
Bioprinted Multi-Cell Type Lung Model for the Study of Viral Inhibitors

 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Virus Preparation
2.2. Bioink Preparation
2.3. Bioprinting
2.4. Rheology
2.5. Cell Viability
2.6. LPS Stimulation
2.7. Viral Growth Assay in the Presence of Oseltamivir
2.8. Immunostaining
2.9. ELISA
2.10. Statistical Analysis
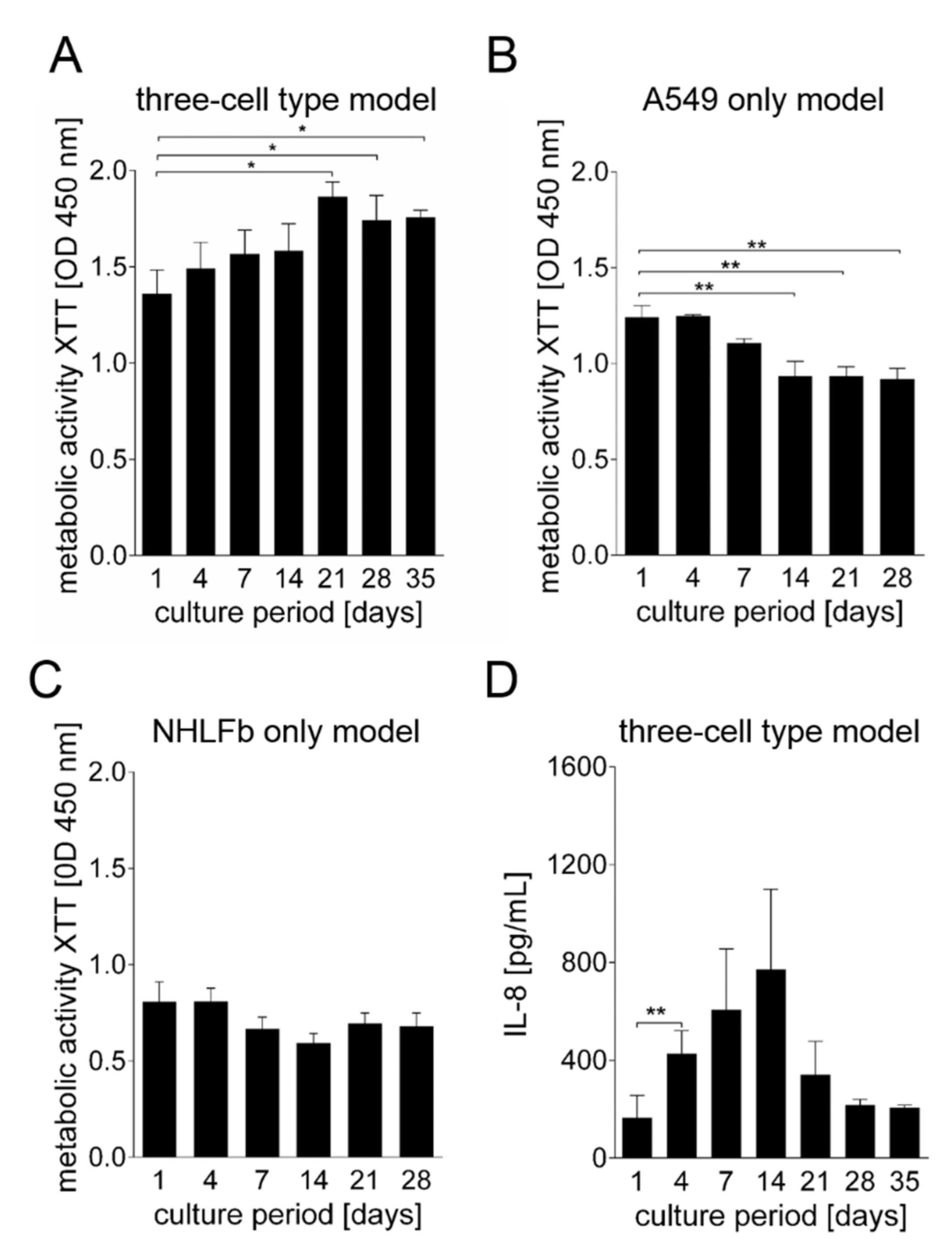
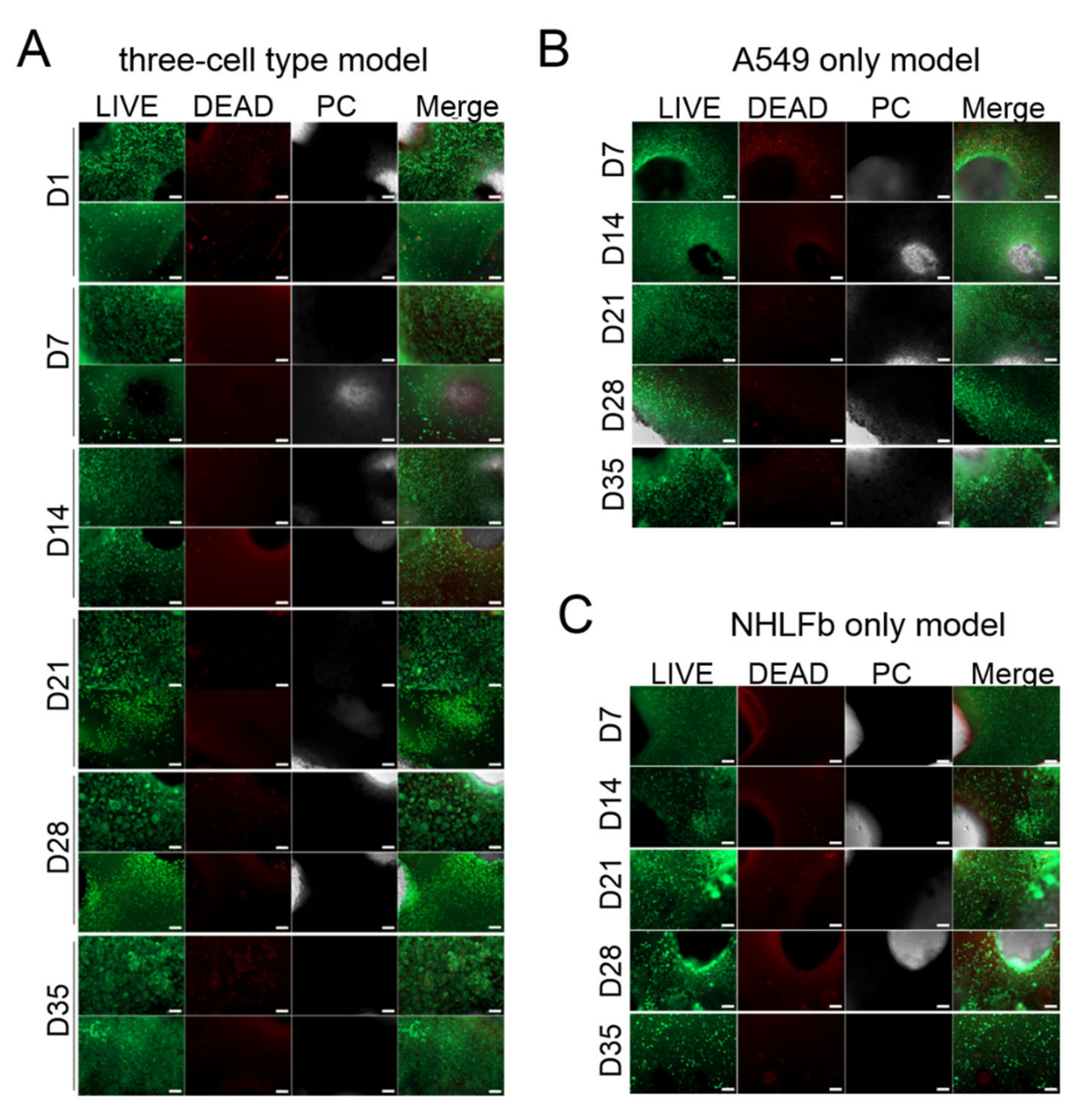
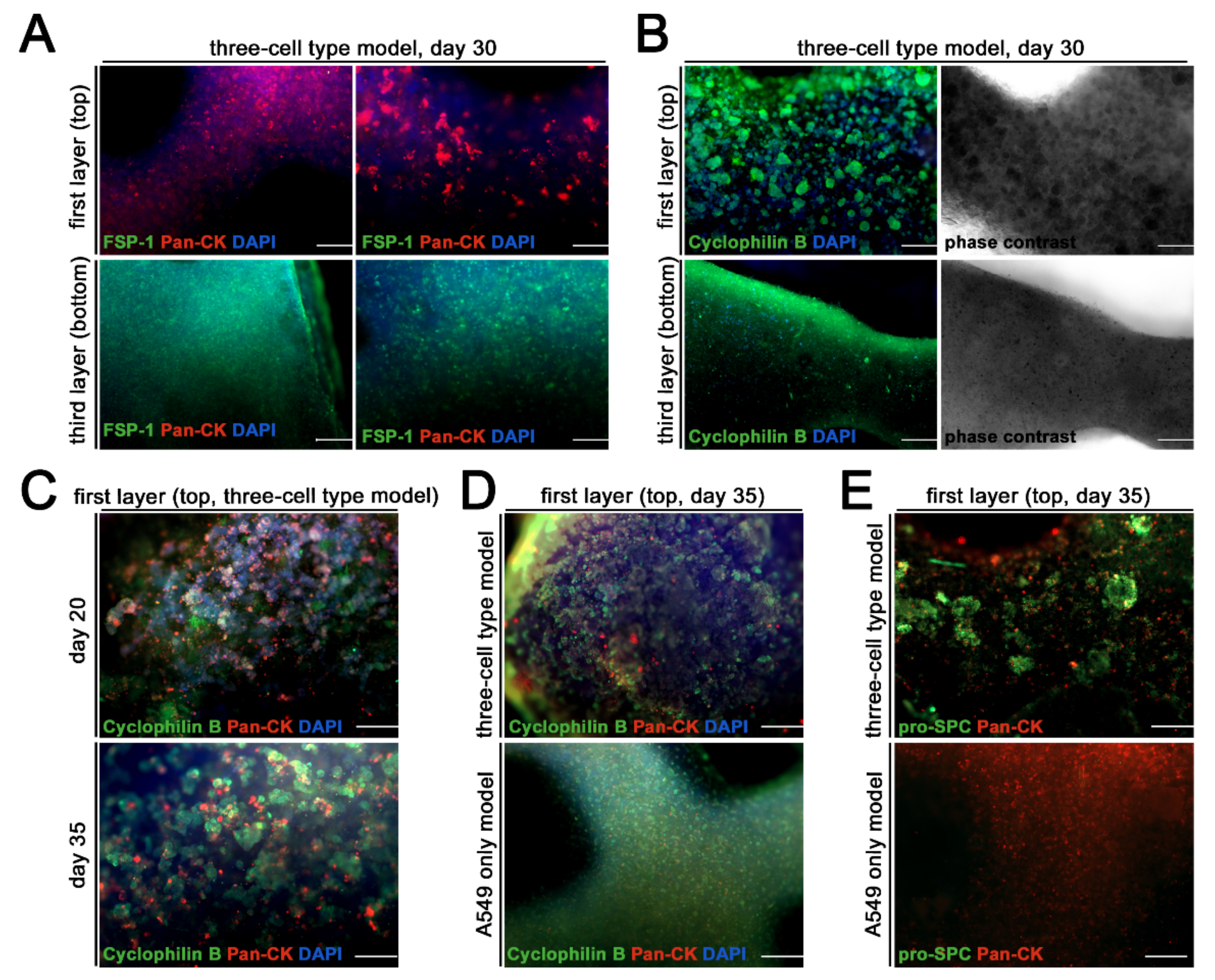
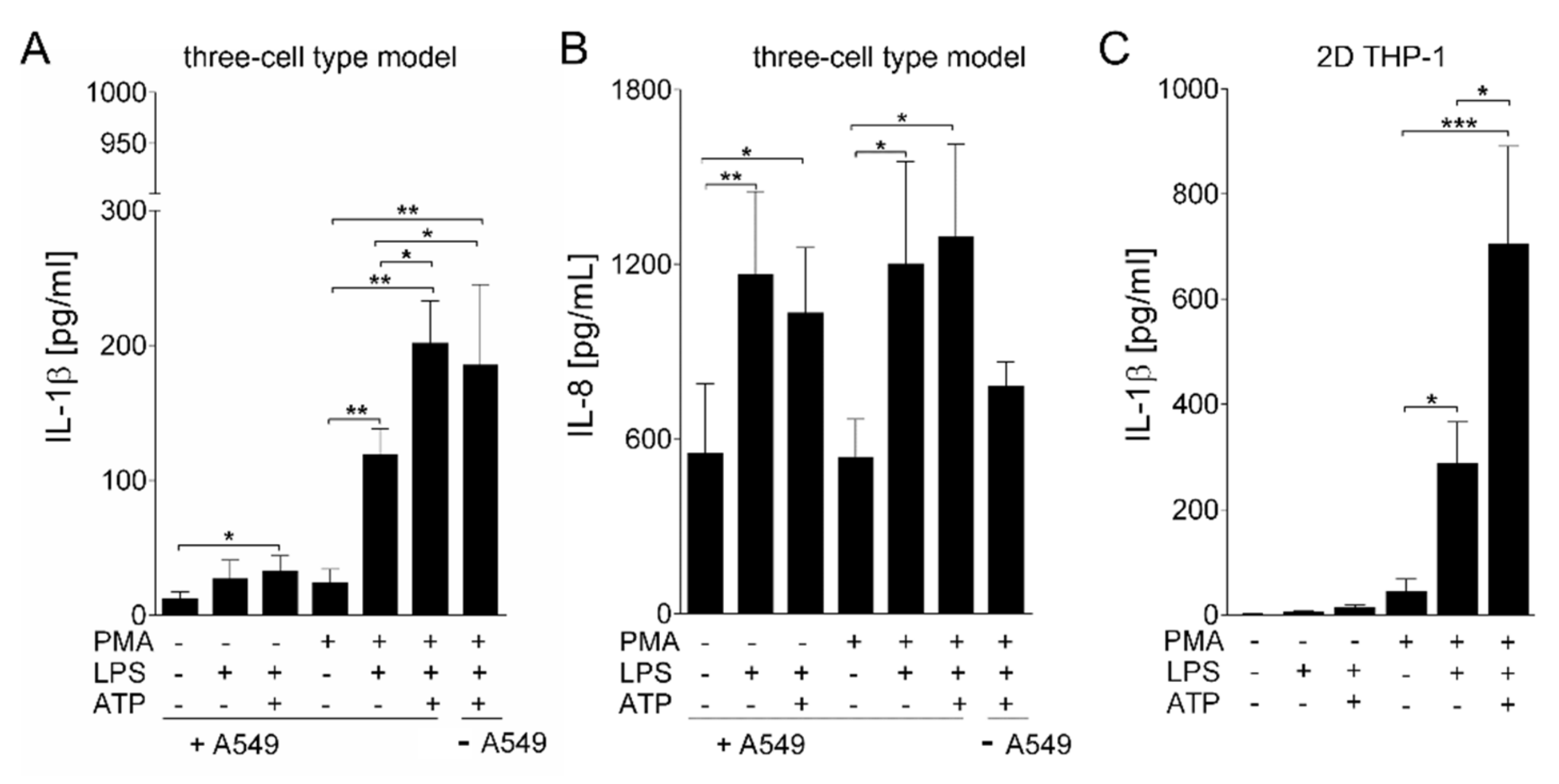
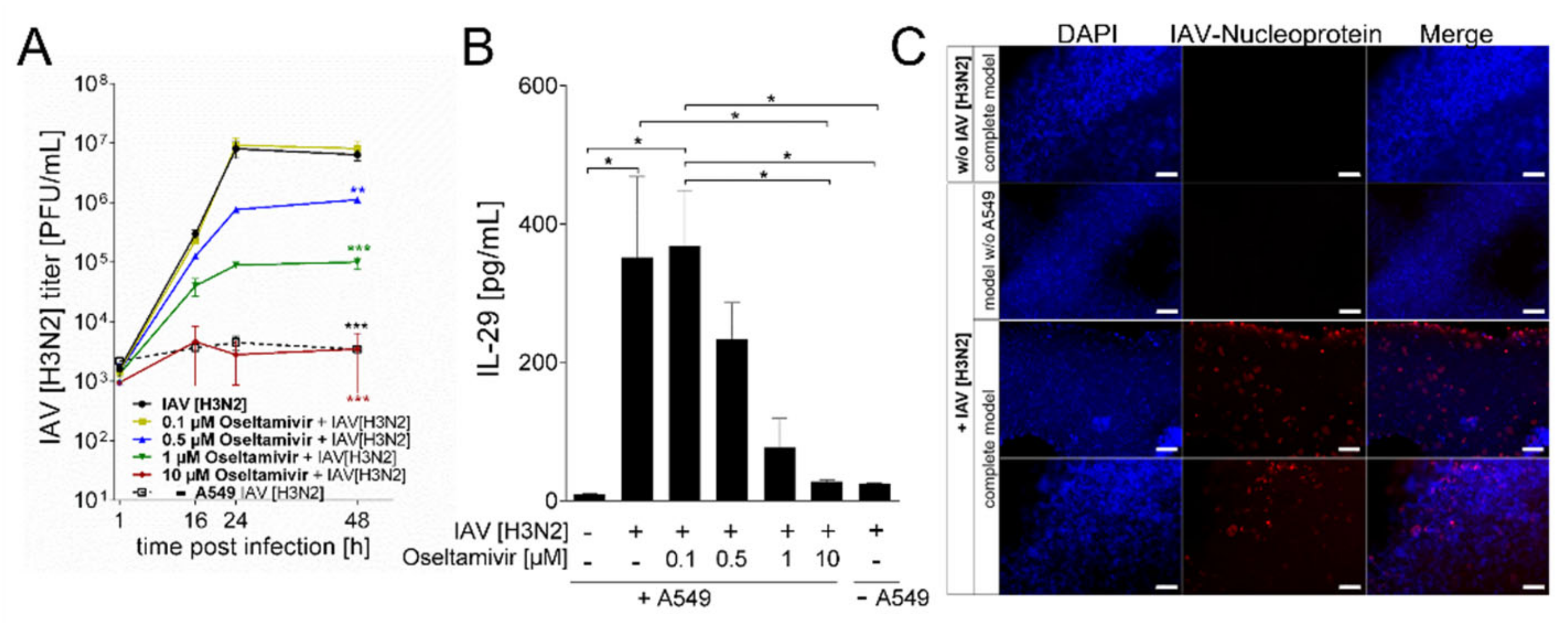
3. Results
Generation and Characterization of the Bioprinted Three Cell Type Lung Model
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bienenstock, J. The lung as an immunologic organ. Annu. Rev. Med. 1984, 35, 49–62. [Google Scholar] [CrossRef]
- Cheng, P.Y.; Palekar, R.; Azziz-Baumgartner, E.; Iuliano, D.; Alencar, A.P.; Bresee, J.; Oliva, O.; de Souza, M.D.M.; Widdowson, M.A. Burden of influenza-associated deaths in the Americas, 2002–2008. Influenza Other Resp. 2015, 9, 13–21. [Google Scholar] [CrossRef]
- Troeger, C.; Forouzanfar, M.; Rao, P.C.; Khalil, I.; Brown, A.; Swartz, S.; Fullman, N.; Mosser, J.; Thompson, R.L.; Reiner, R.C.; et al. Estimates of the global, regional, and national morbidity, mortality, and aetiologies of lower respiratory tract infections in 195 countries: A systematic analysis for the Global Burden of Disease Study 2015. Lancet Infect. Dis. 2017, 17, 1133–1161. [Google Scholar] [CrossRef] [Green Version]
- Osterlund, P.; Pirhonen, J.; Ikonen, N.; Ronkko, E.; Strengell, M.; Makela, S.M.; Broman, M.; Hamming, O.J.; Hartmann, R.; Ziegler, T.; et al. Pandemic H1N1 2009 Influenza A Virus Induces Weak Cytokine Responses in Human Macrophages and Dendritic Cells and Is Highly Sensitive to the Antiviral Actions of Interferons. J. Virol. 2010, 84, 1414–1422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woo, P.C.Y.; Tung, E.T.K.; Chan, K.H.; Lau, C.C.Y.; Lau, S.K.P.; Yuen, K.Y. Cytokine Profiles Induced by the Novel Swine-Origin Influenza A/H1N1 Virus: Implications for Treatment Strategies. J. Infect. Dis. 2010, 201, 346–353. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Zhou, Y.H.; Yang, Z.Q. The cytokine storm of severe influenza and development of immunomodulatory therapy. Cell Mol. Immunol. 2016, 13, 3–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peiris, J.S.M.; Hui, K.P.Y.; Yen, H.L. Host response to influenza virus: Protection versus immunopathology. Curr. Opin. Immunol. 2010, 22, 475–481. [Google Scholar] [CrossRef] [Green Version]
- Tisoncik, J.R.; Korth, M.J.; Simmons, C.P.; Farrar, J.; Martin, T.R.; Katze, M.G. Into the Eye of the Cytokine Storm. Microbiol. Mol. Biol. Rev. 2012, 76, 16–32. [Google Scholar] [CrossRef] [Green Version]
- Unkel, B.; Hoegner, K.; Clausen, B.E.; Lewe-Schlosser, P.; Bodner, J.; Gattenloehner, S.; Janssen, H.; Seeger, W.; Lohmeyer, J.; Herold, S. Alveolar epithelial cells orchestrate DC function in murine viral pneumonia. J. Clin. Investig. 2012, 122, 3652–3664. [Google Scholar] [CrossRef] [PubMed]
- Weinheimer, V.K.; Becher, A.; Tonnies, M.; Holland, G.; Knepper, J.; Bauer, T.T.; Schneider, P.; Neudecker, J.; Ruckert, J.C.; Szymanski, K.; et al. Influenza A Viruses Target Type II Pneumocytes in the Human Lung. J. Infect. Dis. 2012, 206, 1685–1694. [Google Scholar] [CrossRef]
- Herold, S.; Becker, C.; Ridge, K.M.; Budinger, G.R.S. Influenza virus-induced lung injury: Pathogenesis and implications for treatment. Eur. Respir. J. 2015, 45, 1463–1478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hocke, A.C.; Suttorp, N.; Hippenstiel, S. Human lung ex vivo infection models. Cell Tissue Res. 2017, 367, 511–524. [Google Scholar] [CrossRef]
- Kumar, V. Pulmonary Innate Immune Response Determines the Outcome of Inflammation During Pneumonia and Sepsis-Associated Acute Lung Injury. Front. Immunol. 2020, 11, 1722. [Google Scholar] [CrossRef]
