presenting Cells (APCs) that phagocytose malignant or infected cells and process antigen peptides for presentation to B and T lymphocytes for activation [128].
To establish an immune-suppressed environment, the γ134.5 protein can inhibit DC maturation through the abrogation of TBK-1 and IKK activation of IFN and NF-κB, impairing antigen presentation and T lymphocyte activation during HSV infection [18,129]. Similarly, the malignant inhibition of cGAS/STING signaling through epigenetic silencing, mutant p53, or HER2 results in the simultaneous suppression of T lymphocyte infiltration in in vivo tumor models [34,35,78]. The activation of tumor endogenous STING has been identified as an important component for inducing antitumor immune responses [130]. The malignant suppression of cGAS/STING provides an opportunity for oncolytic replication, but the absence of STING signaling can restrict immunogenic cell death that is associated with robust activation of the immune response [130,131,132]. In addition to silencing of antitumor immune activation at the molecular level through cGAS/STING inhibition, malignancy has developed mechanisms of immunosuppression at the cellular level through the overexpression of immune checkpoint molecules (PD1, PDL1, and CTLA4) that normally act to prevent overactive T lymphocyte responses [133]. While this immunosuppressed environment provides an opportunity for oHSV replication, hampered APC activation and T lymphocyte target recognition can lead to ineffective antitumor responses [134]. Thus, many oHSV have been engineered to express cellular cytokines and chemokines to circumvent these crippled innate immune signaling pathways and enhance immune cell stimulation (Table 1).
DCs are major producers of IL-12, which is a cytokine that is capable of enhancing the activation and proliferation of cytotoxic T lymphocytes and NK cells, resulting in increased IFN-γ, a Type II IFN, production from these cell types [135]. IFN-γ can upregulate MHC I expression in tumor cells and transform immunosuppressive M2 macrophage into the immunostimulatory M1 subtype [136]. Although the systemic administration of IL-12 results in serious adverse side effects in humans [136], several oHSV, including M032, encode IL-12 for localized tumor expression (Table 1). M032 has recently entered a Phase I clinical trial for the treatment of glioma [97]. GM-CSF is another cytokine frequently expressed in oHSV. GM-CSF enhances the activation, proliferation, and recruitment of APCs from bone marrow [137]. In addition to α expression of Us11, T-VEC encodes two copies of GM-CSF in an effort to improve APC activation and antigen presentation in an environment replete with tumor antigens from viral oncolysis [100]. APCs ultimately prime cytotoxic T lymphocytes, which are major contributors to the antitumor response [138], and numerous studies have identified positive correlations between infiltrating cytotoxic T lymphocytes and improved therapeutic outcomes [134].
To further examine the immune effectors driving antitumor responses with T-VEC monotherapy, a recently completed Phase II clinical trial analyzed the correlates between immune cell infiltration and objective response rate in individuals with stage IIIB-IVM1a melanoma [139]. The study recorded an objective response rate of 28% and a complete response rate of 14%, which is on par with previous T-VEC melanoma trials [139,140,141]. Intriguingly, there was a 2.4-fold increase in T lymphocyte infiltration in non-injected lesions 6 weeks after initial treatment yet, CD4+ or CD8+ T lymphocyte infiltration did not correlate with the objective response rate [139]. Notably, activated T lymphocytes secreting IFN-γ can upregulate PDL1 expression in the epithelium as a negative feedback mechanism to control hyperactivation [133]. Exploratory analysis revealed detectable PDL1 expression in non-injected lesions of 10/50 patients at baseline, and this increased to 28/50 patients after 6 weeks of T-VEC treatment. This is suggestive of adaptive resistance to T lymphocyte infiltration in which malignancy upregulates immune checkpoints in non-injected lesions of patients treated with T-VEC [139]. This study reinforces combination therapy approaches using immune checkpoint inhibitors (anti-PD1/PDL1) and provides initial evidence to explain the synergistic effects seen when combining T-VEC with checkpoint blockade (Phase 1b: T-VEC in combination with anti-PD1 for advanced melanoma: objective response rate 62%, complete response rate 33%) [142]. In addition, these trials make clinical stage oHSVs such as RP2, ONCR-177, and VG161 particularly exciting as these viruses, among other modifications, encode PD1 antagonist peptides or anti-PD1/CTLA4 monoclonal antibodies, possibly providing a unique angle to combat malignancy in the tumor microenvironment (Table 1).
7. Perspectives
The clinical efficacy of T-VEC has led to the explosive growth of γ134.5-attenuated oHSV development and subsequent clinical trials. While this growth will ultimately be beneficial for cancer therapy, much remains to be improved, especially for patients with advanced disease. A multitude of possible cancer genotypes and phenotypes yields a myriad of possible mechanisms of action for individual oHSVs to exert therapeutic effect. The recent studies regarding the malignant abrogation of IFN responses reviewed here provide a glimpse into the molecular and cellular determinants of tumor susceptibility to γ134.5-attenuated oHSV. Further understanding of the complex layer of virus–host interactions underlying tumor selective replication and antitumor immunity will be critical for development of the next generation of oHSV as well as providing essential information for better patient stratification criteria to improve clinical outcomes.
Author Contributions
Conceptualization, C.K. and B.H.; Writing—Original Draft Preparation, C.K.; Writing—Review and Editing, C.K., E.K. and B.H. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by grants from the National Institute of Cancer (CA252027) and the National Institute of Allergy and Infectious Diseases (AI148148).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Ilkow, C.S.; Swift, S.L.; Bell, J.C.; Diallo, J.-S. From Scourge to Cure: Tumour-Selective Viral Pathogenesis as a New Strategy against Cancer. PLoS Pathog. 2014, 10, e1003836. [Google Scholar] [CrossRef] [Green Version]
