
Emergence of Salmonid Alphavirus Genotype 2 in Norway—Molecular Characterization of Viral Strains Circulating in Norway and Scotland

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Selection of the Viral Strains
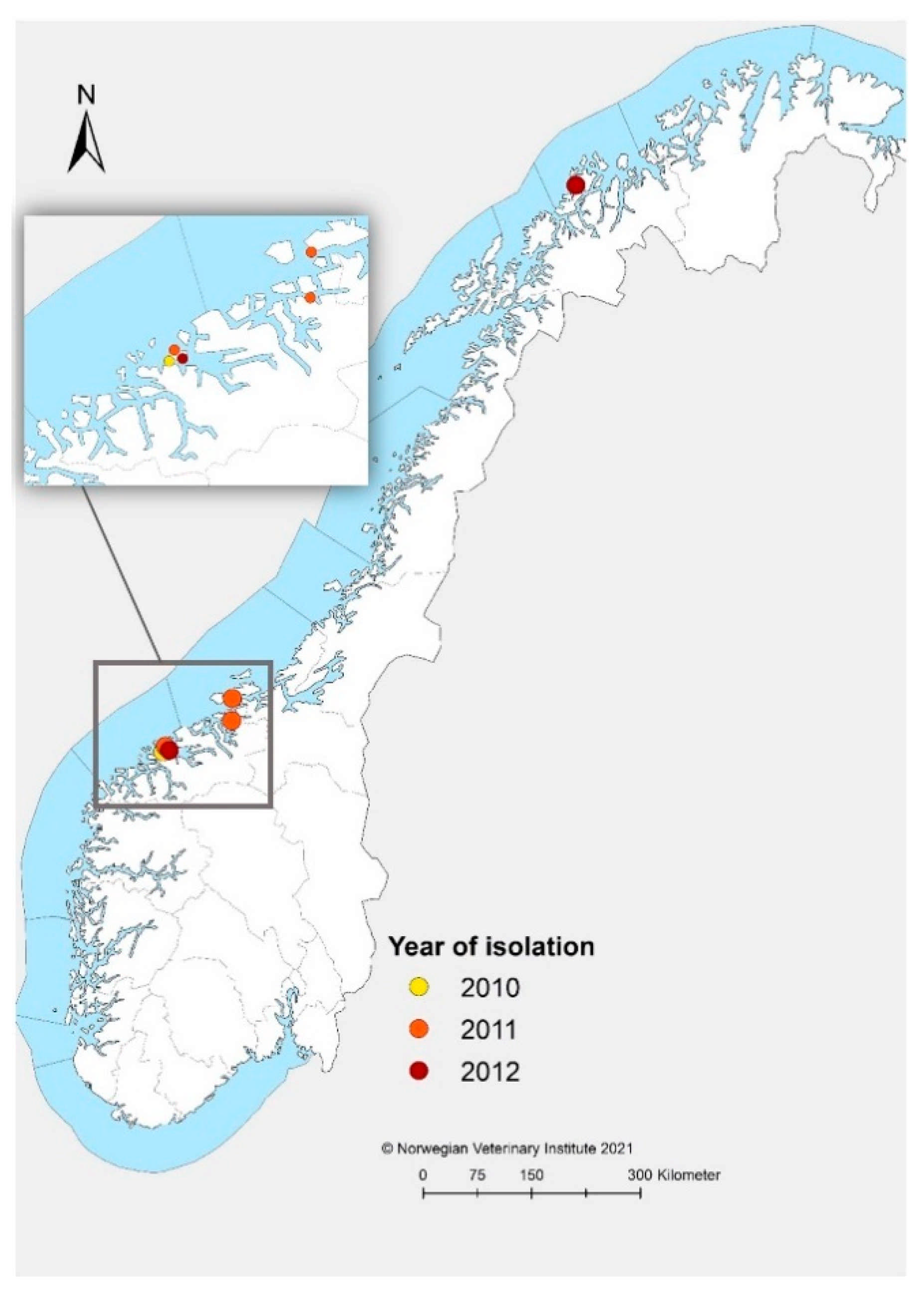
2.2. Origin of Norwegian Strains
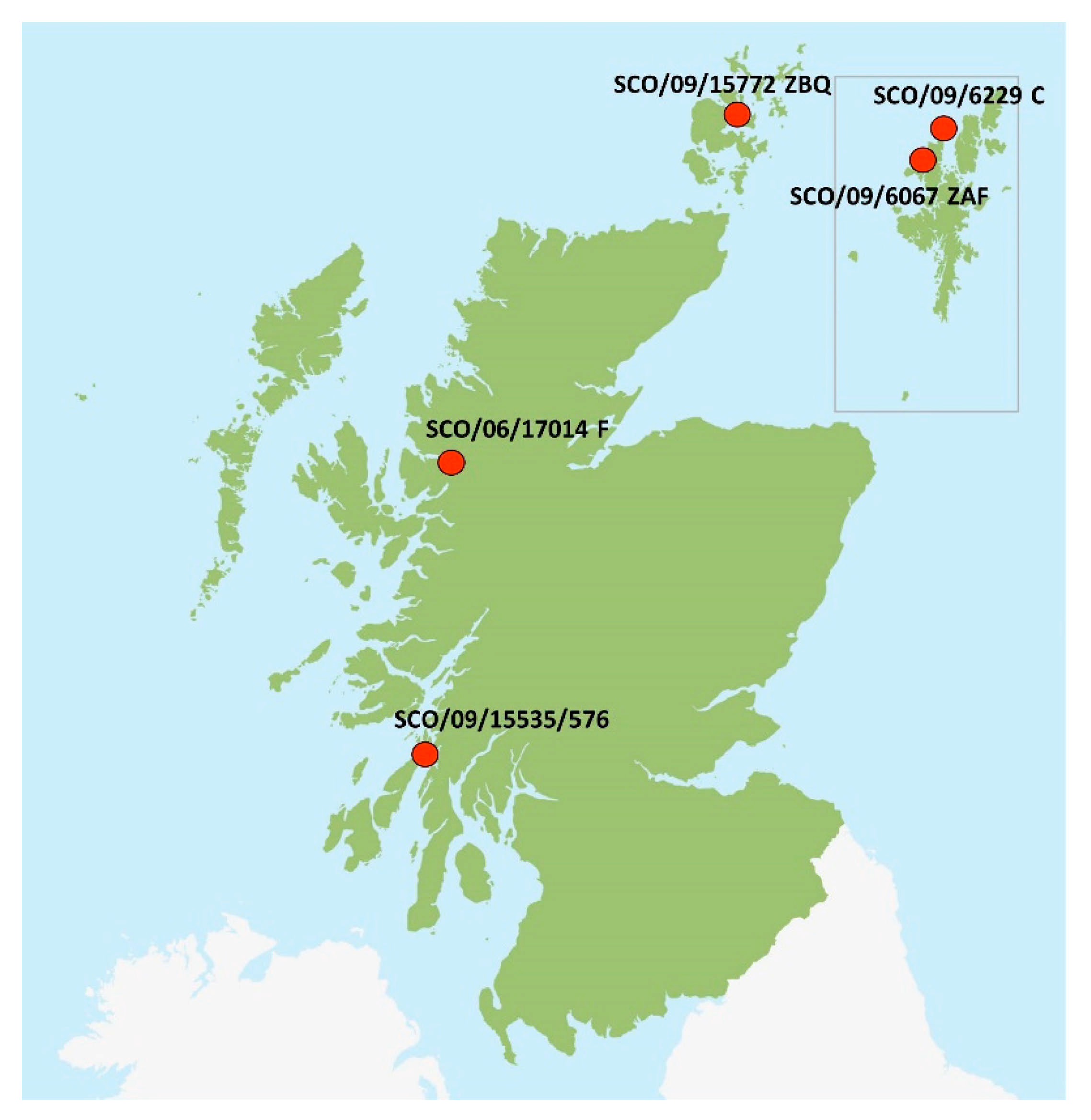
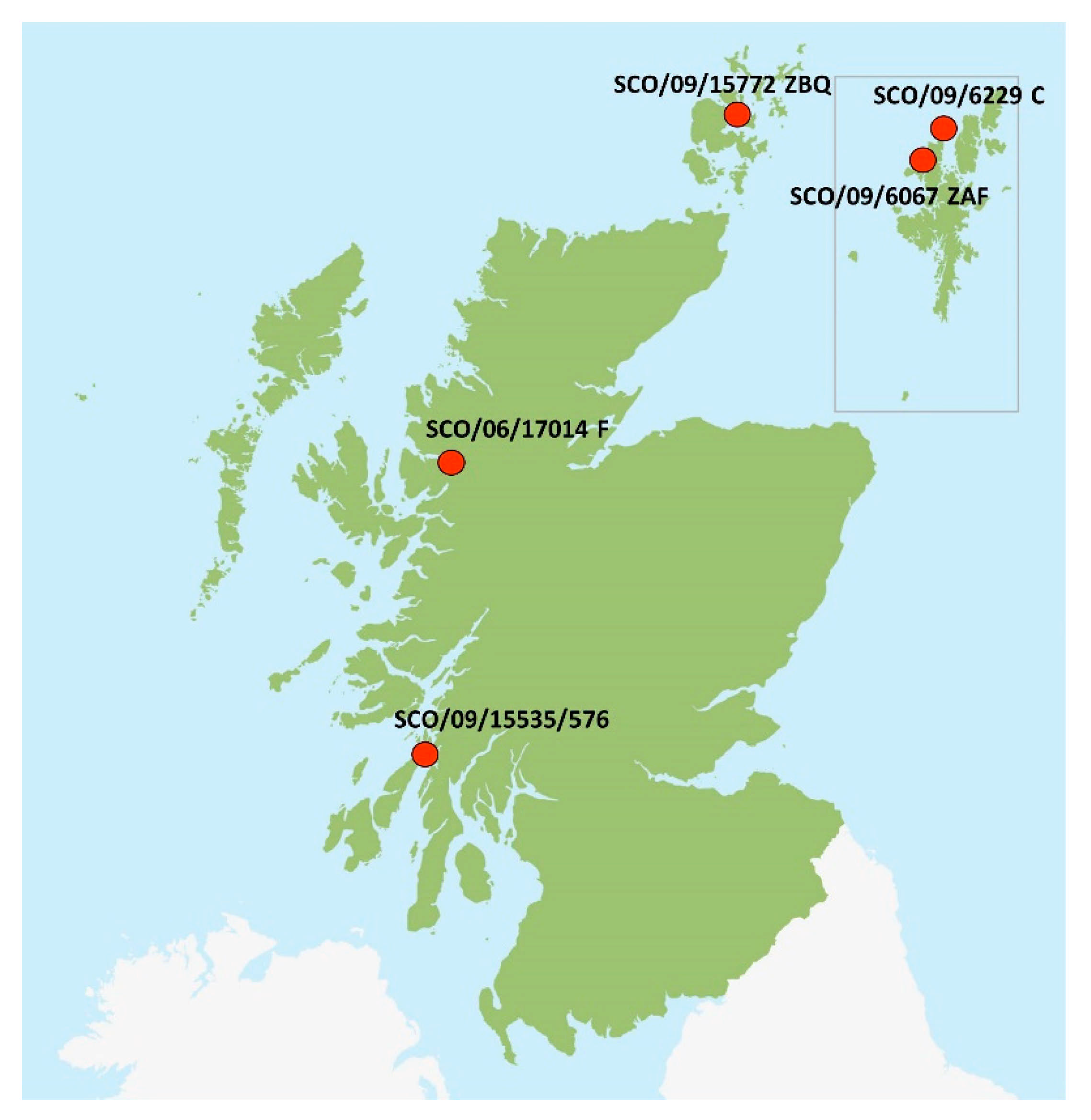
2.3. Origin of Scottish Strains
2.4. Sample Preparation, PCR Amplification of Viral Genome and Sequencing
