
Isolation and Characterisation of the Bundooravirus Genus and Phylogenetic Investigation of the Salasmaviridae Bacteriophages

 ,
,  and
and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacterial Strains and Media
2.2. Isolation and Purification of Phages and DNA Extraction
2.3. Phage DNA Isolation, Genome Sequencing, and Annotation
2.4. Electron Microscopy
2.5. Nucleotide Sequence
2.6. Identification of Phage Resistant B. pumilus Mutants
2.7. Whole Genome Analysis and Clustering
3. Results
3.1. Isolation of Bacteriophages and Their Morphological Features
3.2. Sequencing and Genomic Features of PumA1 and PumA2
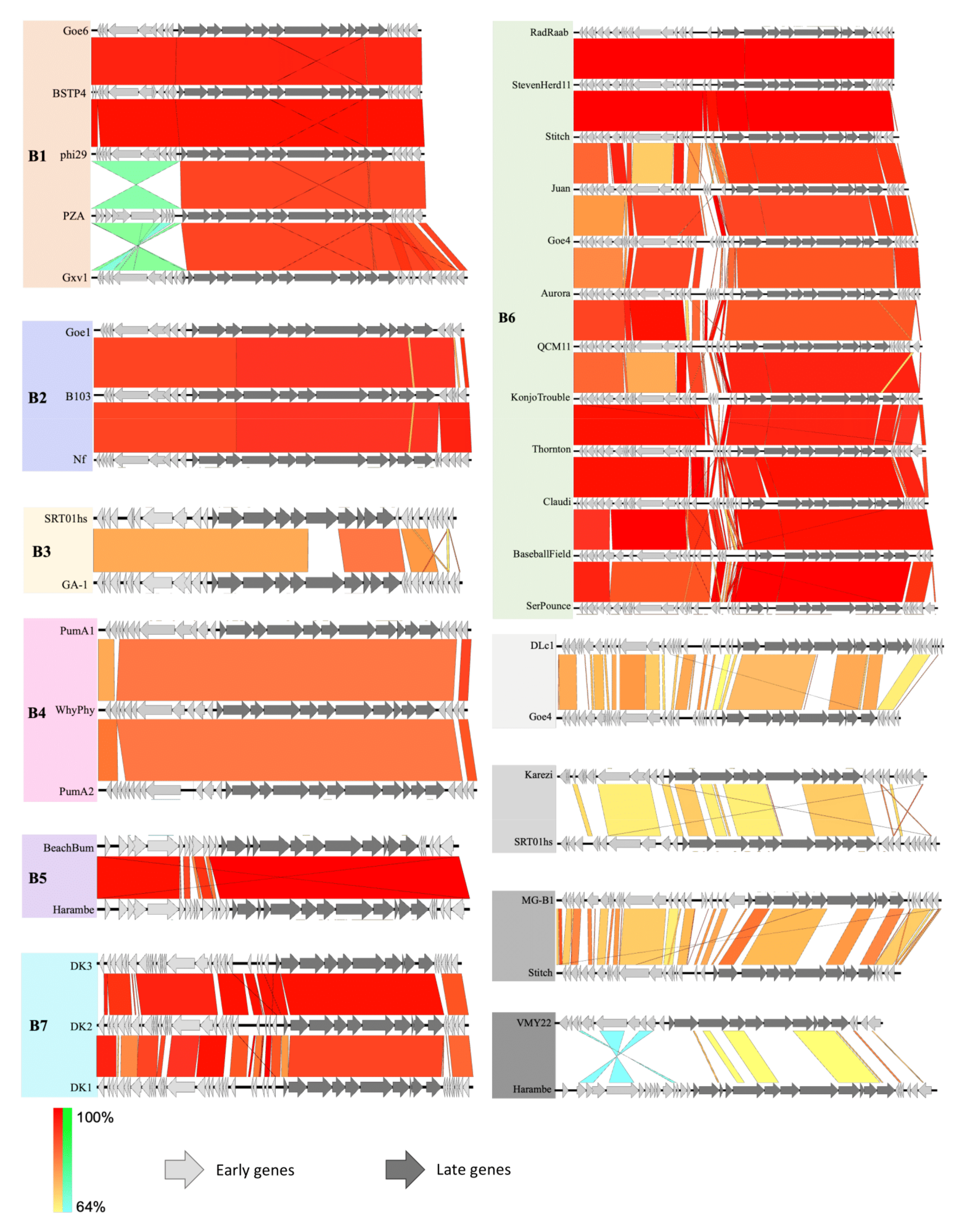
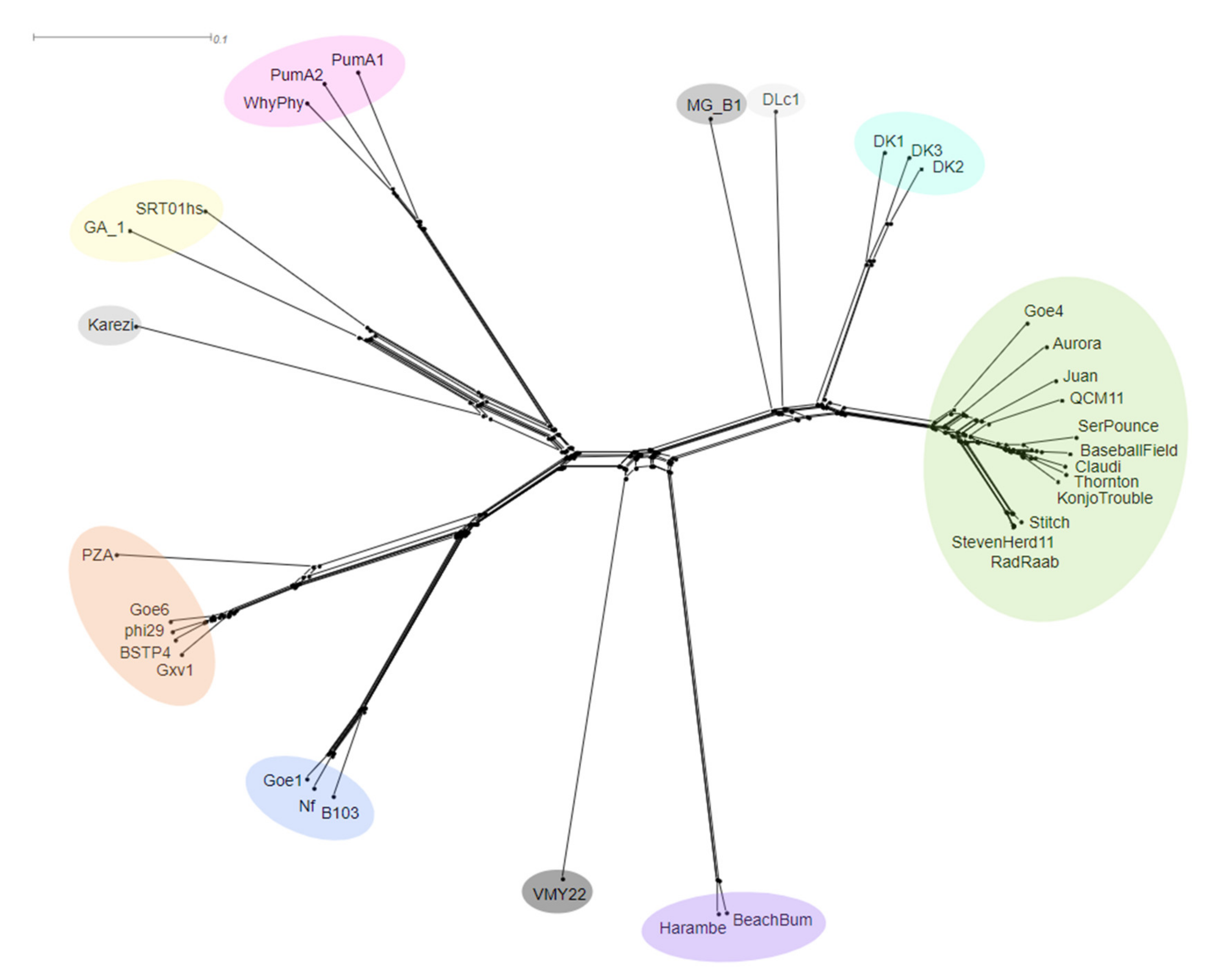
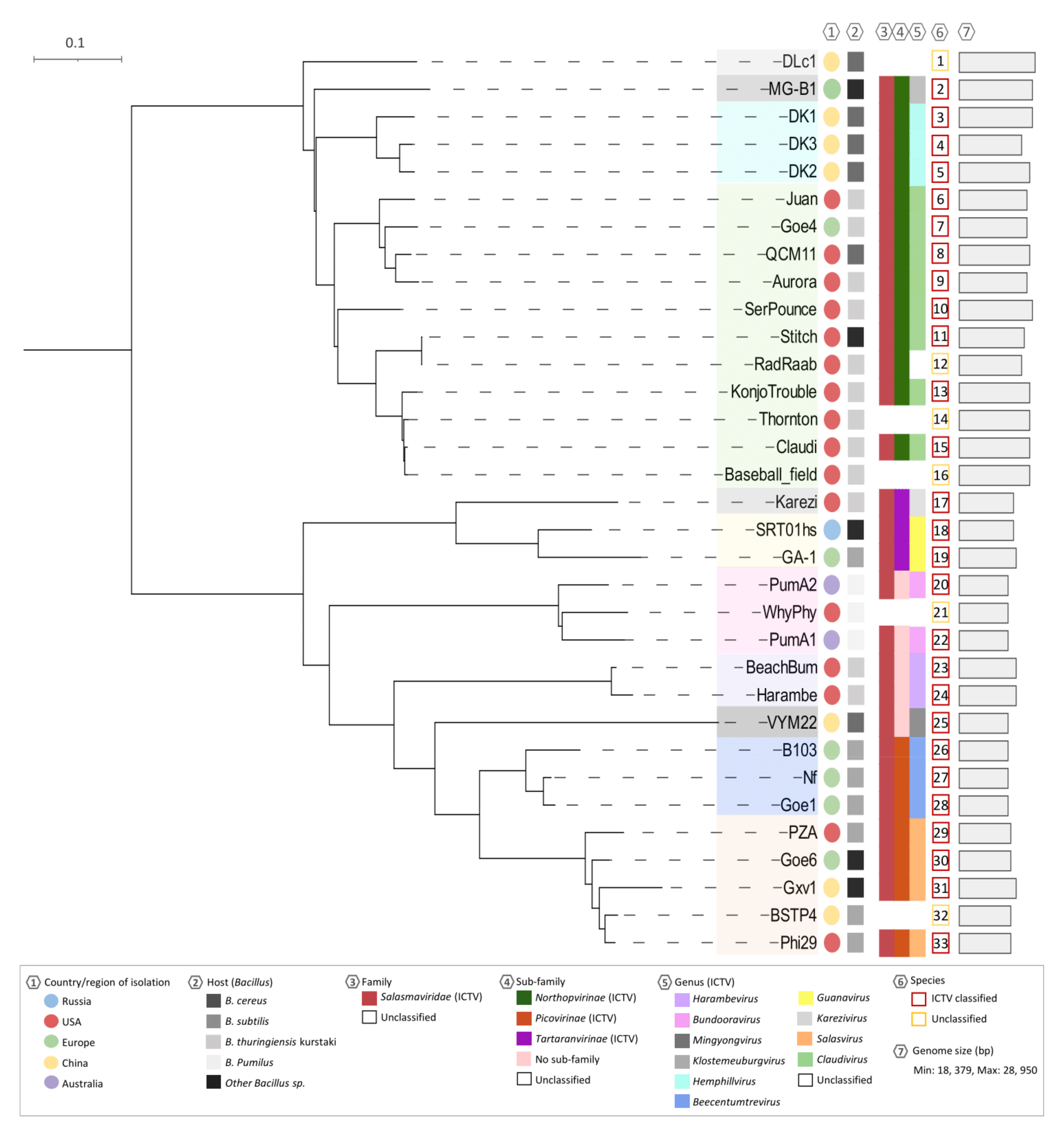
3.3. Whole Genome Comparison and Clustering of phi29-Like Phages
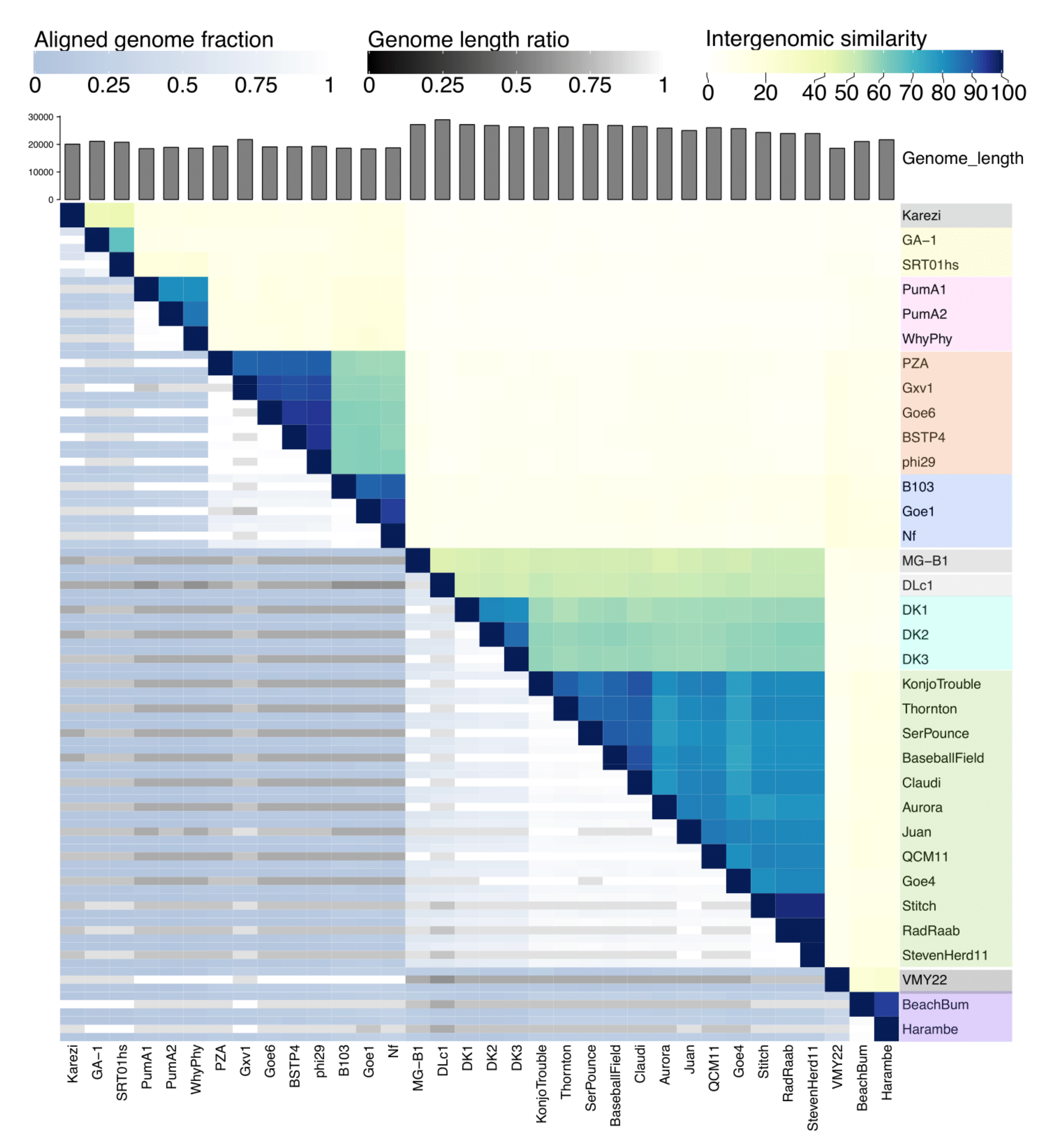
3.4. Dot Plot Analysis and Genomic Identities
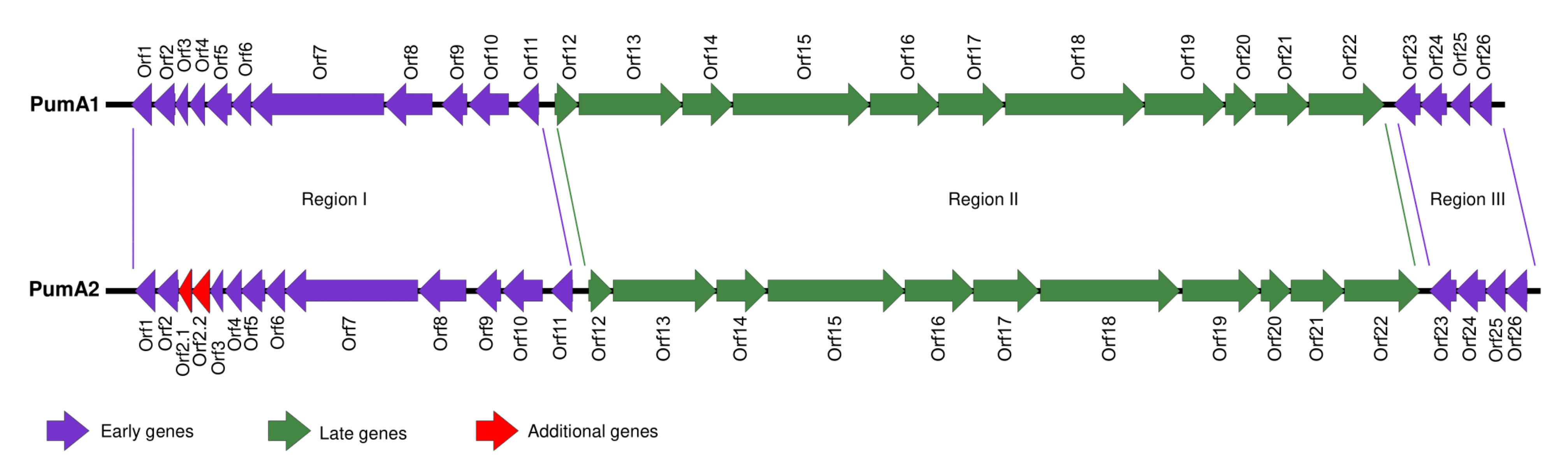
3.5. Genome Map Alignments
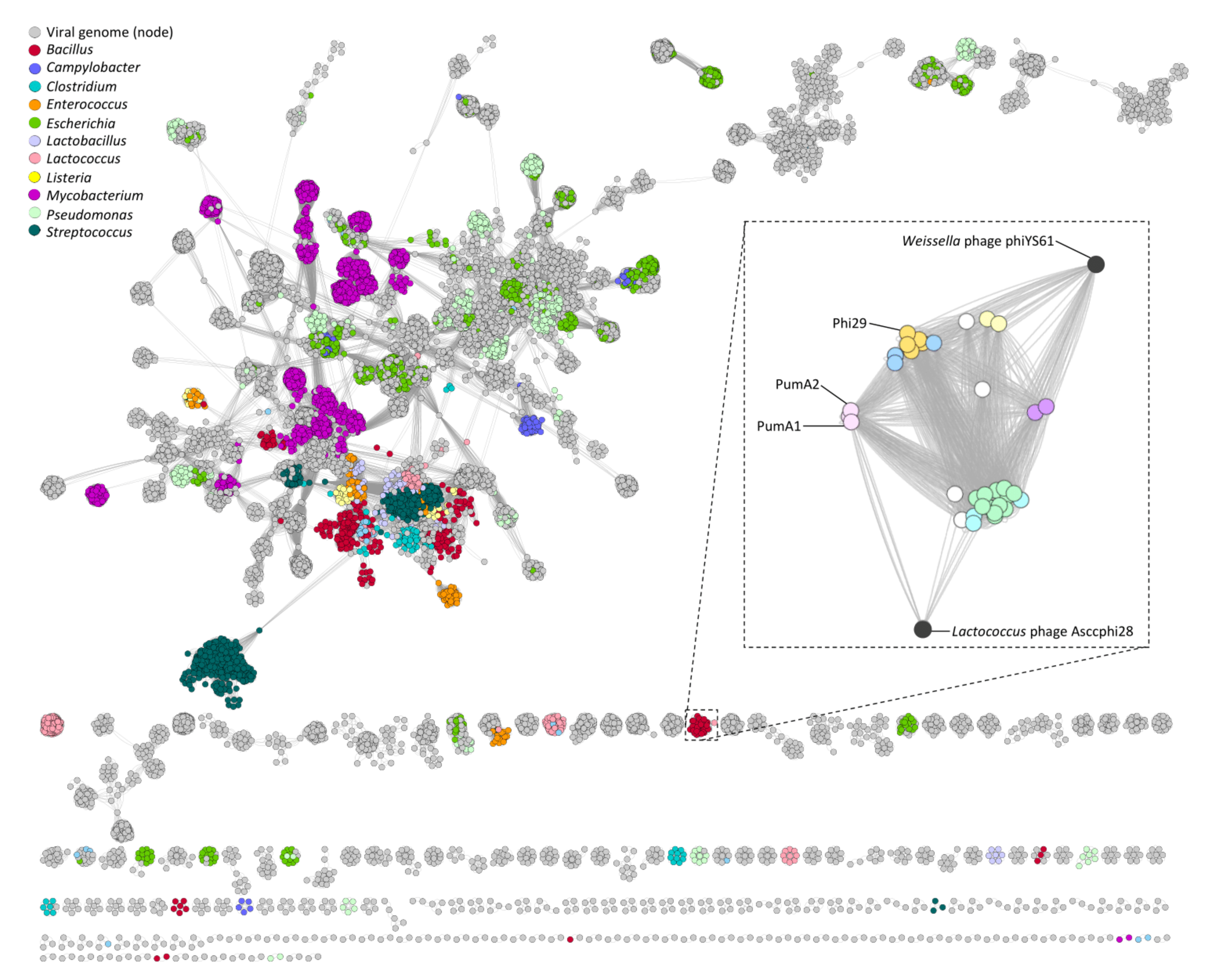
3.6. Gene Sharing Networks
3.7. Candidate Gene Analysis
3.8. PumA1 and PumA2 Host Receptor Site
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Logan, N.A.; Halket, G. Developments in the taxonomy of aerobic, endospore-forming bacteria. In Endospore-forming Soil Bacteria; Springer: Heidelberg, Germany, 2011; pp. 1–29. [Google Scholar]
- Santoyo, G.; Orozco-Mosqueda, M.D.C.; Govindappa, M. Mechanisms of biocontrol and plant growth-promoting activity in soil bacterial species of Bacillus and Pseudomonas: A review. Biocontrol Sci. Technol. 2012, 22, 855–872. [Google Scholar] [CrossRef]
- Glasset, B.; Herbin, S.; Granier, S.A.; Cavalié, L.; Lafeuille, E.; Guérin, C.; Ruimy, R.; Casagrande-Magne, F.; Levast, M.; Chautemps, N.; et al. Bacillus cereus, a serious cause of nosocomial infections: Epidemiologic and genetic survey. PLoS ONE 2018, 13, e0194346. [Google Scholar] [CrossRef]
- Lorenz, L.; Lins, B.; Barrett, J.; Montgomery, A.; Trapani, S.; Schindler, A.; Christie, G.E.; Cresawn, S.G.; Temple, L. Genomic characterization of six novel Bacillus pumilus bacteriophages. Virology 2013, 444, 374–383. [Google Scholar] [CrossRef] [Green Version]
- From, C.; Hormazabal, V.; Granum, P.E. Food poisoning associated with pumilacidin-producing Bacillus pumilus in rice. Int. J. Food Microbiol. 2007, 115, 319–324. [Google Scholar] [CrossRef]
- Kempf, M.J.; Chen, F.; Kern, R.; Venkateswaran, K. Recurrent Isolation of Hydrogen Peroxide-Resistant Spores of Bacillus pumilus from a Spacecraft Assembly Facility. Astrobiology 2005, 5, 391–405. [Google Scholar] [CrossRef]