- Raman, S.S.; Hecht, J.R.; Chan, E. Talimogene laherparepvec: Review of its mechanism of action and clinical efficacy and safety. Immunotherapy 2019, 11, 705–723. [Google Scholar] [CrossRef] [Green Version]
- Andtbacka, R.H.I.; Kaufman, H.L.; Collichio, F.; Amatruda, T.; Senzer, N.; Chesney, J.; Delman, K.A.; Spitler, L.E.; Puzanov, I.; Agarwala, S.S.; et al. Talimogene Laherparepvec Improves Durable Response Rate in Patients with Advanced Melanoma. J. Clin. Oncol. 2015, 33, 2780–2788. [Google Scholar] [CrossRef]
- Andtbacka, R.H.I.; Collichio, F.; Harrington, K.J.; Middleton, M.R.; Downey, G.; Öhrling, K.; Kaufman, H.L. Final analyses of OPTiM: A randomized phase III trial of talimogene laherparepvec versus granulocyte-macrophage colony-stimulating factor in unresectable stage III-IV melanoma. J. Immunother. Cancer 2019, 7, 145. [Google Scholar] [CrossRef] [Green Version]
- Whitley, R.J.; Roizman, B. Herpes simplex virus infections. Lancet 2001, 357, 1513–1518. [Google Scholar] [CrossRef]
- Knipe, D.M.; Cliffe, A. Chromatin control of herpes simplex virus lytic and latent infection. Nat. Rev. Microbiol. 2008, 6, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, A.L.; Diefenbach, R.J.; Miranda-Saksena, M.; Bosnjak, L.; Kim, M.; Jones, C.; Douglas, M.W. The cycle of human herpes simplex virus infection: Virus transport and immune control. J. Infect. Dis. 2006, 194 (Suppl. 1), S11–S18. [Google Scholar] [CrossRef] [Green Version]
- Chou, J.; Kern, E.R.; Whitley, R.J.; Roizman, B. Mapping of herpes simplex virus-1 neurovirulence to gamma 134.5, a gene nonessential for growth in culture. Science 1990, 250, 1262–1266. [Google Scholar] [CrossRef] [PubMed]
- MacLean, A.R.; ul-Fareed, M.; Robertson, L.; Harland, J.; Brown, S.M. Herpes simplex virus type 1 deletion variants 1714 and 1716 pinpoint neurovirulence-related sequences in Glasgow strain 17+ between immediate early gene 1 and the ‘a’ sequence. J. Gen. Virol. 1991, 72, 631–639. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, M.; Chou, J.; Sarmiento, M.; Lerner, R.A.; Roizman, B. Identification by antibody to a synthetic peptide of a protein specified by a diploid gene located in the terminal repeats of the L component of herpes simplex virus genome. J. Virol. 1986, 58, 843–850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chou, J.; Roizman, B. The gamma 1(34.5) gene of herpes simplex virus 1 precludes neuroblastoma cells from triggering total shutoff of protein synthesis characteristic of programed cell death in neuronal cells. Proc. Natl. Acad. Sci. USA 1992, 89, 3266–3270. [Google Scholar] [CrossRef] [Green Version]
- He, B.; Gross, M.; Roizman, B. The gamma(1)34.5 protein of herpes simplex virus 1 complexes with protein phosphatase 1alpha to dephosphorylate the alpha subunit of the eukaryotic translation initiation factor 2 and preclude the shutoff of protein synthesis by double-stranded RNA-activated protein kinase. Proc. Natl. Acad. Sci. USA 1997, 94, 843–848. [Google Scholar] [CrossRef] [Green Version]
- Orvedahl, A.; Alexander, D.; Tallóczy, Z.; Sun, Q.; Wei, Y.; Zhang, W.; Burns, D.; Leib, D.A.; Levine, B. HSV-1 ICP34.5 confers neurovirulence by targeting the Beclin 1 autophagy protein. Cell Host Microbe 2007, 1, 23–35. [Google Scholar] [CrossRef] [Green Version]
- Pan, S.; Liu, X.; Ma, Y.; Cao, Y.; He, B. Herpes Simplex Virus 1 γ134.5 Protein Inhibits STING Activation That Restricts Viral Replication. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Ma, Y.; Voss, K.; van Gent, M.; Chan, Y.K.; Gack, M.U.; Gale, M., Jr.; He, B. The herpesvirus accessory protein γ134.5 facilitates viral replication by disabling mitochondrial translocation of RIG-I. PLoS Pathog. 2021, 17, e1009446. [Google Scholar] [CrossRef]
- Verpooten, D.; Ma, Y.; Hou, S.; Yan, Z.; He, B. Control of TANK-binding kinase 1-mediated signaling by the γ134.5 protein of herpes simplex virus 1. J. Biol. Chem. 2009, 284, 1097–1105. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.; Jin, H.; Valyi-Nagy, T.; Cao, Y.; Yan, Z.; He, B. Inhibition of TANK binding kinase 1 by herpes simplex virus 1 facilitates productive infection. J. Virol. 2012, 86, 2188–2196. [Google Scholar] [CrossRef] [Green Version]
- Jin, H.; Yan, Z.; Ma, Y.; Cao, Y.; He, B. A herpesvirus virulence factor inhibits dendritic cell maturation through protein phosphatase 1 and Ikappa B kinase. J. Virol. 2011, 85, 3397–3407. [Google Scholar] [CrossRef] [Green Version]
- Brown, S.M.; MacLean, A.R.; Aitken, J.D.; Harland, J. ICP34.5 influences herpes simplex virus type 1 maturation and egress from infected cells in vitro. J. Gen. Virol. 1994, 75, 3679–3686. [Google Scholar] [CrossRef]
- Wu, S.; Pan, S.; Zhang, L.; Baines, J.; Roller, R.; Ames, J.; Yang, M.; Wang, J.; Chen, D.; Liu, Y.; et al. Herpes Simplex Virus 1 Induces Phosphorylation and Reorganization of Lamin A/C through the γ134.5 Protein That Facilitates Nuclear Egress. J. Virol. 2016, 90, 10414–10422. [Google Scholar] [CrossRef] [Green Version]
- Chou, J.; Roizman, B. The herpes simplex virus 1 gene for ICP34.5, which maps in inverted repeats, is conserved in several limited-passage isolates but not in strain 17syn+. J. Virol. 1990, 64, 1014–1020. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Qin, C.; Liu, Y.; Rao, Y.; Feng, P. Herpes Simplex Virus and Pattern Recognition Receptors: An Arms Race. Front. Immunol. 2021, 11. [Google Scholar] [CrossRef]
- Ma, Y.; He, B. Recognition of herpes simplex viruses: Toll-like receptors and beyond. J. Mol. Biol. 2014, 426, 1133–1147. [Google Scholar] [CrossRef] [Green Version]
- Rehwinkel, J.; Gack, M.U. RIG-I-like receptors: Their regulation and roles in RNA sensing. Nat. Rev. Immunol. 2020, 20, 537–551. [Google Scholar] [CrossRef]
- Barber, G.N. STING: Infection, inflammation and cancer. Nat. Rev. Immunol. 2015, 15, 760–770. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Bai, X.-C.; Chen, Z.J. Structures and Mechanisms in the cGAS-STING Innate Immunity Pathway. Immunity 2020, 53, 43–53. [Google Scholar] [CrossRef]
- Schoggins, J.W. Interferon-Stimulated Genes: What Do They All Do? Annu. Rev. Virol. 2019, 6, 567–584. [Google Scholar] [CrossRef]