2.5. Data Analysis
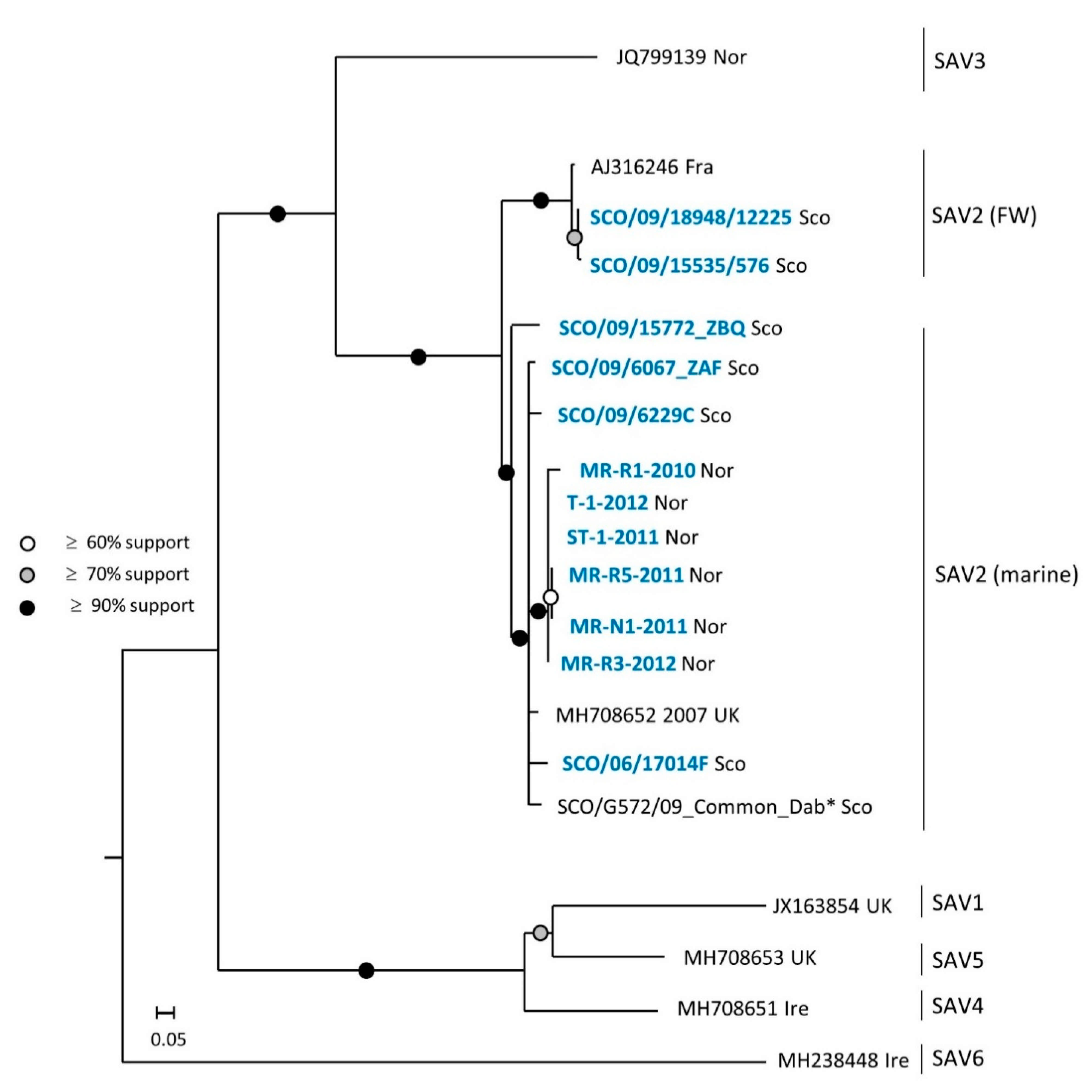
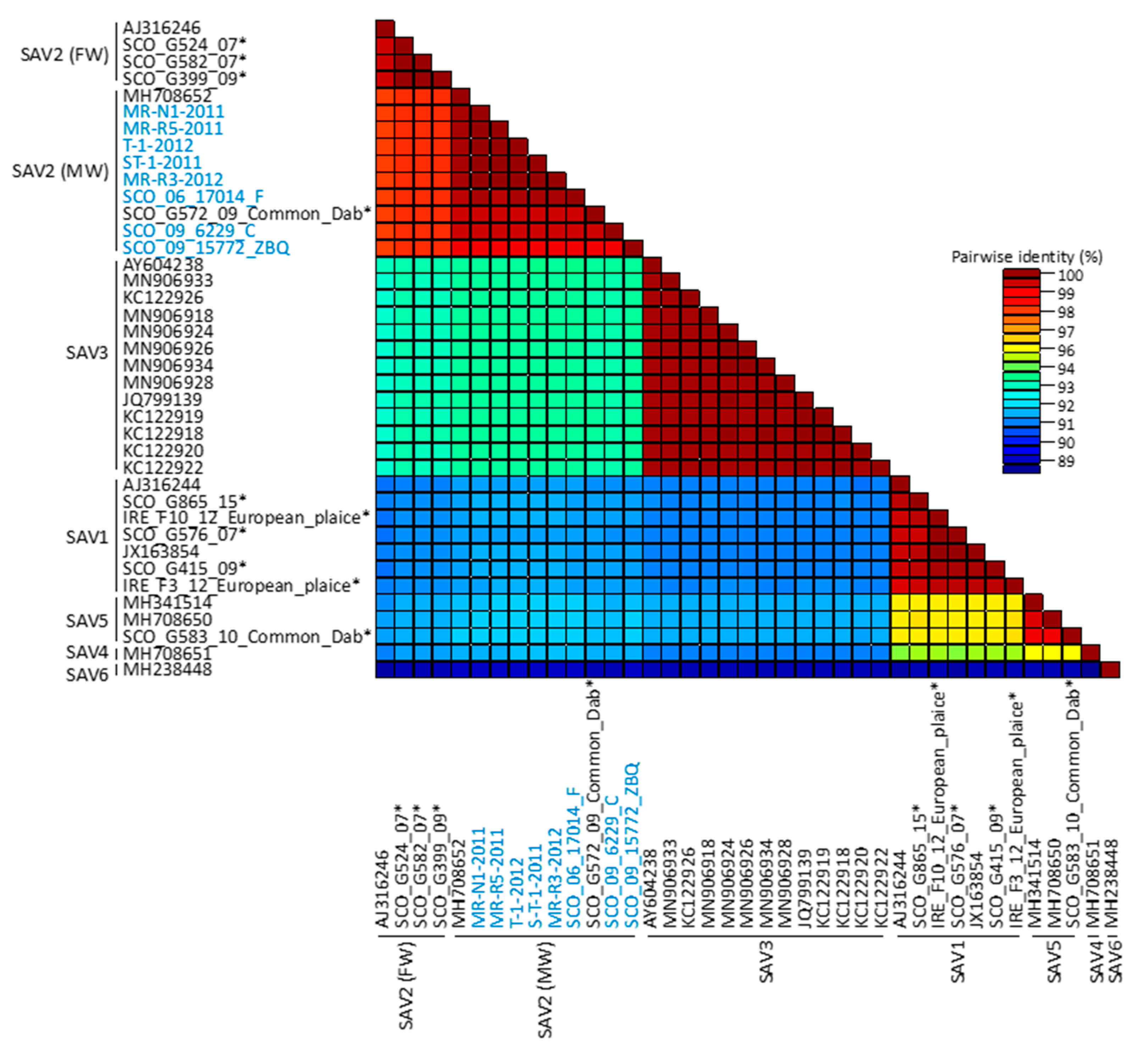
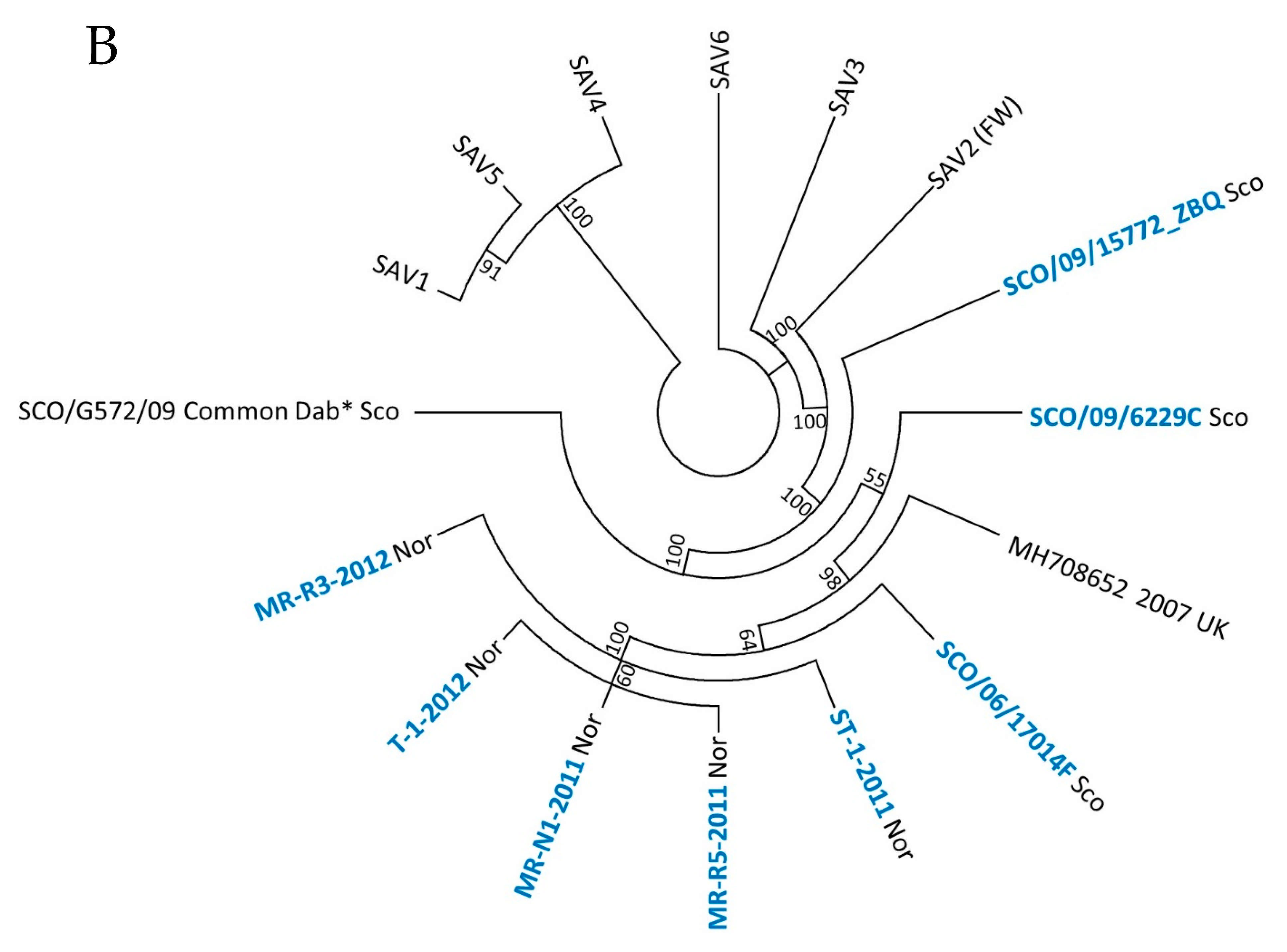
3. Results
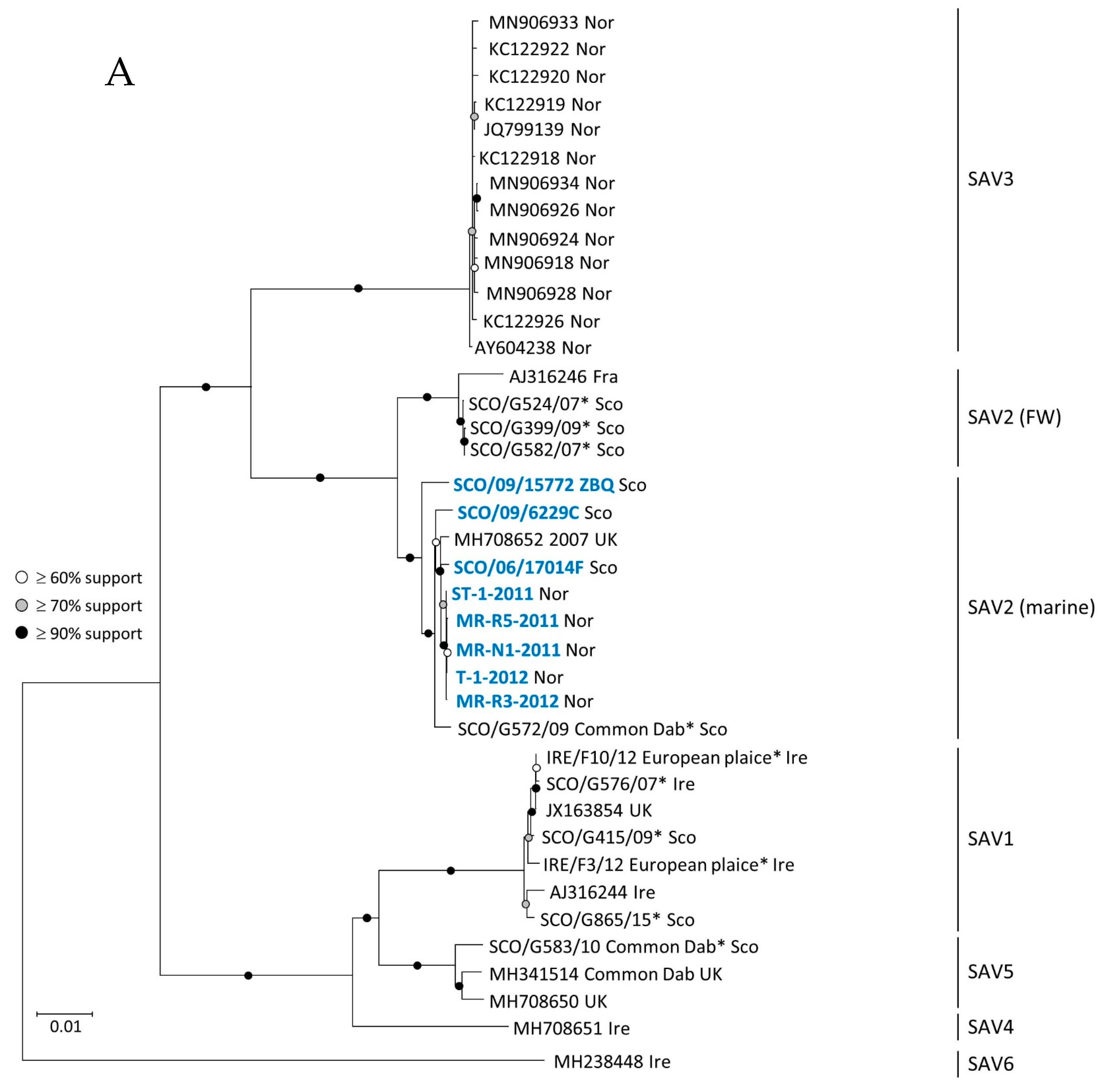
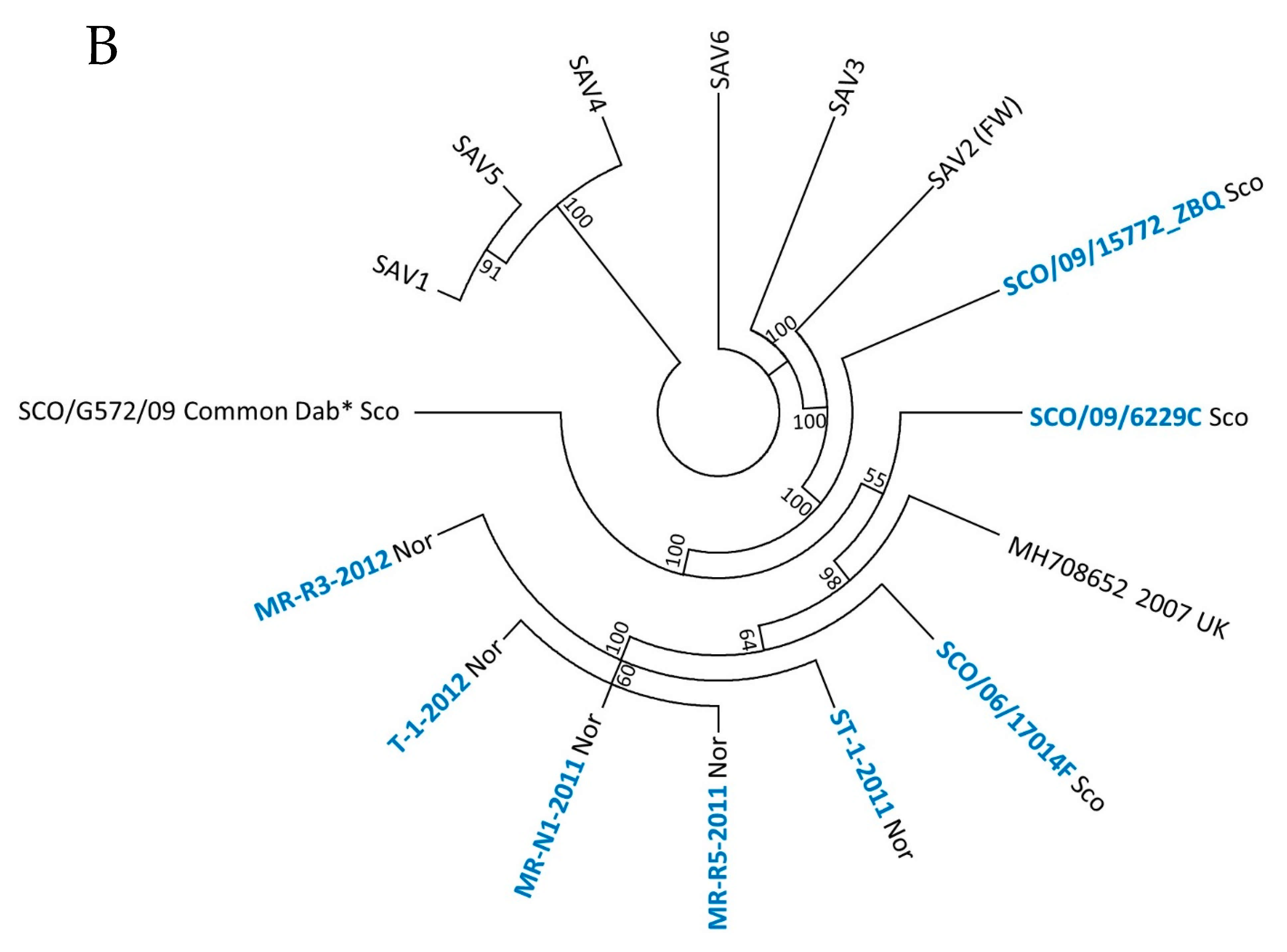
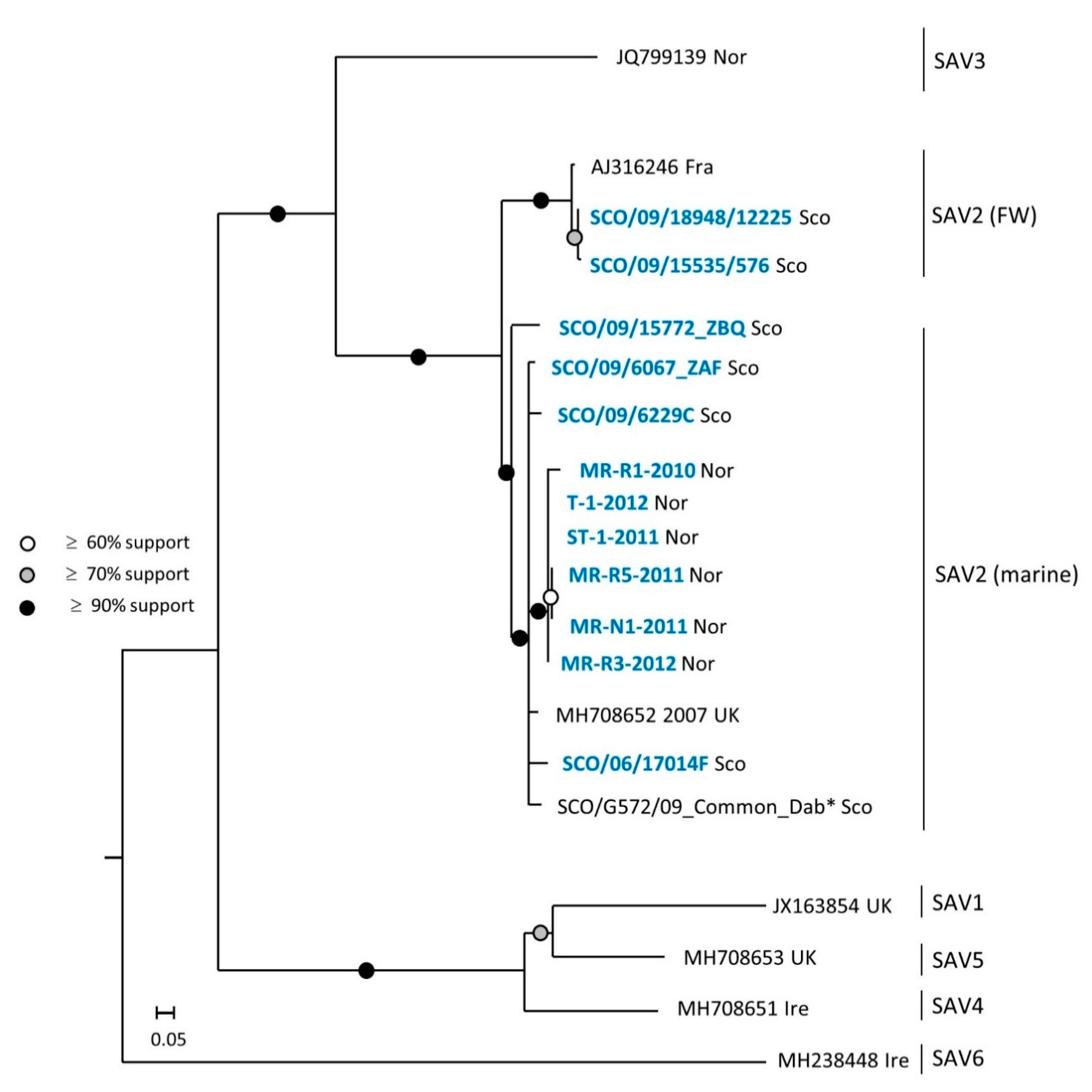
3.1. Phylogeny of SAV
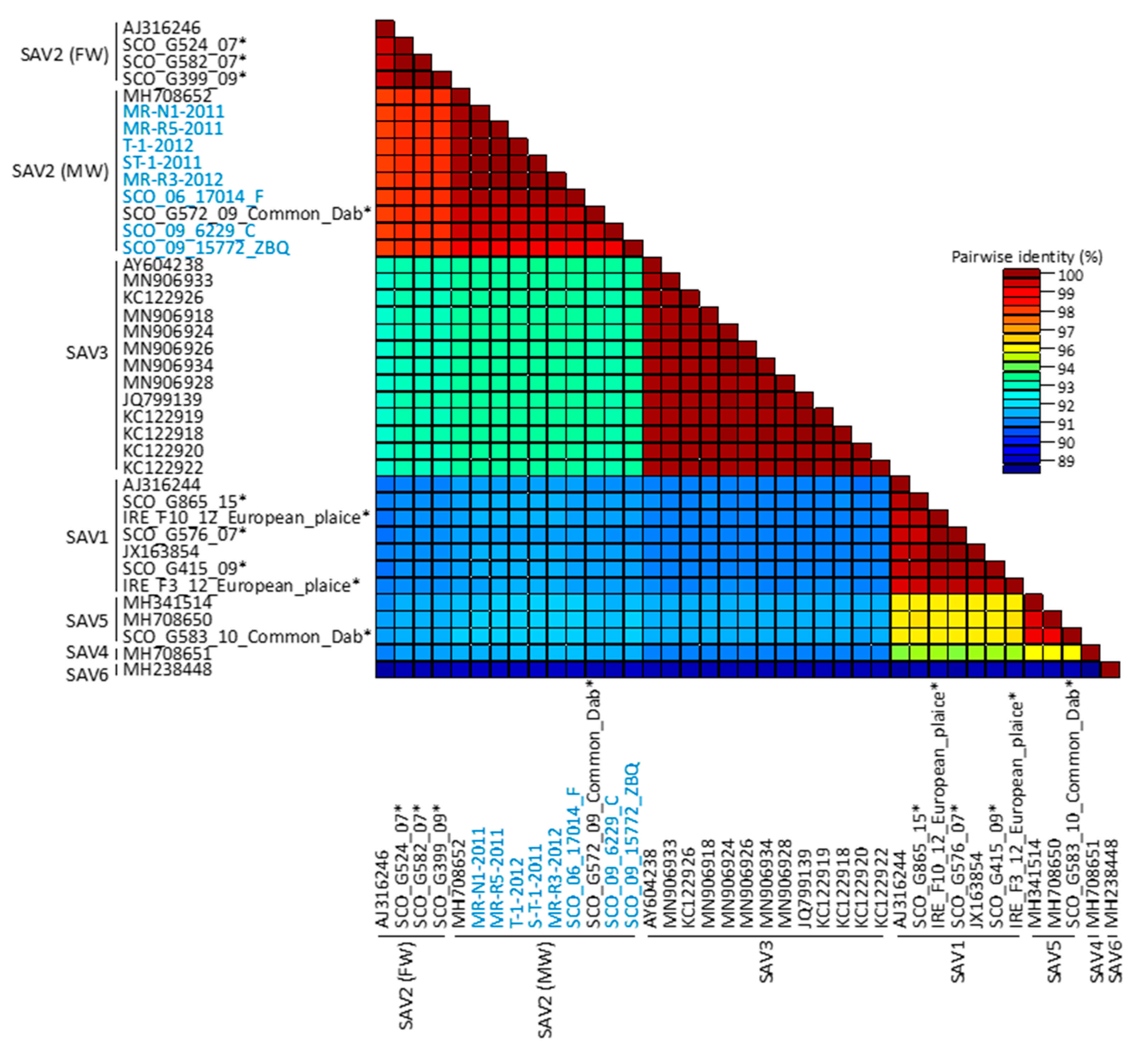
3.2. Genomic Diversity of SAV
3.3. Characteristic of Genomic Deletions in FW SAV2 Strains and SAV3