- Handtke, S.; Schroeter, R.; Jürgen, B.; Methling, K.; Schlüter, R.; Albrecht, D.; van Hijum, S.; Bongaerts, J.; Maurer, K.-H.; Lalk, M.; et al. Bacillus pumilus Reveals a Remarkably High Resistance to Hydrogen Peroxide Provoked Oxidative Stress. PLoS ONE 2014, 9, e85625. [Google Scholar] [CrossRef]
- Rohwer, F. Global Phage Diversity. Cell 2003, 113, 141. [Google Scholar] [CrossRef] [Green Version]
- Schilling, T.; Hoppert, M.; Hertel, R. Genomic Analysis of the Recent Viral Isolate vB_BthP-Goe4 Reveals Increased Diversity of φ29-Like Phages. Viruses 2018, 10, 624. [Google Scholar] [CrossRef] [Green Version]
- Salmond, G.P.C.; Fineran, P. A century of the phage: Past, present and future. Nat. Rev. Genet. 2015, 13, 777–786. [Google Scholar] [CrossRef]
- Levin, B.R.; Bull, J.J. Population and evolutionary dynamics of phage therapy. Nat. Rev. Genet. 2004, 2, 166–173. [Google Scholar] [CrossRef]
- Torres-Barceló, C.; Hochberg, M.E. Evolutionary Rationale for Phages as Complements of Antibiotics. Trends Microbiol. 2016, 24, 249–256. [Google Scholar] [CrossRef]
- Kutateladze, M.; Adamia, R. Bacteriophages as potential new therapeutics to replace or supplement antibiotics. Trends Biotechnol. 2010, 28, 591–595. [Google Scholar] [CrossRef]
- Hatfull, G.F. Bacteriophage genomics. Curr. Opin. Microbiol. 2008, 11, 447–453. [Google Scholar] [CrossRef] [Green Version]
- Hatfull, G.F.; Hendrix, R.W. Bacteriophages and their genomes. Curr. Opin. Virol. 2011, 1, 298–303. [Google Scholar] [CrossRef] [Green Version]
- Tolstoy, I.; Kropinski, A.M.; Brister, J.R. Bacteriophage Taxonomy: An Evolving Discipline. In Bacteriophage Therapy. Methods in Molecular Biology; Humana Press: New York, NY, USA, 2017; pp. 57–71. [Google Scholar]
- Aziz, R.K.; Ackermann, H.-W.; Petty, N.K.; Kropinski, A.M. Essential Steps in Characterizing Bacteriophages: Biology, Taxonomy, and Genome Analysis. In Bacteriophages. Methods in Molecular Biology; Humana Press: New York, NY, USA, 2017; pp. 197–215. [Google Scholar]
- Russell, D.A.; Hatfull, G.F. PhagesDB: The actinobacteriophage database. Bioinformatics 2016, 33, 784–786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mavrich, T.; Hatfull, G.F. Bacteriophage evolution differs by host, lifestyle and genome. Nat. Microbiol. 2017, 2, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grose, J.H.; Jensen, G.L.; Burnett, S.H.; Breakwell, N.P. Correction: Genomic comparison of 93 Bacillus phages reveals 12 clusters, 14 singletons and remarkable diversity. BMC Genom. 2014, 15, 1184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatfull, G.F. Mycobacteriophages: Genes and Genomes. Annu. Rev. Microbiol. 2010, 64, 331–356. [Google Scholar] [CrossRef]