- Cheng, G.; Brett, M.E.; He, B. Val193 and Phe195 of the γ134.5 protein of herpes simplex virus 1 are required for viral resistance to interferon-alpha/beta. Virology 2001, 290, 115–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tallóczy, Z.; Jiang, W.; Virgin, H.W.; Leib, D.A.; Scheuner, D.; Kaufman, R.J.; Eskelinen, E.-L.; Levine, B. Regulation of starvation- and virus-induced autophagy by the eIF2alpha kinase signaling pathway. Proc. Natl. Acad. Sci. USA 2002, 99, 190–195. [Google Scholar] [CrossRef] [Green Version]
- Tallóczy, Z.; Virgin, H.W.; Levine, B. PKR-dependent autophagic degradation of herpes simplex virus type 1. Autophagy 2006, 2, 24–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, K.D.; Mezhir, J.J.; Bickenbach, K.; Veerapong, J.; Charron, J.; Posner, M.C.; Roizman, B.; Weichselbaum, R.R. Activated MEK Suppresses Activation of PKR and Enables Efficient Replication and in vivo Oncolysis by Δ γ134.5 Mutants of Herpes Simplex Virus 1. J. Virol. 2006, 80, 1110–1120. [Google Scholar] [CrossRef] [Green Version]
- Ishikawa, H.; Barber, G.N. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 2008, 455, 674–678. [Google Scholar] [CrossRef]
- Konno, H.; Yamauchi, S.; Berglund, A.; Putney, R.M.; Mulé, J.; Barber, G. Suppression of STING Signaling through Epigenetic Silencing and Missense Mutation Impedes DNA-Damage Mediated Cytokine Production. Oncogene 2018. [Google Scholar] [CrossRef]
- Ghosh, M.; Saha, S.; Bettke, J.; Nagar, R.; Parrales, A.; Iwakuma, T.; van der Velden, A.W.M.; Martinez, L.A. Mutant p53 suppresses innate immune signaling to promote tumorigenesis. Cancer Cell 2021, 39, 494–508.e495. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Zhang, Q.; Zhang, F.; Meng, F.; Liu, S.; Zhou, R.; Wu, Q.; Li, X.; Shen, L.; Huang, J.; et al. HER2 recruits AKT1 to disrupt STING signalling and suppress antiviral defence and antitumour immunity. Nat. Cell Biol. 2019, 21, 1027–1040. [Google Scholar] [CrossRef]
- Sadler, A.J.; Williams, B.R.G. Interferon-inducible antiviral effectors. Nat. Rev. Immunol. 2008, 8, 559–568. [Google Scholar] [CrossRef] [PubMed]
- García, M.A.; Gil, J.; Ventoso, I.; Guerra, S.; Domingo, E.; Rivas, C.; Esteban, M. Impact of Protein Kinase PKR in Cell Biology: From Antiviral to Antiproliferative Action. Microbiol. Mol. Biol. Rev. 2006, 70, 1032–1060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, W.K.; Hovanessian, A.; Brown, R.E.; Clemens, M.J.; Kerr, I.M. Interferon-mediated protein kinase and low-molecular-weight inhibitor of protein synthesis. Nature 1976, 264, 477–480. [Google Scholar] [CrossRef]
- Jacquemont, B.; Roizman, B. RNA synthesis in cells infected with herpes simplex virus. X. Properties of viral symmetric transcripts and of double-stranded RNA prepared from them. J. Virol. 1975, 15, 707–713. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Zhang, C.; Chen, X.; Yu, J.; Wang, Y.; Yang, Y.; Du, M.; Jin, H.; Ma, Y.; He, B.; et al. ICP34.5 protein of herpes simplex virus facilitates the initiation of protein translation by bridging eukaryotic initiation factor 2alpha (eIF2alpha) and protein phosphatase 1. J. Biol. Chem. 2011, 286, 24785–24792. [Google Scholar] [CrossRef] [Green Version]
- Leib, D.A.; Machalek, M.A.; Williams, B.R.; Silverman, R.H.; Virgin, H.W. Specific phenotypic restoration of an attenuated virus by knockout of a host resistance gene. Proc. Natl. Acad. Sci. USA 2000, 97, 6097–6101. [Google Scholar] [CrossRef] [Green Version]
- Verpooten, D.; Feng, Z.; Valyi-Nagy, T.; Ma, Y.; Jin, H.; Yan, Z.; Zhang, C.; Cao, Y.; He, B. Dephosphorylation of eIF2alpha mediated by the γ134.5 protein of herpes simplex virus 1 facilitates viral neuroinvasion. J. Virol. 2009, 83, 12626–12630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilcox, D.R.; Muller, W.J.; Longnecker, R. HSV targeting of the host phosphatase PP1α is required for disseminated disease in the neonate and contributes to pathogenesis in the brain. Proc. Natl. Acad. Sci. USA 2015, 112, E6937–E6944. [Google Scholar] [CrossRef] [Green Version]
- Gal-Ben-Ari, S.; Barrera, I.; Ehrlich, M.; Rosenblum, K. PKR: A Kinase to Remember. Front. Mol. Neurosci. 2019, 11. [Google Scholar] [CrossRef] [Green Version]
- Clemens, M.J. Targets and mechanisms for the regulation of translation in malignant transformation. Oncogene 2004, 23, 3180–3188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gannon, H.S.; Zou, T.; Kiessling, M.K.; Gao, G.F.; Cai, D.; Choi, P.S.; Ivan, A.P.; Buchumenski, I.; Berger, A.C.; Goldstein, J.T.; et al. Identification of ADAR1 adenosine deaminase dependency in a subset of cancer cells. Nat. Commun. 2018, 9, 5450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pang, Q.; Christianson, T.A.; Koretsky, T.; Carlson, H.; David, L.; Keeble, W.; Faulkner, G.R.; Speckhart, A.; Bagby, G.C. Nucleophosmin Interacts with and Inhibits the Catalytic Function of Eukaryotic Initiation Factor 2 Kinase PKR. J. Biol. Chem. 2003, 278, 41709–41717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prior, I.A.; Hood, F.E.; Hartley, J.L. The Frequency of Ras Mutations in Cancer. Cancer Res. 2020, 80, 2969–2974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farassati, F.; Yang, A.-D.; Lee, P.W.K. Oncogenes in Ras signalling pathway dictate host-cell permissiveness to herpes simplex virus 1. Nat. Cell Biol. 2001, 3, 745–750. [Google Scholar] [CrossRef]
- Kwon, J.; Bakhoum, S.F. The Cytosolic DNA-Sensing cGAS–STING Pathway in Cancer. Cancer Discov. 2020, 10, 26–39. [Google Scholar] [CrossRef]
- Horan, K.A.; Hansen, K.; Jakobsen, M.R.; Holm, C.K.; Søby, S.; Unterholzner, L.; Thompson, M.; West, J.A.; Iversen, M.B.; Rasmussen, S.B.; et al. Proteasomal degradation of herpes simplex virus capsids in macrophages releases DNA to the cytosol for recognition by DNA sensors. J. Immunol. 2013, 190, 2311–2319. [Google Scholar] [CrossRef]
- Sun, C.; Luecke, S.; Bodda, C.; Jønsson, K.L.; Cai, Y.; Zhang, B.-C.; Jensen, S.B.; Nordentoft, I.; Jensen, J.M.; Jakobsen, M.R.; et al. Cellular Requirements for Sensing and Elimination of Incoming HSV-1 DNA and Capsids. J. Interferon Cytok. Res. 2019, 39, 191–204. [Google Scholar] [CrossRef]