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- McLoughlin, M.F.; Graham, D.A. Alphavirus infections in salmonids—A review. J. Fish Dis. 2007, 30, 511–531. [Google Scholar] [CrossRef]
- Aunsmo, A.; Valle, P.S.; Sandberg, M.; Midtlyng, P.J.; Bruheim, T. Stochastic modelling of direct costs of pancreas disease (PD) in Norwegian farmed Atlantic salmon (Salmo salar L.). Prev. Vet. Med. 2010, 93, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Sommerset, I.; Walde, C.S.; Bang Jensen, B.; Borno, G.; Haukaas, A.; Brun, E. The Health Situation in Norwegian Aquaculture 2019; Norwegian Veterinary Institute: Oslo, Norway, 2020. [Google Scholar]
- Nelson, R.T.; McLoughlin, M.F.; Rowley, H.M.; Platten, M.A.; McCormick, J.I. Isolation of a toga-like virus from farmed Atlantic salmon Salmo salar with pancreas disease. Dis. Aquat. Org. 1995, 22, 23–32. [Google Scholar] [CrossRef] [Green Version]
- Weston, J.; Villoing, S.; Bremont, M.; Castric, J.; Pfeffer, M.; Jewhurst, V.; McLoughlin, M.; Rodseth, O.; Christie, K.E.; Koumans, J.; et al. Comparison of two aquatic alphaviruses, salmon pancreas disease virus and sleeping disease virus, by using genome sequence analysis, monoclonal reactivity, and cross-infection. J. Virol. 2002, 76, 6155–6163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fringuelli, E.; Rowley, H.M.; Wilson, J.C.; Hunter, R.; Rodger, H.; Graham, D.A. Phylogenetic analyses and molecular epidemiology of European salmonid alphaviruses (SAV) based on partial E2 and nsP3 gene nucleotide sequences. J. Fish Dis. 2008, 31, 811–823. [Google Scholar] [CrossRef]
- OIE. Aquatic Manual, Chapter 2. 3. 6. Available online: https://www.oie.int/en/what-we-do/standards/codes-and-manuals/aquatic-manual-online-access/?id=169&L=1&htmfile=chapitre_salmonid_alphavirus.htm (accessed on 2 August 2021).