- Meijer, W.J.J.; Horcajadas, J.A.; Salas, M. φ29 Family of Phages. Microbiol. Mol. Biol. Rev. 2001, 65, 261–287. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Yuan, X.; Li, N.; Wang, J.; Yu, S.; Zeng, H.; Zhang, J.; Wu, Q.; Ding, Y. Isolation and Characterization of Bacillus cereus Phage vB_BceP-DLc1 Reveals the Largest Member of the Φ29-Like Phages. Microorganisms 2020, 8, 1750. [Google Scholar] [CrossRef]
- Walker, P.J.; Siddell, S.G.; Lefkowitz, E.J.; Mushegian, A.R.; Adriaenssens, E.M.; Dempsey, D.M.; Dutilh, B.E.; Harrach, B.; Harrison, R.L.; Hendrickson, R.C.; et al. Changes to virus taxonomy and the Statutes ratified by the International Committee on Taxonomy of Viruses (2020). Arch. Virol. 2020, 165, 2737–2748. [Google Scholar] [CrossRef]
- Petrovski, S.; Seviour, R.J.; Tillett, D. Genome Sequence and Characterization of the Tsukamurella Bacteriophage TPA2. Appl. Environ. Microbiol. 2011, 77, 1389–1398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17. [Google Scholar] [CrossRef]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Schwartz, S.; Wagner, L.; Miller, W. A Greedy Algorithm for Aligning DNA Sequences. J. Comput. Biol. 2000, 7, 203–214. [Google Scholar] [CrossRef]
- Finn, R.D.; Coggill, P.; Eberhardt, R.; Eddy, S.R.; Mistry, J.; Mitchell, A.; Potter, S.; Punta, M.; Qureshi, M.; Sangrador, A.; et al. The Pfam protein families database: Towards a more sustainable future. Nucleic Acids Res. 2015, 44, D279–D285. [Google Scholar] [CrossRef]
- Laslett, D. ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res. 2004, 32, 11–16. [Google Scholar] [CrossRef]
- Krumsiek, J.; Arnold, R.; Rattei, T. Gepard: A rapid and sensitive tool for creating dotplots on genome scale. Bioinformatics 2007, 23, 1026–1028. [Google Scholar] [CrossRef] [Green Version]
- Moraru, C.; Varsani, A.; Kropinski, A. VIRIDIC—A Novel Tool to Calculate the Intergenomic Similarities of Prokaryote-Infecting Viruses. Viruses 2020, 12, 1268. [Google Scholar] [CrossRef]
- Sullivan, M.J.; Petty, N.; Beatson, S.A. Easyfig: A genome comparison visualizer. Bioinformatics 2011, 27, 1009–1010. [Google Scholar] [CrossRef]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; López, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef]
- Huson, D.H. SplitsTree: Analyzing and visualizing evolutionary data. Bioinformatics 1998, 14, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Bolduc, B.; Bin Jang, H.; Doulcier, G.; You, Z.-Q.; Roux, S.; Sullivan, M.B. vConTACT: An iVirus tool to classify double-stranded DNA viruses that infect Archaea and Bacteria. PeerJ 2017, 5, e3243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopes, C.T.; Franz, M.; Kazi, F.; Donaldson, S.L.; Morris, Q.; Bader, G.D. Cytoscape Web: An interactive web-based network browser. Bioinformatics 2010, 26, 2347–2348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meier-Kolthof, J.P.; Göker, M. VICTOR: Genome-based phylogeny and classification of prokaryotic viruses. Bioinformatics 2017, 33, 3396–3404. [Google Scholar] [CrossRef] [Green Version]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef]