- Barnett, K.C.; Coronas-Serna, J.M.; Zhou, W.; Ernandes, M.J.; Cao, A.; Kranzusch, P.J.; Kagan, J.C. Phosphoinositide Interactions Position cGAS at the Plasma Membrane to Ensure Efficient Distinction between Self- and Viral DNA. Cell 2019, 176, 1432–1446.e1411. [Google Scholar] [CrossRef] [Green Version]
- Civril, F.; Deimling, T.; de Oliveira Mann, C.C.; Ablasser, A.; Moldt, M.; Witte, G.; Hornung, V.; Hopfner, K.-P. Structural mechanism of cytosolic DNA sensing by cGAS. Nature 2013, 498, 332–337. [Google Scholar] [CrossRef] [Green Version]
- Luecke, S.; Holleufer, A.; Christensen, M.H.; Jønsson, K.L.; Boni, G.A.; Sørensen, L.K.; Johannsen, M.; Jakobsen, M.R.; Hartmann, R.; Paludan, S.R. cGAS is activated by DNA in a length-dependent manner. EMBO Rep. 2017, 18, 1707–1715. [Google Scholar] [CrossRef]
- Xie, W.; Lama, L.; Adura, C.; Tomita, D.; Glickman, J.F.; Tuschl, T.; Patel, D.J. Human cGAS catalytic domain has an additional DNA-binding interface that enhances enzymatic activity and liquid-phase condensation. Proc. Natl. Acad. Sci. USA 2019, 116, 11946–11955. [Google Scholar] [CrossRef] [Green Version]
- Zhou, W.; Whiteley, A.T.; de Oliveira Mann, C.C.; Morehouse, B.R.; Nowak, R.P.; Fischer, E.S.; Gray, N.S.; Mekalanos, J.J.; Kranzusch, P.J. Structure of the Human cGAS-DNA Complex Reveals Enhanced Control of Immune Surveillance. Cell 2018, 174, 300–311.e11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woodward, J.J.; Iavarone, A.T.; Portnoy, D.A. c-di-AMP Secreted by Intracellular Listeria monocytogenes Activates a Host Type I Interferon Response. Science 2010, 328, 1703–1705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ergun, S.L.; Fernandez, D.; Weiss, T.M.; Li, L. STING Polymer Structure Reveals Mechanisms for Activation, Hyperactivation, and Inhibition. Cell 2019, 178, 290–301.e210. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, H.; Ma, Z.; Barber, G.N. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 2009, 461, 788–792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukai, K.; Konno, H.; Akiba, T.; Uemura, T.; Waguri, S.; Kobayashi, T.; Barber, G.N.; Arai, H.; Taguchi, T. Activation of STING requires palmitoylation at the Golgi. Nat. Commun. 2016, 7, 11932. [Google Scholar] [CrossRef]
- Zhang, C.; Shang, G.; Gui, X.; Zhang, X.; Bai, X.-C.; Chen, Z.J. Structural basis of STING binding with and phosphorylation by TBK1. Nature 2019, 567, 394–398. [Google Scholar] [CrossRef] [PubMed]
- Su, C.; Zheng, C. Herpes Simplex Virus 1 Abrogates the cGAS/STING-Mediated Cytosolic DNA-Sensing Pathway via Its Virion Host Shutoff Protein, UL41. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bodda, C.; Reinert, L.S.; Fruhwürth, S.; Richardo, T.; Sun, C.; Zhang, B.-C.; Kalamvoki, M.; Pohlmann, A.; Mogensen, T.H.; Bergström, P.; et al. HSV1 VP1-2 deubiquitinates STING to block type I interferon expression and promote brain infection. J. Exp. Med. 2020, 217. [Google Scholar] [CrossRef]
- Christensen, M.H.; Jensen, S.B.; Miettinen, J.J.; Luecke, S.; Prabakaran, T.; Reinert, L.S.; Mettenleiter, T.; Chen, Z.J.; Knipe, D.M.; Sandri-Goldin, R.M.; et al. HSV-1 ICP27 targets the TBK1-activated STING signalsome to inhibit virus-induced type I IFN expression. EMBO J. 2016, 35, 1385–1399. [Google Scholar] [CrossRef]
- Deschamps, T.; Kalamvoki, M. Evasion of the STING DNA-Sensing Pathway by VP11/12 of Herpes Simplex Virus 1. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.; You, H.; Su, C.; Li, Y.; Chen, S.; Zheng, C. Herpes Simplex Virus 1 Tegument Protein VP22 Abrogates cGAS/STING-Mediated Antiviral Innate Immunity. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [Green Version]
- You, H.; Lin, Y.; Lin, F.; Yang, M.; Li, J.; Zhang, R.; Huang, Z.; Shen, Q.; Tang, R.; Zheng, C. β-Catenin Is Required for the cGAS/STING Signaling Pathway but Antagonized by the Herpes Simplex Virus 1 US3 Protein. J. Virol. 2020, 94. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dou, Z.; Ghosh, K.; Vizioli, M.G.; Zhu, J.; Sen, P.; Wangensteen, K.J.; Simithy, J.; Lan, Y.; Lin, Y.; Zhou, Z.; et al. Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature 2017, 550, 402–406. [Google Scholar] [CrossRef] [Green Version]
- Mackenzie, K.J.; Carroll, P.; Martin, C.-A.; Murina, O.; Fluteau, A.; Simpson, D.J.; Olova, N.; Sutcliffe, H.; Rainger, J.K.; Leitch, A.; et al. cGAS surveillance of micronuclei links genome instability to innate immunity. Nature 2017, 548, 461–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishak, C.A.; Classon, M.; De Carvalho, D.D. Deregulation of Retroelements as an Emerging Therapeutic Opportunity in Cancer. Trends Cancer 2018, 4, 583–597. [Google Scholar] [CrossRef]
- Crasta, K.; Ganem, N.J.; Dagher, R.; Lantermann, A.B.; Ivanova, E.V.; Pan, Y.; Nezi, L.; Protopopov, A.; Chowdhury, D.; Pellman, D. DNA breaks and chromosome pulverization from errors in mitosis. Nature 2012, 482, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Kitajima, S.; Ivanova, E.; Guo, S.; Yoshida, R.; Campisi, M.; Sundararaman, S.K.; Tange, S.; Mitsuishi, Y.; Thai, T.C.; Masuda, S.; et al. Suppression of STING Associated with LKB1 Loss in KRAS-Driven Lung Cancer. Cancer Discov. 2019, 9, 34–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, S.; Kelly, T.K.; Jones, P.A. Epigenetics in cancer. Carcinogenesis 2010, 31, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Xia, T.; Konno, H.; Barber, G.N. Recurrent Loss of STING Signaling in Melanoma Correlates with Susceptibility to Viral Oncolysis. Cancer Res. 2016, 76, 6747–6759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, T.; Konno, H.; Ahn, J.; Barber, G.N. Deregulation of STING Signaling in Colorectal Carcinoma Constrains DNA Damage Responses and Correlates with Tumorigenesis. Cell Rep. 2016, 14, 282–297. [Google Scholar] [CrossRef] [Green Version]
- Falahat, R.; Berglund, A.; Putney, R.M.; Perez-Villarroel, P.; Aoyama, S.; Pilon-Thomas, S.; Barber, G.N.; Mulé, J.J. Epigenetic reprogramming of tumor cell-intrinsic STING function sculpts antigenicity and T cell recognition of melanoma. Proc. Natl. Acad. Sci. USA 2021, 118. [Google Scholar] [CrossRef]
- Wu, L.; Cao, J.; Cai, W.L.; Lang, S.M.; Horton, J.R.; Jansen, D.J.; Liu, Z.Z.; Chen, J.F.; Zhang, M.; Mott, B.T.; et al. KDM5 histone demethylases repress immune response via suppression of STING. PLoS Biol. 2018, 16, e2006134. [Google Scholar] [CrossRef] [Green Version]