- Kadioglu, A.; Weiser, J.N.; Paton, J.C.; Andrew, P.W. The role of Streptococcus pneumoniae virulence factors in host respiratory colonization and disease. Nat. Rev. Microbiol. 2008, 6, 288–301. [Google Scholar] [CrossRef] [PubMed]
- Fatykhova, D.; Rabes, A.; Machnik, C.; Guruprasad, K.; Pache, F.; Berg, J.; Toennies, M.; Bauer, T.T.; Schneider, P.; Schimek, M.; et al. Serotype 1 and 8 Pneumococci Evade Sensing by Inflammasomes in Human Lung Tissue. PLoS ONE 2015, 10, e0137108. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Yin, S.; Chen, Y.; Wu, Y.; Zheng, W.; Dong, H.; Bai, Y.; Qin, Y.; Li, J.; Feng, S.; et al. LPSinduced proinflammatory cytokine expression in human airway epithelial cells and macrophages via NFkappaB, STAT3 or AP1 activation. Mol. Med. Rep. 2018, 17, 5484–5491. [Google Scholar] [PubMed] [Green Version]
- Xu, F.; Droemann, D.; Rupp, J.; Shen, H.; Wu, X.; Goldmann, T.; Hippenstiel, S.; Zabel, P.; Dalhoff, K. Modulation of the inflammatory response to Streptococcus pneumoniae in a model of acute lung tissue infection. Am. J. Respir. Cell Mol. Biol. 2008, 39, 522–529. [Google Scholar] [CrossRef]
- Tumpey, T.M.; Garcia-Sastre, A.; Taubenberger, J.K.; Palese, P.; Swayne, D.E.; Pantin-Jackwood, M.J.; Schultz-Cherry, S.; Solorzano, A.; Van Rooijen, N.; Katz, J.M.; et al. Pathogenicity of influenza viruses with genes from the 1918 pandemic virus: Functional roles of alveolar macrophages and neutrophils in limiting virus replication and mortality in mice. J. Virol. 2005, 79, 14933–14944. [Google Scholar] [CrossRef] [Green Version]
- Irvin, C.G.; Bates, J.H. Measuring the lung function in the mouse: The challenge of size. Respir. Res. 2003, 4, 4. [Google Scholar] [CrossRef] [Green Version]
- Perrin, S. Make mouse studies work. Nature 2014, 507, 423–425. [Google Scholar] [CrossRef]
- Radigan, K.A.; Misharin, A.V.; Chi, M.; Budinger, G.R.S. Modeling human influenza infection in the laboratory. Infect. Drug Resist. 2015, 8, 311–320. [Google Scholar] [CrossRef] [Green Version]