- Redondo, R.A.F.; Kupczok, A.; Stift, G.; Bollback, J.P. Complete Genome Sequence of the Novel Phage MG-B1 Infecting Bacillus weihenstephanensis. Genome Announc. 2013, 1. [Google Scholar] [CrossRef] [Green Version]
- Ji, X.; Zhang, C.; Fang, Y.; Zhang, Q.; Lin, L.; Tang, B.; Wei, Y. Isolation and characterization of glacier VMY22, a novel lytic cold-active bacteriophage of Bacillus cereus. Virol. Sin. 2015, 30, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Zhang, T.; Jin, M.; Zeng, R. Characterization of Bacillus phage Gxv1, a novel lytic Salasvirus phage isolated from deep-sea seamount sediments. Mar. Life Sci. Technol. 2020, 3, 13–19. [Google Scholar] [CrossRef]
- Kong, L.; Ding, Y.; Wu, Q.; Wang, J.; Zhang, J.; Li, H.-Y.; Yu, S.; Yu, P.; Gao, T.; Zeng, H.; et al. Genome sequencing and characterization of three Bacillus cereus-specific phages, DK1, DK2, and DK3. Arch. Virol. 2019, 164, 1927–1929. [Google Scholar] [CrossRef]
- Willms, I.M.; Hertel, R. Phage vB_BsuP-Goe1: The smallest identified lytic phage of Bacillus subtilis. FEMS Microbiol. Lett. 2016, 363, fnw208. [Google Scholar] [CrossRef] [Green Version]
- Pecenkova, T.; Benes, V.; Paces, J.; Vlček, Č.; Pačes, V. Bacteriophage B103: Complete DNA sequence of its genome and relationship to other Bacillus phages. Gene 1997, 199, 157–163. [Google Scholar] [CrossRef]
- Bradley, D.E. The Isolation and morphology of Some New Bacteriophages Specific for Bacillus and Acetobacter species. J. Gen. Microbiol. 1965, 41, 233–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castilla-Llorente, V.; Salas, M.; Meijer, W.J.J. Different responses to Spo0A-mediated suppression of the related Bacillus subtilis phages Nf and φ29. Environ. Microbiol. 2009, 11, 1137–1149. [Google Scholar] [CrossRef] [PubMed]
- Pačes, V.; Viček, Č.; Urbánek, P.; Hostomský, Z. Nucleotide sequence of the major early region of Bacillus subtilis phage PZA, a close relative of φ29. Gene 1985, 38, 45–56. [Google Scholar] [CrossRef]
- Anderson, D.L.; Hickman, D.D.; Reilly, B.E. Structure of Bacillus subtilis Bacteriophage φ29 and the Length of φ29 Deoxyribonucleic Acid. J. Bacteriol. 1966, 91, 2081–2089. [Google Scholar] [CrossRef] [Green Version]
- Erill, I.; Caruso, S.M. Complete Genome Sequences of Three phi29-like Bacillus cereus Group Podoviridae. Genome Announc. 2017, 5, e00701-17. [Google Scholar] [CrossRef] [Green Version]