- Lo Cigno, I.; Calati, F.; Borgogna, C.; Zevini, A.; Albertini, S.; Martuscelli, L.; De Andrea, M.; Hiscott, J.; Landolfo, S.; Gariglio, M. Human Papillomavirus E7 Oncoprotein Subverts Host Innate Immunity via SUV39H1-Mediated Epigenetic Silencing of Immune Sensor Genes. J. Virol. 2020, 94. [Google Scholar] [CrossRef]
- Olivier, M.; Hollstein, M.; Hainaut, P. TP53 Mutations in Human Cancers: Origins, Consequences, and Clinical Use. Cold Spring Harb. Perspect. Biol. 2010, 2. [Google Scholar] [CrossRef] [Green Version]
- Ozaki, T.; Nakagawara, A. Role of p53 in Cell Death and Human Cancers. Cancers 2011, 3, 994–1013. [Google Scholar] [CrossRef] [PubMed]
- Siemers, N.O.; Holloway, J.L.; Chang, H.; Chasalow, S.D.; Ross-MacDonald, P.B.; Voliva, C.F.; Szustakowski, J.D. Genome-wide association analysis identifies genetic correlates of immune infiltrates in solid tumors. PLoS ONE 2017, 12, e0179726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Z.; Liu, Z.; Li, M.; Chen, C.; Wang, X. Immunogenomics Analysis Reveals that TP53 Mutations Inhibit Tumor Immunity in Gastric Cancer. Transl. Oncol. 2018, 11, 1171–1187. [Google Scholar] [CrossRef]
- Lyu, H.; Li, M.; Jiang, Z.; Liu, Z.; Wang, X. Correlate the TP53 Mutation and the HRAS Mutation with Immune Signatures in Head and Neck Squamous Cell Cancer. Comput. Struct. Biotechnol. J. 2019, 17, 1020–1030. [Google Scholar] [CrossRef]
- Roskoski, R. The ErbB/HER family of protein-tyrosine kinases and cancer. Pharmacol. Res. 2014, 79, 34–74. [Google Scholar] [CrossRef]
- Pahuja, K.B.; Nguyen, T.T.; Jaiswal, B.S.; Prabhash, K.; Thaker, T.M.; Senger, K.; Chaudhuri, S.; Kljavin, N.M.; Antony, A.; Phalke, S.; et al. Actionable Activating Oncogenic ERBB2/HER2 Transmembrane and Juxtamembrane Domain Mutations. Cancer Cell 2018, 34, 792–806.e795. [Google Scholar] [CrossRef] [Green Version]
- Chung, S.M.; Advani, S.J.; Bradley, J.D.; Kataoka, Y.; Vashistha, K.; Yan, S.Y.; Markert, J.M.; Gillespie, G.Y.; Whitley, R.J.; Roizman, B.; et al. The use of a genetically engineered herpes simplex virus (R7020) with ionizing radiation for experimental hepatoma. Gene. Ther. 2002, 9, 75–80. [Google Scholar] [CrossRef] [Green Version]
- Andreansky, S.; Soroceanu, L.; Flotte, E.R.; Chou, J.; Markert, J.M.; Gillespie, G.Y.; Roizman, B.; Whitley, R.J. Evaluation of genetically engineered herpes simplex viruses as oncolytic agents for human malignant brain tumors. Cancer Res. 1997, 57, 1502–1509. [Google Scholar] [PubMed]
- Bennett, J.J.; Delman, K.A.; Burt, B.M.; Mariotti, A.; Malhotra, S.; Zager, J.; Petrowsky, H.; Mastorides, S.; Federoff, H.; Fong, Y. Comparison of safety, delivery, and efficacy of two oncolytic herpes viruses (G207 and NV1020) for peritoneal cancer. Cancer Gene. Ther. 2002, 9, 935–945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chou, J.; Poon, A.P.; Johnson, J.; Roizman, B. Differential response of human cells to deletions and stop codons in the gamma(1)34.5 gene of herpes simplex virus. J. Virol. 1994, 68, 8304–8311. [Google Scholar] [CrossRef] [Green Version]
- De Queiroz, N.M.G.P.; Xia, T.; Konno, H.; Barber, G.N. Ovarian Cancer Cells Commonly Exhibit Defective STING Signaling Which Affects Sensitivity to Viral Oncolysis. Mol. Cancer Res. 2019, 17, 974–986. [Google Scholar] [CrossRef] [PubMed]
- Meignier, B.; Longnecker, R.; Roizman, B. In Vivo behavior of genetically engineered herpes simplex viruses R7017 and R7020: Construction and evaluation in rodents. J. Infect. Dis. 1988, 158, 602–614. [Google Scholar] [CrossRef] [PubMed]
- Mineta, T.; Rabkin, S.D.; Yazaki, T.; Hunter, W.D.; Martuza, R.L. Attenuated multi-mutated herpes simplex virus-1 for the treatment of malignant gliomas. Nat. Med. 1995, 1, 938–943. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.; Kuncheria, L.; Roulstone, V.; Kyula, J.N.; Mansfield, D.; Bommareddy, P.K.; Smith, H.; Kaufman, H.L.; Harrington, K.J.; Coffin, R.S. Development of a new fusion-enhanced oncolytic immunotherapy platform based on herpes simplex virus type 1. J. Immunother. Cancer 2019, 7, 214. [Google Scholar] [CrossRef] [Green Version]
- Peters, C.; Grandi, P.; Nigim, F. Updates on Oncolytic Virus Immunotherapy for Cancers. Mol. Ther. Oncolytics 2019, 12, 259–262. [Google Scholar] [CrossRef] [Green Version]
- Patel, D.M.; Foreman, P.M.; Nabors, L.B.; Riley, K.O.; Gillespie, G.Y.; Markert, J.M. Design of a Phase I Clinical Trial to Evaluate M032, a Genetically Engineered HSV-1 Expressing IL-12, in Patients with Recurrent/Progressive Glioblastoma Multiforme, Anaplastic Astrocytoma, or Gliosarcoma. Hum. Gene. Ther. Clin. Dev. 2016, 27, 69–78. [Google Scholar] [CrossRef]
- Chouljenko, D.V.; Ding, J.; Lee, I.F.; Murad, Y.M.; Bu, X.; Liu, G.; Delwar, Z.; Sun, Y.; Yu, S.; Samudio, I.; et al. Induction of Durable Antitumor Response by a Novel Oncolytic Herpesvirus Expressing Multiple Immunomodulatory Transgenes. Biomedicines 2020, 8, 484. [Google Scholar] [CrossRef] [PubMed]
- Haines, B.B.; Denslow, A.; Grzesik, P.; Lee, J.S.; Farkaly, T.; Hewett, J.; Wambua, D.; Kong, L.; Behera, P.; Jacques, J.; et al. ONCR-177, an Oncolytic HSV-1 Designed to Potently Activate Systemic Antitumor Immunity. Cancer Immunol. Res. 2021, 9, 291–308. [Google Scholar] [CrossRef]
- Liu, B.L.; Robinson, M.; Han, Z.Q.; Branston, R.H.; English, C.; Reay, P.; McGrath, Y.; Thomas, S.K.; Thornton, M.; Bullock, P.; et al. ICP34.5 deleted herpes simplex virus with enhanced oncolytic, immune stimulating, and anti-tumour properties. Gene Ther. 2003, 10, 292–303. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Q.; Zhang, W.; Ning, Z.; Zhuang, X.; Lu, H.; Liang, J.; Li, J.; Zhang, Y.; Dong, Y.; Zhang, Y.; et al. A Novel Oncolytic Herpes Simplex Virus Type 2 Has Potent Anti-Tumor Activity. PLoS ONE 2014, 9, e93103. [Google Scholar] [CrossRef] [PubMed]
- Todo, T.; Martuza, R.L.; Rabkin, S.D.; Johnson, P.A. Oncolytic herpes simplex virus vector with enhanced MHC class I presentation and tumor cell killing. Proc. Natl. Acad. Sci. USA 2001, 98, 6396–6401. [Google Scholar] [CrossRef] [Green Version]