- Zscheppang, K.; Berg, J.; Hedtrich, S.; Verheyen, L.; Wagner, D.E.; Suttorp, N.; Hippenstiel, S.; Hocke, A.C. Human Pulmonary 3D Models For Translational Research. Biotechnol. J. 2018, 13, 1700341. [Google Scholar] [CrossRef]
- Bouvier, N.M.; Lowen, A.C. Animal Models for Influenza Virus Pathogenesis and Transmission. Viruses 2010, 2, 1530–1563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazzoleni, G.; Di Lorenzo, D.; Steimberg, N. Modelling tissues in 3D: The next future of pharmaco-toxicology and food research? Genes Nutr. 2009, 4, 13–22. [Google Scholar] [CrossRef] [Green Version]
- Gretebeck, L.M.; Subbarao, K. Animal models for SARS and MERS coronaviruses. Curr. Opin. Virol. 2015, 13, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Sutton, T.C.; Subbarao, K. Development of animal models against emerging coronaviruses: From SARS to MERS coronavirus. Virology 2015, 479–480, 247–258. [Google Scholar] [CrossRef] [Green Version]
- Mestas, J.; Hughes, C.C.W. Of mice and not men: Differences between mouse and human immunology. J. Immunol. 2004, 172, 2731–2738. [Google Scholar] [CrossRef] [Green Version]
- Vanderburgh, J.; Sterling, J.A.; Guelcher, S.A. 3D Printing of Tissue Engineered Constructs for In Vitro Modeling of Disease Progression and Drug Screening. Ann. Biomed. Eng. 2017, 45, 164–179. [Google Scholar] [CrossRef] [Green Version]
- Berg, J.; Zscheppang, K.; Fatykhova, D.; Tonnies, M.; Bauer, T.T.; Schneider, P.; Neudecker, J.; Ruckert, J.C.; Eggeling, S.; Schimek, M.; et al. Tyk2 as a target for immune regulation in human viral/bacterial pneumonia. Eur. Respir. J. 2017, 50, 1601953. [Google Scholar] [CrossRef]
- Nicholas, B.; Staples, K.J.; Moese, S.; Meldrum, E.; Ward, J.; Dennison, P.; Havelock, T.; Hinks, T.S.; Amer, K.; Woo, E.; et al. A novel lung explant model for the ex vivo study of efficacy and mechanisms of anti-influenza drugs. J. Immunol. 2015, 194, 6144–6154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhowmick, R.; Gappa-Fahlenkamp, H. Cells and Culture Systems Used to Model the Small Airway Epithelium. Lung 2016, 194, 419–428. [Google Scholar] [CrossRef] [PubMed]