- Duperier, J.; Bulpitt, M.; Bispo, F.; Greguske, E. Genome Annotations of Two Bacillus Phages, Tomato and BaseballField. Microbiol. Resour. Announc. 2021, 10, e01196-20. [Google Scholar] [CrossRef]
- Yoshikawa, H.; Ito, J. Terminal proteins and short inverted terminal repeats of the small Bacillus bacteriophage genomes. Proc. Natl. Acad. Sci. USA 1981, 78, 2596–2600. [Google Scholar] [CrossRef] [Green Version]
- Carstens, E.B. Ratification vote on taxonomic proposals to the International Committee on Taxonomy of Viruses (2009). Arch. Virol. 2009, 155, 133–146. [Google Scholar] [CrossRef] [PubMed]
- Paez, J.G. Genome coverage and sequence fidelity of φ29 polymerase-based multiple strand displacement whole genome amplification. Nucleic Acids Res. 2004, 32, e71. [Google Scholar] [CrossRef] [PubMed]
- Mencia, M.; Monsalve, M.; Rojo, F.; Salas, M. Transcription activation by phage phi29 protein p4 is mediated by interaction with the alpha subunit of Bacillus subtilis RNA polymerase. Proc. Natl. Acad. Sci. USA 1996, 93, 6616–6620. [Google Scholar] [CrossRef] [Green Version]
- Badia, D.; Camacho, A.; Pérez-Lago, L.; Escandón, C.; Salas, M.; Coll, M. The Structure of Phage ϕ29 Transcription Regulator p4-DNA Complex Reveals an N-Hook Motif for DNA Binding. Mol. Cell 2006, 22, 73–81. [Google Scholar] [CrossRef]
- Hawley, L.A.; Reilly, B.E.; Hagen, E.W.; Anderson, D.L. Viral Protein Synthesis in Bacteriophage φ29-Infected Bacillus subtilis. J. Virol. 1973, 12, 1149–1159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Wang, D.; Gui, M.; Xiang, Y. Structural assembly of the tailed bacteriophage φ29. Nat. Commun. 2019, 10, 2366. [Google Scholar] [CrossRef] [Green Version]
- Fu, C.-Y.; Prevelige, P.E. In vitro incorporation of the phage Phi29 connector complex. Virology 2009, 394, 149–153. [Google Scholar] [CrossRef] [Green Version]
- E Tosi, M.; E Reilly, B.; Anderson, D.L. Morphogenesis of bacteriophage phi29 of Bacillus subtilis: Cleavage and assembly of the neck appendage protein. J. Virol. 1975, 16, 1282–1295. [Google Scholar] [CrossRef] [Green Version]
- Tao, Y.; Olson, N.H.; Xu, W.; Anderson, D.L.; Rossmann, M.G.; Baker, T.S. Assembly of a Tailed Bacterial Virus and Its Genome Release Studied in Three Dimensions. Cell 1998, 95, 431–437. [Google Scholar] [CrossRef] [Green Version]