- Cassady, K.A. Human cytomegalovirus TRS1 and IRS1 gene products block the double-stranded-RNA-activated host protein shutoff response induced by herpes simplex virus type 1 infection. J. Virol. 2005, 79, 8707–8715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiocca, E.A.; Nakashima, H.; Kasai, K.; Fernandez, S.A.; Oglesbee, M. Preclinical Toxicology of rQNestin34.5v.2: An Oncolytic Herpes Virus with Transcriptional Regulation of the ICP34.5 Neurovirulence Gene. Mol. Ther. Methods Clin. Dev. 2020, 17, 871–893. [Google Scholar] [CrossRef]
- Johnson, P.A.; MacLean, C.; Marsden, H.S.; Dalziel, R.G.; Everett, R.D. The product of gene US11 of herpes simplex virus type 1 is expressed as a true late gene. J. Gen. Virol. 1986, 67, 871–883. [Google Scholar] [CrossRef]
- Cassady, K.A.; Gross, M.; Roizman, B. The herpes simplex virus US11 protein effectively compensates for the γ134.5 gene if present before activation of protein kinase R by precluding its phosphorylation and that of the alpha subunit of eukaryotic translation initiation factor 2. J. Virol. 1998, 72, 8620–8626. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Matrenec, R.; Gack, M.U.; He, B. Disassembly of the TRIM23-TBK1 Complex by the Us11 Protein of Herpes Simplex Virus 1 Impairs Autophagy. J. Virol. 2019, 93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xing, J.; Wang, S.; Lin, R.; Mossman, K.L.; Zheng, C. Herpes Simplex Virus 1 Tegument Protein US11 Downmodulates the RLR Signaling Pathway via Direct Interaction with RIG-I and MDA-5. J. Virol. 2012, 86, 3528–3540. [Google Scholar] [CrossRef] [Green Version]
- Mulvey, M.; Camarena, V.; Mohr, I. Full resistance of herpes simplex virus type 1-infected primary human cells to alpha interferon requires both the Us11 and gamma(1)34.5 gene products. J. Virol. 2004, 78, 10193–10196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohr, I.; Gluzman, Y. A herpesvirus genetic element which affects translation in the absence of the viral GADD34 function. EMBO J. 1996, 15, 4759–4766. [Google Scholar] [CrossRef]
- Mohr, I. Neutralizing innate host defenses to control viral translation in HSV-1 infected cells. Int. Rev. Immunol. 2004, 23, 199–220. [Google Scholar] [CrossRef]
- Hardcastle, J.; Kurozumi, K.; Dmitrieva, N.; Sayers, M.P.; Ahmad, S.; Waterman, P.; Weissleder, R.; Chiocca, E.A.; Kaur, B. Enhanced antitumor efficacy of vasculostatin (Vstat120) expressing oncolytic HSV-1. Mol. Ther. 2010, 18, 285–294. [Google Scholar] [CrossRef] [PubMed]
- Hakki, M.; Marshall, E.E.; De Niro, K.L.; Geballe, A.P. Binding and nuclear relocalization of protein kinase R by human cytomegalovirus TRS1. J. Virol. 2006, 80, 11817–11826. [Google Scholar] [CrossRef] [Green Version]
- Cassady, K.A.; Saunders, U.; Shimamura, M. Δγ₁134.5 herpes simplex viruses encoding human cytomegalovirus IRS1 or TRS1 induce interferon regulatory factor 3 phosphorylation and an interferon-stimulated gene response. J. Virol. 2012, 86, 610–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, A.C.; Parker, J.N.; Gillespie, G.Y.; Lakeman, F.D.; Meleth, S.; Markert, J.M.; Cassady, K.A. Enhanced antiglioma activity of chimeric HCMV/HSV-1 oncolytic viruses. Gene. Ther. 2007, 14, 1045–1054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cassady, K.A.; Bauer, D.F.; Roth, J.; Chambers, M.R.; Shoeb, T.; Coleman, J.; Prichard, M.; Gillespie, G.Y.; Markert, J.M. Pre-clinical Assessment of C134, a Chimeric Oncolytic Herpes Simplex Virus, in Mice and Non-human Primates. Mol. Ther. Oncolytics 2017, 5, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Chung, R.Y.; Saeki, Y.; Chiocca, E.A. B-myb promoter retargeting of herpes simplex virus gamma34.5 gene-mediated virulence toward tumor and cycling cells. J. Virol. 1999, 73, 7556–7564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kambara, H.; Okano, H.; Chiocca, E.A.; Saeki, Y. An oncolytic HSV-1 mutant expressing ICP34.5 under control of a nestin promoter increases survival of animals even when symptomatic from a brain tumor. Cancer Res. 2005, 65, 2832–2839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakashima, H.; Nguyen, T.; Kasai, K.; Passaro, C.; Ito, H.; Goins, W.F.; Shaikh, I.; Erdelyi, R.; Nishihara, R.; Nakano, I.; et al. Toxicity and Efficacy of a Novel GADD34-expressing Oncolytic HSV-1 for the Treatment of Experimental Glioblastoma. Clin. Cancer Res. 2018, 24, 2574–2584. [Google Scholar] [CrossRef] [Green Version]
- Adusumilli, P.S.; Chan, M.-K.; Chun, Y.S.; Hezel, M.; Chou, T.-C.; Rusch, V.W.; Fong, Y. Cisplatin-induced GADD34 upregulation potentiates oncolytic viral therapy in the treatment of malignant pleural mesothelioma. Cancer Biol. Ther. 2006, 5, 48–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jarnagin, W.R.; Zager, J.S.; Hezel, M.; Stanziale, S.F.; Adusumilli, P.S.; Gonen, M.; Ebright, M.I.; Culliford, A.; Gusani, N.J.; Fong, Y. Treatment of cholangiocarcinoma with oncolytic herpes simplex virus combined with external beam radiation therapy. Cancer Gene. Ther. 2006, 13, 326–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kennedy, E.M.; Farkaly, T.; Grzesik, P.; Lee, J.; Denslow, A.; Hewett, J.; Bryant, J.; Behara, P.; Goshert, C.; Wambua, D.; et al. Design of an Interferon-Resistant Oncolytic HSV-1 Incorporating Redundant Safety Modalities for Improved Tolerability. Mol. Ther. Oncolytics 2020, 18, 476–490. [Google Scholar] [CrossRef] [PubMed]
- Menotti, L.; Cerretani, A.; Hengel, H.; Campadelli-Fiume, G. Construction of a Fully Retargeted Herpes Simplex Virus 1 Recombinant Capable of Entering Cells Solely via Human Epidermal Growth Factor Receptor 2. J. Virol. 2008, 82, 10153–10161. [Google Scholar] [CrossRef] [Green Version]
- De Lucia, M.; Cotugno, G.; Bignone, V.; Garzia, I.; Nocchi, L.; Langone, F.; Petrovic, B.; Sasso, E.; Pepe, S.; Froechlich, G.; et al. Retargeted and Multi-cytokine-Armed Herpes Virus Is a Potent Cancer Endovaccine for Local and Systemic Anti-tumor Treatment. Mol. Ther. Oncolytics 2020, 19, 253–264. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; He, B. Selective Editing of Herpes Simplex Virus 1 Enables Interferon Induction and Viral Replication That Destroy Malignant Cells. J. Virol. 2019, 93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanai, R.; Zaupa, C.; Sgubin, D.; Antoszczyk, S.J.; Martuza, R.L.; Wakimoto, H.; Rabkin, S.D. Effect of γ34.5 deletions on oncolytic herpes simplex virus activity in brain tumors. J. Virol. 2012, 86, 4420–4431. [Google Scholar] [CrossRef] [Green Version]