- Hjortaas, M.J.; Skjelstad, H.R.; Taksdal, T.; Olsen, A.B.; Johansen, R.; Bang-Jensen, B.; Orpetveit, I.; Sindre, H. The first detections of subtype 2-related salmonid alphavirus (SAV2) in Atlantic salmon, Salmo salar L., in Norway. J. Fish Dis. 2013, 36, 71–74. [Google Scholar] [CrossRef] [PubMed]
- Hodneland, K.; Bratland, A.; Christie, K.E.; Endresen, C.; Nylund, A. New subtype of salmonid alphavirus (SAV), Togaviridae, from Atlantic salmon Salmo salar and rainbow trout Oncorhynchus mykiss in Norway. Dis. Aquat. Org. 2005, 66, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Graham, D.A.; Fringuelli, E.; Rowley, H.M.; Cockerill, D.; Cox, D.I.; Turnbull, T.; Rodger, H.; Morris, D.; Mc Loughlin, M.F. Geographical distribution of salmonid alphavirus subtypes in marine farmed Atlantic salmon, Salmo salar L., in Scotland and Ireland. J. Fish Dis. 2012, 35, 755–765. [Google Scholar] [CrossRef]
- Gallagher, M.D.; Matejusova, I.; Ruane, N.M.; Macqueen, D.J. Genome-wide target enriched viral sequencing reveals extensive ‘hidden’ salmonid alphavirus diversity in farmed and wild fish populations. Aquaculture 2020, 522, 735117. [Google Scholar] [CrossRef]
- Bruno, D.W.; Noguera, P.A.; Black, J.; Murray, W.; Macqueen, D.J.; Matejusova, I. Identification of a wild reservoir of salmonid alphavirus in common dab Limanda limanda, with emphasis on virus culture and sequencing. Aquac. Environ. Interact. 2014, 5, 89–98. [Google Scholar] [CrossRef] [Green Version]
- Snow, M.; Black, J.; Matejusova, I.; McIntosh, R.; Baretto, E.; Wallace, I.S.; Bruno, D.W. Detection of salmonid alphavirus RNA in wild marine fish: Implications for the origins of salmon pancreas disease in aquaculture. Dis. Aquat. Org. 2010, 91, 177–188. [Google Scholar] [CrossRef]
- Tighe, A.J.; Gallagher, M.D.; Carlsson, J.; Matejusova, I.; Swords, F.; Macqueen, D.J.; Ruane, N.M. Nanopore whole genome sequencing and partitioned phylogenetic analysis supports a new salmonid alphavirus genotype (SAV7). Dis. Aquat. Org. 2020, 142, 203–211. [Google Scholar] [CrossRef]
- Ruane, N.M.; Swords, D.; Morrissey, T.; Geary, M.; Hickey, C.; Collins, E.M.; Geoghegan, F.; Swords, F. Isolation of salmonid alphavirus subtype 6 from wild-caught ballan wrasse, Labrus bergylta (Ascanius). J. Fish Dis. 2018, 41, 1643–1651. [Google Scholar] [CrossRef] [PubMed]
- Boucher, P.; Laurencin, F.B. Sleeping disease and pancreas disease: Comparative histopathology and acquired cross-protection. J. Fish Dis. 1996, 19, 303–310. [Google Scholar] [CrossRef]
- Hjortaas, M.J.; Jensen, B.B.; Taksdal, T.; Olsen, A.B.; Lillehaug, A.; Trettenes, E.; Sindre, H. Genetic characterization of salmonid alphavirus in Norway. J. Fish Dis. 2016, 39, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Grad, Y.H.; Kirkcaldy, R.D.; Trees, D.; Dordel, J.; Harris, S.R.; Goldstein, E.; Weinstock, H.; Parkhill, J.; Hanage, W.P.; Bentley, S.; et al. Genomic epidemiology of Neisseria gonorrhoeae with reduced susceptibility to cefixime in the USA: A retrospective observational study. Lancet Infect. Dis. 2014, 14, 220–226. [Google Scholar] [CrossRef] [Green Version]
- Faria, N.R.; Rambaut, A.; Suchard, M.A.; Baele, G.; Bedford, T.; Ward, M.J.; Tatem, A.J.; Sousa, J.D.; Arinaminpathy, N.; Pépin, J.; et al. HIV epidemiology. The early spread and epidemic ignition of HIV-1 in human populations. Science 2014, 346, 56–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Worobey, M.; Pekar, J.; Larsen, B.B.; Nelson, M.I.; Hill, V.; Joy, J.B.; Rambaut, A.; Suchard, M.A.; Wertheim, J.O.; Lemey, P. The emergence of SARS-CoV-2 in Europe and North America. Science 2020, 370, 564–570. [Google Scholar] [CrossRef]
- Weston, J.H.; Graham, D.A.; Branson, E.; Rowley, H.M.; Walker, I.W.; Jewhurst, V.A.; Jewhurst, H.L.; Todd, D. Nucleotide sequence variation in salmonid alphaviruses from outbreaks of salmon pancreas disease and sleeping disease. Dis. Aquat. Org. 2005, 66, 105–111. [Google Scholar] [CrossRef] [Green Version]
- Jansen, M.D.; Taksdal, T.; Wasmuth, M.A.; Gjerset, B.; Brun, E.; Olsen, A.B.; Breck, O.; Sandberg, M. Salmonid alphavirus (SAV) and pancreas disease (PD) in Atlantic salmon, Salmo salar L., in freshwater and seawater sites in Norway from 2006 to 2008. J. Fish Dis. 2010, 33, 391–402. [Google Scholar] [CrossRef]
- Karlsen, M.; Gjerset, B.; Hansen, T.; Rambaut, A. Multiple introductions of salmonid alphavirus from a wild reservoir have caused independent and self-sustainable epizootics in aquaculture. J. Gen. Virol. 2014, 95, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, M.D.; Karlsen, M.; Petterson, E.; Haugland, Ø.; Matejusova, I.; Macqueen, D.J. Genome Sequencing of SAV3 Reveals Repeated Seeding Events of Viral Strains in Norwegian Aquaculture. Front. Microbiol. 2020, 11, 740. [Google Scholar] [CrossRef]
- Gallagher, M.D.; Matejusova, I.; Nguyen, L.; Ruane, N.M.; Falk, K.; Macqueen, D.J. Nanopore sequencing for rapid diagnostics of salmonid RNA viruses. Sci. Rep. 2018, 8, 16307. [Google Scholar] [CrossRef] [Green Version]
- Petterson, E.; Stormoen, M.; Evensen, Ø.; Mikalsen, A.B.; Haugland, Ø. Natural infection of Atlantic salmon (Salmo salar L.) with salmonid alphavirus 3 generates numerous viral deletion mutants. J. Gen. Virol. 2013, 94, 1945–1954. [Google Scholar] [CrossRef]