- Steiner, M.; Lubitz, W.; Bläsi, U. The missing link in phage lysis of gram-positive bacteria: Gene 14 of Bacillus subtilis phage phi29 encodes the functional homolog of lambda S protein. J. Bacteriol. 1993, 175, 1038–1042. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Guo, P. Magnesium-induced conformational change of packaging RNA for procapsid recognition and binding during phage phi29 DNA encapsidation. J. Virol. 1997, 71, 495–500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatfull, G.F.; Jacobs-Sera, D.; Lawrence, J.G.; Pope, W.H.; Russell, D.A.; Ko, C.-C.; Weber, R.J.; Patel, M.C.; Germane, K.; Edgar, R.H.; et al. Comparative Genomic Analysis of 60 Mycobacteriophage Genomes: Genome Clustering, Gene Acquisition, and Gene Size. J. Mol. Biol. 2010, 397, 119–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lima-Mendez, G.; van Helden, J.; Toussaint, A.; Leplae, R. Reticulate Representation of Evolutionary and Functional Relationships between Phage Genomes. Mol. Biol. Evol. 2008, 25, 762–777. [Google Scholar] [CrossRef] [Green Version]
- Kotsonis, S.E.; Powell, I.B.; Pillidge, C.J.; Limsowtin, G.K.Y.; Hillier, A.J.; Davidson, B.E. Characterization and Genomic Analysis of Phage Asccφ28, a Phage of the Family Podoviridae Infecting Lactococcus lactis. Appl. Environ. Microbiol. 2008, 74, 3453–3460. [Google Scholar] [CrossRef] [Green Version]
- Kleppen, H.P.; Holo, H.; Jeon, S.-R.; Nes, I.F.; Yoon, S.-S. Novel Podoviridae Family Bacteriophage Infecting Weissella cibaria Isolated from Kimchi. Appl. Environ. Microbiol. 2012, 78, 7299–7308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, F.E. Requirement of glucosylated teichoic acid for adsorption of phage in Bacillus subtilis 168. Proc. Natl. Acad. Sci. USA 1967, 58, 2377–2384. [Google Scholar] [CrossRef] [Green Version]
- Bhavsar, A.P.; Erdman, L.K.; Schertzer, J.W.; Brown, E.D. Teichoic Acid Is an Essential Polymer in Bacillus subtilis That Is Functionally Distinct from Teichuronic Acid. J. Bacteriol. 2004, 186, 7865–7873. [Google Scholar] [CrossRef] [Green Version]
- Kawai, Y.; Marles-Wright, J.; Cleverley, R.M.; Emmins, R.; Ishikawa, S.; Kuwano, M.; Heinz, N.; Bui, N.K.; Hoyland, C.N.; Ogasawara, N.; et al. A widespread family of bacterial cell wall assembly proteins. EMBO J. 2011, 30, 4931–4941. [Google Scholar] [CrossRef] [Green Version]
- Hanson, C.A.; Marston, M.F.; Martiny, J. Biogeographic Variation in Host Range Phenotypes and Taxonomic Composition of Marine Cyanophage Isolates. Front. Microbiol. 2016, 7, 983. [Google Scholar] [CrossRef] [Green Version]
- Focardi, A.; Ostrowski, M.; Goossen, K.; Brown, M.V.; Paulsen, I. Investigating the Diversity of Marine Bacteriophage in Contrasting Water Masses Associated with the East Australian Current (EAC) System. Viruses 2020, 12, 317. [Google Scholar] [CrossRef] [Green Version]
- Sousa, J.A.M.D.; Pfeifer, E.; Touchon, M.; Rocha, E.P.C. Causes and Consequences of Bacteriophage Diversification via Genetic Exchanges across Lifestyles and Bacterial Taxa. Mol. Biol. Evol. 2021, 38, 2497–2512. [Google Scholar] [CrossRef]