- Jing, X.; Cerveny, M.; Yang, K.; He, B. Replication of herpes simplex virus 1 depends on the γ134.5 functions that facilitate virus response to interferon and egress in the different stages of productive infection. J. Virol. 2004, 78, 7653–7666. [Google Scholar] [CrossRef] [Green Version]
- Iwasaki, A.; Medzhitov, R. Control of adaptive immunity by the innate immune system. Nat. Immunol. 2015, 16, 343–353. [Google Scholar] [CrossRef]
- Jin, H.; Ma, Y.; Prabhakar, B.S.; Feng, Z.; Valyi-Nagy, T.; Yan, Z.; Verpooten, D.; Zhang, C.; Cao, Y.; He, B. The γ134.5 protein of herpes simplex virus 1 is required to interfere with dendritic cell maturation during productive infection. J. Virol. 2009, 83, 4984–4994. [Google Scholar] [CrossRef] [Green Version]
- Froechlich, G.; Caiazza, C.; Gentile, C.; D’Alise, A.M.; De Lucia, M.; Langone, F.; Leoni, G.; Cotugno, G.; Scisciola, V.; Nicosia, A.; et al. Integrity of the Antiviral STING-mediated DNA Sensing in Tumor Cells Is Required to Sustain the Immunotherapeutic Efficacy of Herpes Simplex Oncolytic Virus. Cancers 2020, 12, 3407. [Google Scholar] [CrossRef]
- Workenhe, S.T.; Simmons, G.; Pol, J.G.; Lichty, B.D.; Halford, W.P.; Mossman, K.L. Immunogenic HSV-mediated Oncolysis Shapes the Antitumor Immune Response and Contributes to Therapeutic Efficacy. Mol. Ther. 2014, 22, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Falahat, R.; Perez-Villarroel, P.; Mailloux, A.W.; Zhu, G.; Pilon-Thomas, S.; Barber, G.N.; Mulé, J.J. STING Signaling in Melanoma Cells Shapes Antigenicity and Can Promote Antitumor T-cell Activity. Cancer Immunol. Res. 2019, 7, 1837–1848. [Google Scholar] [CrossRef] [PubMed]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [Green Version]
- Barnes, T.A.; Amir, E. HYPE or HOPE: The prognostic value of infiltrating immune cells in cancer. Br. J. Cancer 2017, 117, 451–460. [Google Scholar] [CrossRef] [Green Version]
- Trinchieri, G. Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat. Rev. Immunol. 2003, 3, 133–146. [Google Scholar] [CrossRef]
- Berraondo, P.; Etxeberria, I.; Ponz-Sarvise, M.; Melero, I. Revisiting Interleukin-12 as a Cancer Immunotherapy Agent. Clin. Cancer Res. 2018, 24, 2716–2718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y.; Liu, C.H.; Roberts, A.I.; Das, J.; Xu, G.; Ren, G.; Zhang, Y.; Zhang, L.; Yuan, Z.R.; Tan, H.S.W.; et al. Granulocyte-macrophage colony-stimulating factor (GM-CSF) and T-cell responses: What we do and don’t know. Cell Res. 2006, 16, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Raskov, H.; Orhan, A.; Christensen, J.P.; Gögenur, I. Cytotoxic CD8 + T cells in cancer and cancer immunotherapy. Br. J. Cancer 2021, 124, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Malvehy, J.; Samoylenko, I.; Schadendorf, D.; Gutzmer, R.; Grob, J.-J.; Sacco, J.J.; Gorski, K.S.; Anderson, A.; Pickett, C.A.; Liu, K.; et al. Talimogene laherparepvec upregulates immune-cell populations in non-injected lesions: Findings from a phase II, multicenter, open-label study in patients with stage IIIB-IVM1c melanoma. J. Immunother. Cancer 2021, 9. [Google Scholar] [CrossRef]
- Senzer, N.N.; Kaufman, H.L.; Amatruda, T.; Nemunaitis, M.; Reid, T.; Daniels, G.; Gonzalez, R.; Glaspy, J.; Whitman, E.; Harrington, K.; et al. Phase II Clinical Trial of a Granulocyte-Macrophage Colony-Stimulating Factor–Encoding, Second-Generation Oncolytic Herpesvirus in Patients With Unresectable Metastatic Melanoma. J. Clin. Oncol. 2009, 27, 5763–5771. [Google Scholar] [CrossRef]
- Andtbacka, R.H.I.; Ross, M.; Puzanov, I.; Milhem, M.; Collichio, F.; Delman, K.A.; Amatruda, T.; Zager, J.S.; Cranmer, L.; Hsueh, E.; et al. Patterns of Clinical Response with Talimogene Laherparepvec (T-VEC) in Patients with Melanoma Treated in the OPTiM Phase III Clinical Trial. Ann. Surg. Oncol. 2016, 23, 4169–4177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribas, A.; Dummer, R.; Puzanov, I.; VanderWalde, A.; Andtbacka, R.H.I.; Michielin, O.; Olszanski, A.J.; Malvehy, J.; Cebon, J.; Fernandez, E.; et al. Oncolytic Virotherapy Promotes Intratumoral T Cell Infiltration and Improves Anti-PD-1 Immunotherapy. Cell 2017, 170, 1109–1119.e1110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
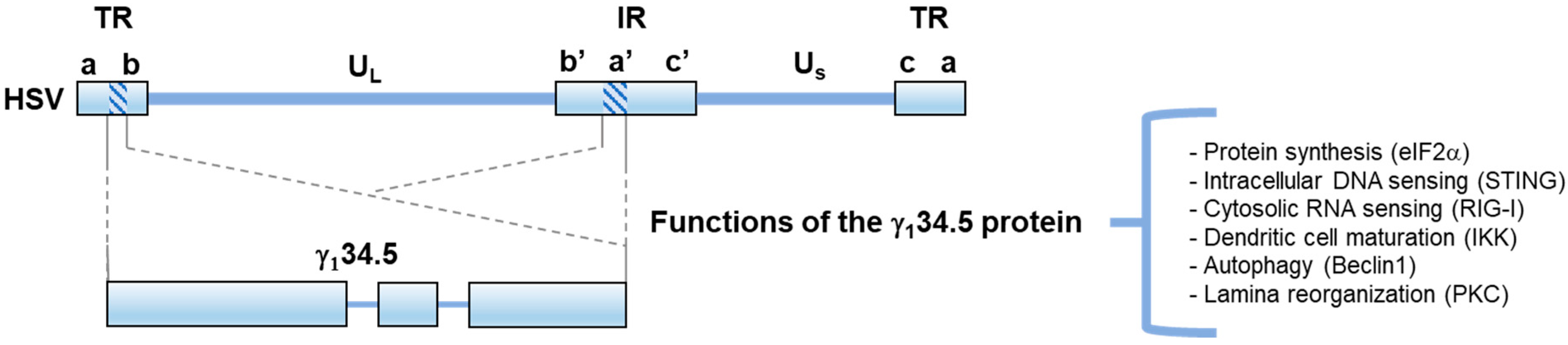
Figure 1.
Schematic diagram of the HSV genome and the location of the γ134.5 gene. The two covalently linked components of HSV-1 DNA, L and S, each consist of unique sequences, UL and US, respectively, flanked by inverted repeats (TR and IR). The reiterated sequences flanking UL, are designated as ab and b’a’, whereas the repeats flanking US, are designated a’c’ and ca. The expanded portions beneath denote the γ134.5 gene, which encodes a virulence factor with a large amino-terminal domain, a variable linker region, and a carboxyl-terminal domain. Known functions of γ134.5 are outlined. The C-terminal domain of γ134.5 mediates the dephosphorylation of host eIF2, preventing shut-off of protein synthesis. The N terminal domain of γ134.5 targets STING and Beclin 1, inhibiting host DNA sensing and autophagy, respectively. Intact γ134.5 is needed to modulate RIG-I, IKK, and PKC, inhibiting RNA sensing, DC maturation, and enabling laminar reorganization/viral egress.
Figure 1.
Schematic diagram of the HSV genome and the location of the γ134.5 gene. The two covalently linked components of HSV-1 DNA, L and S, each consist of unique sequences, UL and US, respectively, flanked by inverted repeats (TR and IR). The reiterated sequences flanking UL, are designated as ab and b’a’, whereas the repeats flanking US, are designated a’c’ and ca. The expanded portions beneath denote the γ134.5 gene, which encodes a virulence factor with a large amino-terminal domain, a variable linker region, and a carboxyl-terminal domain. Known functions of γ134.5 are outlined. The C-terminal domain of γ134.5 mediates the dephosphorylation of host eIF2, preventing shut-off of protein synthesis. The N terminal domain of γ134.5 targets STING and Beclin 1, inhibiting host DNA sensing and autophagy, respectively. Intact γ134.5 is needed to modulate RIG-I, IKK, and PKC, inhibiting RNA sensing, DC maturation, and enabling laminar reorganization/viral egress.