- Jansen, M.D.; Wasmuth, M.A.; Olsen, A.B.; Gjerset, B.; Modahl, I.; Breck, O.; Haldorsen, R.N.; Hjelmeland, R.; Taksdal, T. Pancreas disease (PD) in sea-reared Atlantic salmon, Salmo salar L., in Norway; a prospective, longitudinal study of disease development and agreement between diagnostic test results. J. Fish Dis. 2010, 33, 723–736. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Dudley, J.; Nei, M.; Kumar, S. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol. Biol. Evol. 2007, 24, 1596–1599. [Google Scholar] [CrossRef]
- Guindon, S.; Dufayard, J.-F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New Algorithms and Methods to Estimate Maximum-Likelihood Phylogenies: Assessing the Performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015, 1, vev003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murray, A.G.; Smith, R.J.; Stagg, R.M. Shipping and the spread of infectious salmon anemia in Scottish aquaculture. Emerg. Infect. Dis. 2002, 8, 1–5. [Google Scholar] [CrossRef]
- Götte, B.; Liu, L.; McInerney, G.M. The Enigmatic Alphavirus Non-Structural Protein 3 (nsP3) Revealing Its Secrets at Last. Viruses 2018, 10, 105. [Google Scholar] [CrossRef] [Green Version]
- Strauss, E.G.; Levinson, R.; Rice, C.M.; Dalrymple, J.; Strauss, J.H. Nonstructural proteins nsP3 and nsP4 of Ross River and O’Nyong-nyong viruses: Sequence and comparison with those of other alphaviruses. Virology 1988, 164, 265–274. [Google Scholar] [CrossRef]
- Neuvonen, M.; Kazlauskas, A.; Martikainen, M.; Hinkkanen, A.; Ahola, T.; Saksela, K. SH3 domain-mediated recruitment of host cell amphiphysins by alphavirus nsP3 promotes viral RNA replication. PLoS Pathog. 2011, 7, e1002383. [Google Scholar] [CrossRef] [Green Version]
- LaStarza, M.W.; Lemm, J.A.; Rice, C.M. Genetic analysis of the nsP3 region of Sindbis virus: Evidence for roles in minus-strand and subgenomic RNA synthesis. J. Virol. 1994, 68, 5781–5791. [Google Scholar] [CrossRef] [Green Version]
- Galbraith, S.E.; Sheahan, B.J.; Atkins, G.J. Deletions in the hypervariable domain of the nsP3 gene attenuate Semliki Forest virus virulence. J. Gen. Virol. 2006, 87, 937–947. [Google Scholar] [CrossRef] [PubMed]
- Davis, N.L.; Willis, L.V.; Smith, J.F.; Johnston, R.E. In vitro synthesis of infectious venezuelan equine encephalitis virus RNA from a cDNA clone: Analysis of a viable deletion mutant. Virology 1989, 171, 189–204. [Google Scholar] [CrossRef]
- Panas, M.D.; Schulte, T.; Thaa, B.; Sandalova, T.; Kedersha, N.; Achour, A.; McInerney, G.M. Viral and Cellular Proteins Containing FGDF Motifs Bind G3BP to Block Stress Granule Formation. PLoS Pathog. 2015, 11, e1004659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulte, T.; Liu, L.; Panas, M.D.; Thaa, B.; Dickson, N.; Götte, B.; Achour, A.; McInerney, G.M. Combined structural, biochemical and cellular evidence demonstrates that both FGDF motifs in alphavirus nsP3 are required for efficient replication. Open Biol. 2016, 6, 160078. [Google Scholar] [CrossRef] [Green Version]
- Ari, Ş.; Arikan, M. Next-Generation Sequencing: Advantages, Disadvantages, and Future. In Plant Omics: Trends and Applications; Hakeem, K.R., Tombuloğlu, H., Tombuloğlu, G., Eds.; Springer International Publishing: Cham, Switzerland, 2016; pp. 109–135. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain | Geographic Origin | Date of Sampling | Host | Site | Acc No | Genotype | Sequence Length (nt) |
|---|---|---|---|---|---|---|---|
| Norwegian | |||||||
| MR-R1-2010 | Møre and Romsdal | June 2010 | A. salmon | Marine | * | Marine SAV2 | 2252 and 990 |
| MR-R5-2011 | Møre and Romsdal | Oct 2011 | A. salmon | Marine | MZ395641 | Marine SAV2 | 11,766 |
| MR-N1-2011 | Møre and Romsdal | Dec 2011 | A. salmon | Marine | MZ395642 | Marine SAV2 | 11,658 |
| ST-1-2011 | Trøndelag | Dec 2011 | A. salmon | Marine | MZ395643 | Marine SAV2 | 11,719 |
| T-1-2012 | Troms | Jan 2012 | A. salmon | Marine | MZ395644 | Marine SAV2 | 11,753 |
| MR-R3-2012 | Møre and Romsdal | June 2012 | A. salmon | Marine | MZ395645 | Marine SAV2 | 11,751 |
| Scottish | |||||||
| SCO/09/6067 ZAF | West Shetland Island | 2009 | A. salmon | Marine | MZ395646 | Marine SAV2 | 8575 |
| SCO/09/6229C | North Shetland Island | 2009 | A. salmon | Marine | MZ395647 | Marine SAV2 | 11,594 |
| SCO/09/15772 ZBQ | Orkney | 2009 | A. salmon | Marine | MZ395648 | Marine SAV2 | 11,409 |
| SCO/06/17014F | Northwest Scotland | 2006 | A. salmon | Marine | MZ395649 | Marine SAV2 | 11,595 |
| SCO/09/15535/576 | Argyll and Bute | 2009 | A. salmon | Marine | MZ395650 | FW SAV2 | 1770 |
| SCO/09/18948/12225 | Unknown | 2002 | R. trout | Fresh water | MZ395651 | FW SAV2 | 1912 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hjortaas, M.J.; Fringuelli, E.; Monjane, A.L.; Mikalsen, A.B.; Jonassen, C.M.; Savage, P.; Sindre, H. Emergence of Salmonid Alphavirus Genotype 2 in Norway—Molecular Characterization of Viral Strains Circulating in Norway and Scotland. Viruses 2021, 13, 1556. https://0-doi-org.brum.beds.ac.uk/10.3390/v13081556
Hjortaas MJ, Fringuelli E, Monjane AL, Mikalsen AB, Jonassen CM, Savage P, Sindre H. Emergence of Salmonid Alphavirus Genotype 2 in Norway—Molecular Characterization of Viral Strains Circulating in Norway and Scotland. Viruses. 2021; 13(8):1556. https://0-doi-org.brum.beds.ac.uk/10.3390/v13081556
Chicago/Turabian StyleHjortaas, Monika J., Elena Fringuelli, Adérito L. Monjane, Aase B. Mikalsen, Christine M. Jonassen, Paul Savage, and Hilde Sindre. 2021. "Emergence of Salmonid Alphavirus Genotype 2 in Norway—Molecular Characterization of Viral Strains Circulating in Norway and Scotland" Viruses 13, no. 8: 1556. https://0-doi-org.brum.beds.ac.uk/10.3390/v13081556