- Dion, M.B.; Oechslin, F.; Moineau, S. Phage diversity, genomics and phylogeny. Nat. Rev. Genet. 2020, 18, 125–138. [Google Scholar] [CrossRef] [PubMed]
- Salifu, S.P.; Rello, A.V.; Campbell, S.A.; Inglis, N.F.; Scortti, M.; Foley, S.; Vázquez-Boland, J.A. Genome and proteome analysis of phage E3 infecting the soil-borne actinomyceteRhodococcus equi. Environ. Microbiol. Rep. 2013, 5, 170–178. [Google Scholar] [CrossRef] [PubMed]
- Roucourt, B.; Lavigne, R. The role of interactions between phage and bacterial proteins within the infected cell: A diverse and puzzling interactome. Environ. Microbiol. 2009, 11, 2789–2805. [Google Scholar] [CrossRef] [PubMed]
- Kupczok, A.; Neve, H.; Huang, K.D.; Hoeppner, M.P.; Heller, K.J.; Franz, C.M.A.P.; Dagan, T. Rates of Mutation and Recombination in Siphoviridae Phage Genome Evolution over Three Decades. Mol. Biol. Evol. 2018, 35, 1147–1159. [Google Scholar] [CrossRef] [PubMed]
- Hua, J.; Huet, A.; Lopez, C.A.; Toropova, K.; Pope, W.H.; Duda, R.L.; Hendrix, R.W.; Conway, J.F. Capsids and Genomes of Jumbo-Sized Bacteriophages Reveal the Evolutionary Reach of the HK97 Fold. mBio 2017, 8, e01579-17. [Google Scholar] [CrossRef] [Green Version]
- Reilly, B.E.; Nelson, R.A.; Anderson, D.L. Morphogenesis of Bacteriophage φ29 of Bacillus subtilis: Mapping and Functional Analysis of the Head Fiber Gene. J. Virol. 1977, 24, 363–377. [Google Scholar] [CrossRef] [Green Version]
- Xiang, Y.; Rossmann, M.G. Structure of bacteriophage φ29 head fibers has a supercoiled triple repeating helix-turn-helix motif. Proc. Natl. Acad. Sci. USA 2011, 108, 4806–4810. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bacteriophage | Country Isolated | Accession Number | Year Isolated | Genome Size (kb) | Reference | Host |
|---|---|---|---|---|---|---|
| PumA1 | Australia | MN524844 | 2017 | 18,446 | This study | B. pumilus |
| PumA2 | Australia | MN524845 | 2017 | 18,932 | This study | B. pumilus |
| MG-B1 | Austria | KC685370 | 2011 | 27,190 | [41] | B. weihenstephanensis |
| VMY22 | China | KT780304 | 2015 | 18,609 | [42] | B. cereus |
| Gxv1 | China | MT459794 | 2020 | 21,781 | [43] | Bacillus sp. |
| DK2 | China | MK284527 | 2018 | 23,357 | [44] | B. cereus |
| DK3 | China | MK284528 | 2018 | 26,865 | [44] | B. cereus |
| DK1 | China | MK284526 | 2018 | 27,180 | [44] | B. cereus |
| DLc1 | China | MW012634 | 2020 | 28,950 | [23] | B. cereus |
| Goe1 | Germany | KU831549 | 2014 | 18,379 | [45] | B. subtilis |
| Goe6 | Germany | MF407276 | 2017 | 19,105 | Unpublished | B. velezensis |