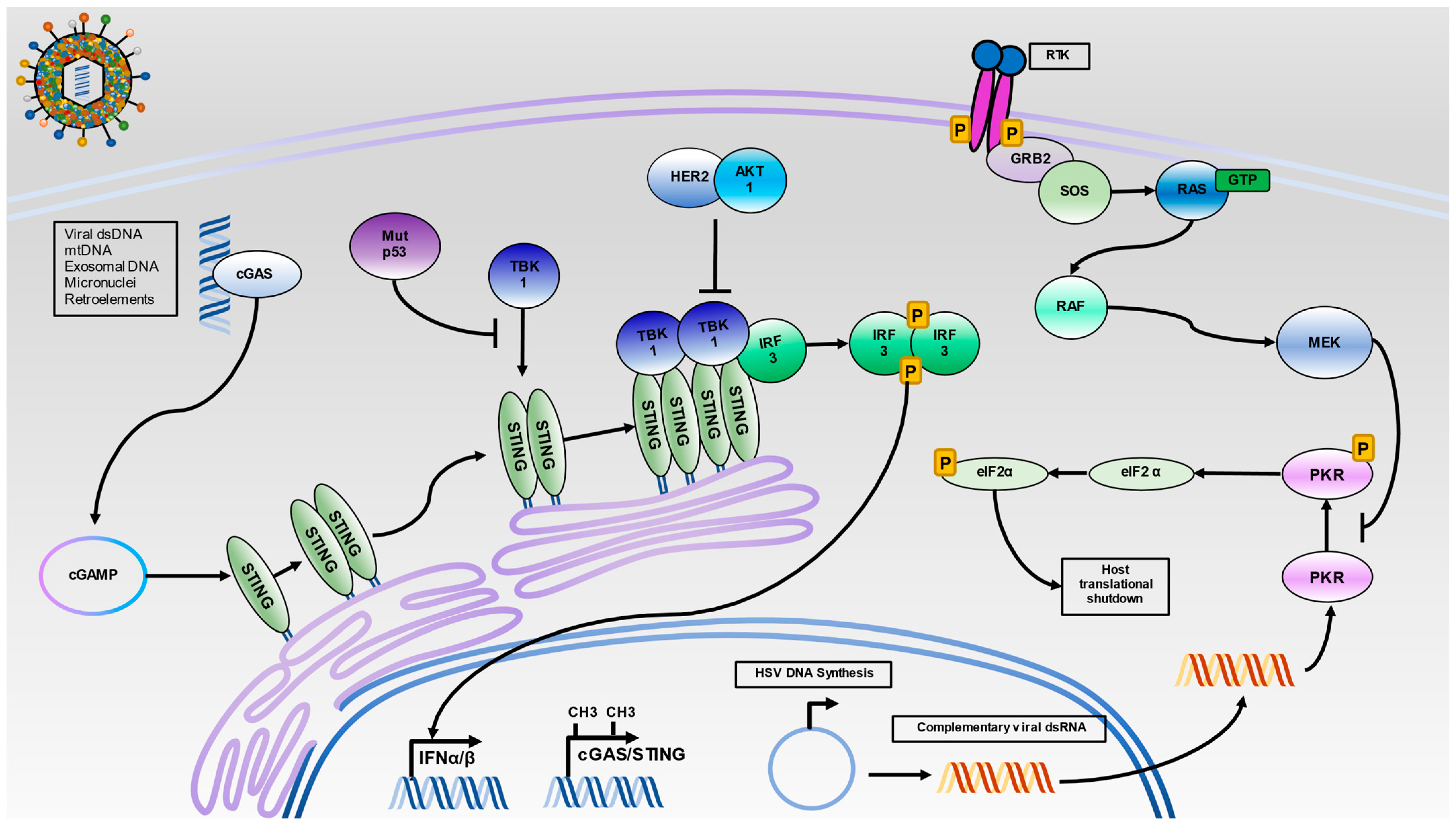
Figure 2.
Intracellular Nucleic Acid Sensing by the STING and PKR Pathways. HSV infection results in the release of capsid into cells where viral dsDNA can activate cGAS. Additionally, dsDNA from several sources, including mitochondrial DNA (mtDNA), exosomal DNA, micronuclei, and reactivated retroelements can serve to activate cGAS. Once activated, cGAS will synthesize the cyclic dinucleotide c-GMP-AMP (cGAMP), which activates STING. Then, STING translocates from the endoplasmic reticulum to the Golgi apparatus. Oligomerized STING facilitates the trans-autophosphorylation of TBK-1 and subsequent recruitment of IRF3 to the STING signaling complex. The phosphorylation of IRF3 results in its dimerization and translocation to the nucleus for initiation of IFNα and IFNβ production. As infection progresses, viral DNA will circularize in the nucleus and undergo rolling circle replication. The onset of viral replication is thought to produce highly complementary viral dsRNA, which can activate PKR. The activation of PKR results in dimerization and trans-autophosphorylation. Then, activated PKR can phosphorylate eIF2α and shut down protein translation. In malignant cells, several mechanisms to silence intracellular nucleic acid sensing may operate. Hypermethylation of cGAS/STING promoters, inhibition of the TBK1-STING interaction by mutant p53, and TBK1 dissociation from STING by HER2 via AKT1 can prevent IRF3 activation. Additionally, unregulated Ras signaling can lead to constitutive activation of MEK1/2, which can inhibit PKR activation.
Figure 2.
Intracellular Nucleic Acid Sensing by the STING and PKR Pathways. HSV infection results in the release of capsid into cells where viral dsDNA can activate cGAS. Additionally, dsDNA from several sources, including mitochondrial DNA (mtDNA), exosomal DNA, micronuclei, and reactivated retroelements can serve to activate cGAS. Once activated, cGAS will synthesize the cyclic dinucleotide c-GMP-AMP (cGAMP), which activates STING. Then, STING translocates from the endoplasmic reticulum to the Golgi apparatus. Oligomerized STING facilitates the trans-autophosphorylation of TBK-1 and subsequent recruitment of IRF3 to the STING signaling complex. The phosphorylation of IRF3 results in its dimerization and translocation to the nucleus for initiation of IFNα and IFNβ production. As infection progresses, viral DNA will circularize in the nucleus and undergo rolling circle replication. The onset of viral replication is thought to produce highly complementary viral dsRNA, which can activate PKR. The activation of PKR results in dimerization and trans-autophosphorylation. Then, activated PKR can phosphorylate eIF2α and shut down protein translation. In malignant cells, several mechanisms to silence intracellular nucleic acid sensing may operate. Hypermethylation of cGAS/STING promoters, inhibition of the TBK1-STING interaction by mutant p53, and TBK1 dissociation from STING by HER2 via AKT1 can prevent IRF3 activation. Additionally, unregulated Ras signaling can lead to constitutive activation of MEK1/2, which can inhibit PKR activation.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Kangas, C.; Krawczyk, E.; He, B. Oncolytic HSV: Underpinnings of Tumor Susceptibility. Viruses 2021, 13, 1408. https://0-doi-org.brum.beds.ac.uk/10.3390/v13071408
AMA Style
Kangas C, Krawczyk E, He B. Oncolytic HSV: Underpinnings of Tumor Susceptibility. Viruses. 2021; 13(7):1408. https://0-doi-org.brum.beds.ac.uk/10.3390/v13071408
Chicago/Turabian StyleKangas, Chase, Eric Krawczyk, and Bin He. 2021. "Oncolytic HSV: Underpinnings of Tumor Susceptibility" Viruses 13, no. 7: 1408. https://0-doi-org.brum.beds.ac.uk/10.3390/v13071408
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.