- Baker, B.M.; Chen, C.S. Deconstructing the third dimension—How 3D culture microenvironments alter cellular cues. J. Cell Sci. 2012, 125, 3015–3024. [Google Scholar] [CrossRef] [Green Version]
- Bonnier, F.; Keating, M.E.; Wrobel, T.P.; Majzner, K.; Baranska, M.; Garcia-Munoz, A.; Blanco, A.; Byrne, H.J. Cell viability assessment using the Alamar blue assay: A comparison of 2D and 3D cell culture models. Toxicol. Vitr. 2015, 29, 124–131. [Google Scholar] [CrossRef] [Green Version]
- Duval, K.; Grover, H.; Han, L.H.; Mou, Y.; Pegoraro, A.F.; Fredberg, J.; Chen, Z. Modeling Physiological Events in 2D vs. 3D Cell Culture. Physiology 2017, 32, 266–277. [Google Scholar] [CrossRef]
- Koban, R.; Neumann, M.; Nelson, P.P.; Ellerbrok, H. Differential Efficacy of Novel Antiviral Substances in 3D and Monolayer Cell Culture. Viruses 2020, 12, 1294. [Google Scholar] [CrossRef]
- Gungor-Ozkerim, P.S.; Inci, I.; Zhang, Y.S.; Khademhosseini, A.; Dokmeci, M.R. Bioinks for 3D bioprinting: An overview. Biomater. Sci. 2018, 6, 915–946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mandrycky, C.; Wang, Z.; Kim, K.; Kim, D.H. 3D bioprinting for engineering complex tissues. Biotechnol. Adv. 2016, 34, 422–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, S.V.; De Coppi, P.; Atala, A. Opportunities and challenges of translational 3D bioprinting. Nat. Biomed. Eng. 2020, 4, 370–380. [Google Scholar] [CrossRef] [PubMed]
- Berg, J.; Hiller, T.; Kissner, M.S.; Qazi, T.H.; Duda, G.N.; Hocke, A.C.; Hippenstiel, S.; Elomaa, L.; Weinhart, M.; Fahrenson, C.; et al. Optimization of cell-laden bioinks for 3D bioprinting and efficient infection with influenza A virus. Sci. Rep. 2018, 8, 13877. [Google Scholar] [CrossRef] [PubMed]
- Berg, J.; Kurreck, J. Clean Bioprinting—Fabrication of 3D Organ Models Devoid of Animal Components. Altex Altern. Anim. Exp. 2021, 38, 269–288. [Google Scholar]