| Goe4 | Germany | MH817022 | 2018 | 25,722 | [9] | B. thuringiensis kurstaki |
| B103 | Prague | X99260 | 1981 | 18,630 | [46] | B. subtilis |
| SRT01hs | Russia | MN857617 | 2020 | 20,784 | Unpublished | B. altitudinis |
| GA-1 | Scotland | X96987 | 1965 | 21,129 | [47] | B. subtilis |
| BSTP4 | South Korea | MW354668 | 2020 | 19,145 | Unpublished | B. subtilis |
| Nf | Spain | EU622808 | 2008 | 18,753 | [48] | B. subtilis |
| PZA | USA | PZACG | 1976 | 19,366 | [49] | B. subtilis |
| Phi29 | USA | EU771092 | 1965 | 19,828 | [50] | B. subtilis |
| Karezi | USA | MN082625 | 2013 | 20,083 | Unpublished | B. thuringiensis kurstaki |
| BeachBum | USA | KY921761 | 2016 | 21,054 | [51] | B. thuringiensis kutstaki |
| Harambe | USA | KY821088 | 2016 | 21,684 | [51] | B. thuringiensis kutstaki |
| RadRaab | USA | MF156580 | 2016 | 23,946 | Unpublished | B. thuringiensis kurstaki |
| StevenHerd11 | USA | MK084630 | 2017 | 23,953 | Unpublished | B. thuringiensis kurstaki |
| Stitch | USA | KX349901 | 2012 | 24,320 | Unpublished | Bacillus sp. |
| Juan | USA | MF156577 | 2016 | 25,032 | Unpublished | B. thuringiensis kurstaki |
| Aurora | USA | KX349899 | 2010 | 25,908 | Unpublished | B. thuringiensis kurstaki |
| QCM11 | USA | KX961631 | 2016 | 26,054 | Unpublished | B. cereus group |
| KonjoTrouble | USA | MF156578 | 2016 | 26,061 | Unpublished | B. thuringiensis kurstaki |
| Claudi | USA | KX349900 | 2014 | 26,504 | Unpublished | B. thuringiensis kurstaki |
| SerPounce | USA | KY947509 | 2016 | 27,206 | [51] | B. thuringiensis kurstaki |
| WhyPhy | USA | MW419775 | 2020 | 18,642 | Unpublished | B. pumilus |
| Thornton | USA | MW348917 | 2017 | 26,319 | Unpublished | B. thuringiensis kurstaki |
| Baseball_field | USA | MT777452 | 2015 | 26, 863 | [52] | B. thuringiensis kurstaki |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stanton, C.R.; Rice, D.T.F.; Beer, M.; Batinovic, S.; Petrovski, S. Isolation and Characterisation of the Bundooravirus Genus and Phylogenetic Investigation of the Salasmaviridae Bacteriophages. Viruses 2021, 13, 1557. https://0-doi-org.brum.beds.ac.uk/10.3390/v13081557
Stanton CR, Rice DTF, Beer M, Batinovic S, Petrovski S. Isolation and Characterisation of the Bundooravirus Genus and Phylogenetic Investigation of the Salasmaviridae Bacteriophages. Viruses. 2021; 13(8):1557. https://0-doi-org.brum.beds.ac.uk/10.3390/v13081557
Chicago/Turabian StyleStanton, Cassandra R., Daniel T. F. Rice, Michael Beer, Steven Batinovic, and Steve Petrovski. 2021. "Isolation and Characterisation of the Bundooravirus Genus and Phylogenetic Investigation of the Salasmaviridae Bacteriophages" Viruses 13, no. 8: 1557. https://0-doi-org.brum.beds.ac.uk/10.3390/v13081557