- Hiller, T.; Berg, J.; Elomaa, L.; Rohrs, V.; Ullah, I.; Schaar, K.; Dietrich, A.C.; Al-Zeer, M.A.; Kurtz, A.; Hocke, A.C.; et al. Generation of a 3D Liver Model Comprising Human Extracellular Matrix in an Alginate/Gelatin-Based Bioink by Extrusion Bioprinting for Infection and Transduction Studies. Int. J. Mol. Sci 2018, 19, 3129. [Google Scholar] [CrossRef] [Green Version]
- Fantini, V.; Bordoni, M.; Scocozza, F.; Conti, M.; Scarian, E.; Carelli, S.; Di Giulio, A.M.; Marconi, S.; Pansarasa, O.; Auricchio, F.; et al. Bioink Composition and Printing Parameters for 3D Modeling Neural Tissue. Cells 2019, 8, 830. [Google Scholar] [CrossRef] [Green Version]
- Park, J.H.; Yoon, J.K.; Lee, J.B.; Shin, Y.M.; Lee, K.W.; Bae, S.W.; Lee, J.; Yu, J.; Jung, C.R.; Youn, Y.N.; et al. Experimental Tracheal Replacement Using 3-dimensional Bioprinted Artificial Trachea with Autologous Epithelial Cells and Chondrocytes. Sci. Rep. 2019, 9, 2103. [Google Scholar] [CrossRef]
- Pati, F.; Jang, J.; Ha, D.H.; Won Kim, S.; Rhie, J.W.; Shim, J.H.; Kim, D.H.; Cho, D.W. Printing three-dimensional tissue analogues with decellularized extracellular matrix bioink. Nat. Commun. 2014, 5, 3935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Z.; Su, X.; Xu, Y.; Kong, B.; Sun, W.; Mi, S. Bioprinting three-dimensional cell-laden tissue constructs with controllable degradation. Sci. Rep. 2016, 6, 24474. [Google Scholar] [CrossRef] [Green Version]
- Bauernfeind, F.G.; Horvath, G.; Stutz, A.; Alnemri, E.S.; MacDonald, K.; Speert, D.; Fernandes-Alnemri, T.; Wu, J.; Monks, B.G.; Fitzgerald, K.A.; et al. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J. Immunol. 2009, 183, 787–791. [Google Scholar] [CrossRef]
- Netea, M.G.; Simon, A.; van de Veerdonk, F.; Kullberg, B.J.; Van der Meer, J.W.; Joosten, L.A. IL-1beta processing in host defense: Beyond the inflammasomes. PLoS Pathog. 2010, 6, e1000661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mota, C.; Camarero-Espinosa, S.; Baker, M.B.; Wieringa, P.; Moroni, L. Bioprinting: From Tissue and Organ Development to in Vitro Models. Chem. Rev. 2020, 120, 10547–10607. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Starly, B.; Daly, A.C.; Burdick, J.A.; Groll, J.; Skeldon, G.; Shu, W.; Sakai, Y.; Shinohara, M.; Nishikawa, M.; et al. The bioprinting roadmap. Biofabrication 2020, 12, 022002. [Google Scholar] [CrossRef]
- Iverson, E.; Kaler, L.; Agostino, E.L.; Song, D.; Duncan, G.A.; Scull, M.A. Leveraging 3D Model Systems to Understand Viral Interactions with the Respiratory Mucosa. Viruses 2020, 12, 1425. [Google Scholar] [CrossRef] [PubMed]
- Weinhart, M.; Hocke, A.; Hippenstiel, S.; Kurreck, J.; Hedtrich, S. 3D organ models-Revolution in pharmacological research? Pharmacol. Res. 2019, 139, 446–451. [Google Scholar] [CrossRef] [PubMed]
- Koban, R.; Lam, T.; Schwarz, F.; Kloke, L.; Burge, S.; Ellerbrok, H.; Neumann, M. Simplified Bioprinting-Based 3D Cell Culture Infection Models for Virus Detection. Viruses 2020, 12, 1298. [Google Scholar] [CrossRef] [PubMed]
- Burgess, J.K.; Mauad, T.; Tjin, G.; Karlsson, J.C.; Westergren-Thorsson, G. The extracellular matrix—The under-recognized element in lung disease? J. Pathol. 2016, 240, 397–409. [Google Scholar] [CrossRef]
- Hasan, S.; Sebo, P.; Osicka, R. A guide to polarized airway epithelial models for studies of host-pathogen interactions. FEBS J. 2018, 285, 4343–4358. [Google Scholar] [CrossRef] [Green Version]
- Cooper, J.R.; Abdullatif, M.B.; Burnett, E.C.; Kempsell, K.E.; Conforti, F.; Tolley, H.; Collins, J.E.; Davies, D.E. Long Term Culture of the A549 Cancer Cell Line Promotes Multilamellar Body Formation and Differentiation towards an Alveolar Type II Pneumocyte Phenotype. PLoS ONE 2016, 11, e0164438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuchs, S.; Hollins, A.J.; Laue, M.; Schaefer, U.F.; Roemer, K.; Gumbleton, M.; Lehr, C.M. Differentiation of human alveolar epithelial cells in primary culture: Morphological characterization and synthesis of caveolin-1 and surfactant protein-C. Cell Tissue Res. 2003, 311, 31–45. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Oberley-Deegan, R.; Wang, S.; Nikrad, M.; Funk, C.J.; Hartshorn, K.L.; Mason, R.J. Differentiated human alveolar type II cells secrete antiviral IL-29 (IFN-lambda 1) in response to influenza A infection. J. Immunol. 2009, 182, 1296–1304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, A.; Kim, J.H.; Ranjan, P.; Metcalfe, M.G.; Cao, W.; Mishina, M.; Gangappa, S.; Guo, Z.; Boyden, E.S.; Zaki, S.; et al. Influenza virus exploits tunneling nanotubes for cell-to-cell spread. Sci. Rep. 2017, 7, 40360. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Liu, M.; Chen, F.; Chen, F.; Tian, Y.; Huang, Q.; Liu, S.; Yang, J. A Small-Molecule Compound Has Anti-influenza A Virus Activity by Acting as a “PB2 Inhibitor”. Mol. Pharm. 2018, 15, 4110–4120. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bioink 1 | Bioink 2 | |
|---|---|---|
| Alginate (% w/v) | 2 | 3 |
| Gelatin (% w/v) | 3 | 3 |
| Collagen I (mg/mL) | 0.5 | 0.5 |
| CaSO4 (M) | 0.03 | 0.05 |
| Fibroblasts (cells/mL) | - | 2.5 × 107 |
| THP-1 (cells/mL) | - | 3.5 × 106 |
| A549 cells (cell/mL) | 1.5 × 107 | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Berg, J.; Weber, Z.; Fechler-Bitteti, M.; Hocke, A.C.; Hippenstiel, S.; Elomaa, L.; Weinhart, M.; Kurreck, J. Bioprinted Multi-Cell Type Lung Model for the Study of Viral Inhibitors. Viruses 2021, 13, 1590. https://0-doi-org.brum.beds.ac.uk/10.3390/v13081590
Berg J, Weber Z, Fechler-Bitteti M, Hocke AC, Hippenstiel S, Elomaa L, Weinhart M, Kurreck J. Bioprinted Multi-Cell Type Lung Model for the Study of Viral Inhibitors. Viruses. 2021; 13(8):1590. https://0-doi-org.brum.beds.ac.uk/10.3390/v13081590
Chicago/Turabian StyleBerg, Johanna, Zia Weber, Mona Fechler-Bitteti, Andreas C. Hocke, Stefan Hippenstiel, Laura Elomaa, Marie Weinhart, and Jens Kurreck. 2021. "Bioprinted Multi-Cell Type Lung Model for the Study of Viral Inhibitors" Viruses 13, no. 8: 1590. https://0-doi-org.brum.beds.ac.uk/10.3390/v13081590