
Detection and Complete Genome Analysis of Circoviruses and Cycloviruses in the Small Indian Mongoose (Urva auropunctata): Identification of Novel Species




Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Statement
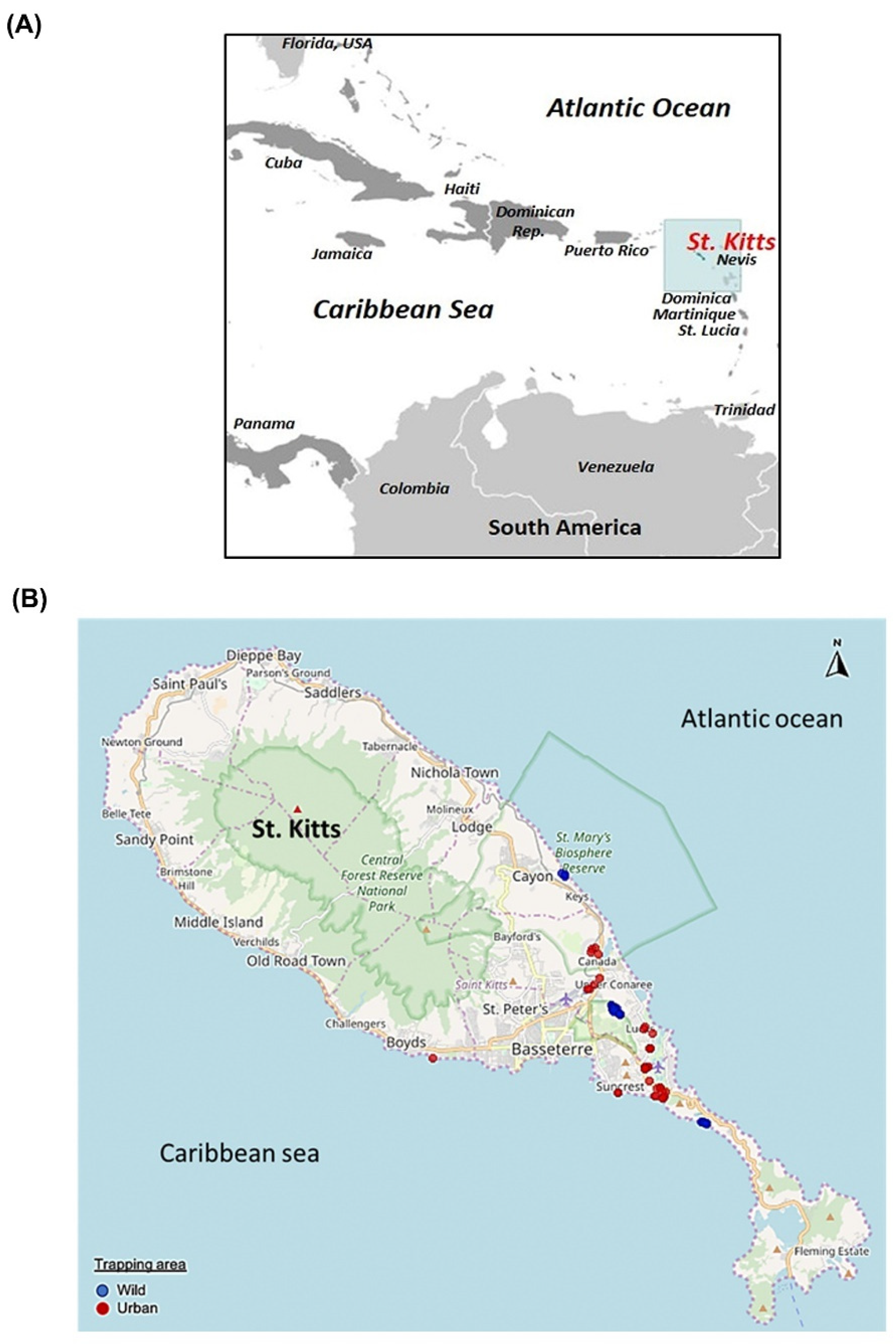
2.2. Sample Collection
2.3. Amplification of Viral DNA
2.4. Nucleotide Sequencing
2.5. Sequence Analysis
2.6. GenBank Accession Numbers
3. Results and Discussion
3.1. Detection of Circoviruses and Cycloviruses in the Small Indian Mongoose
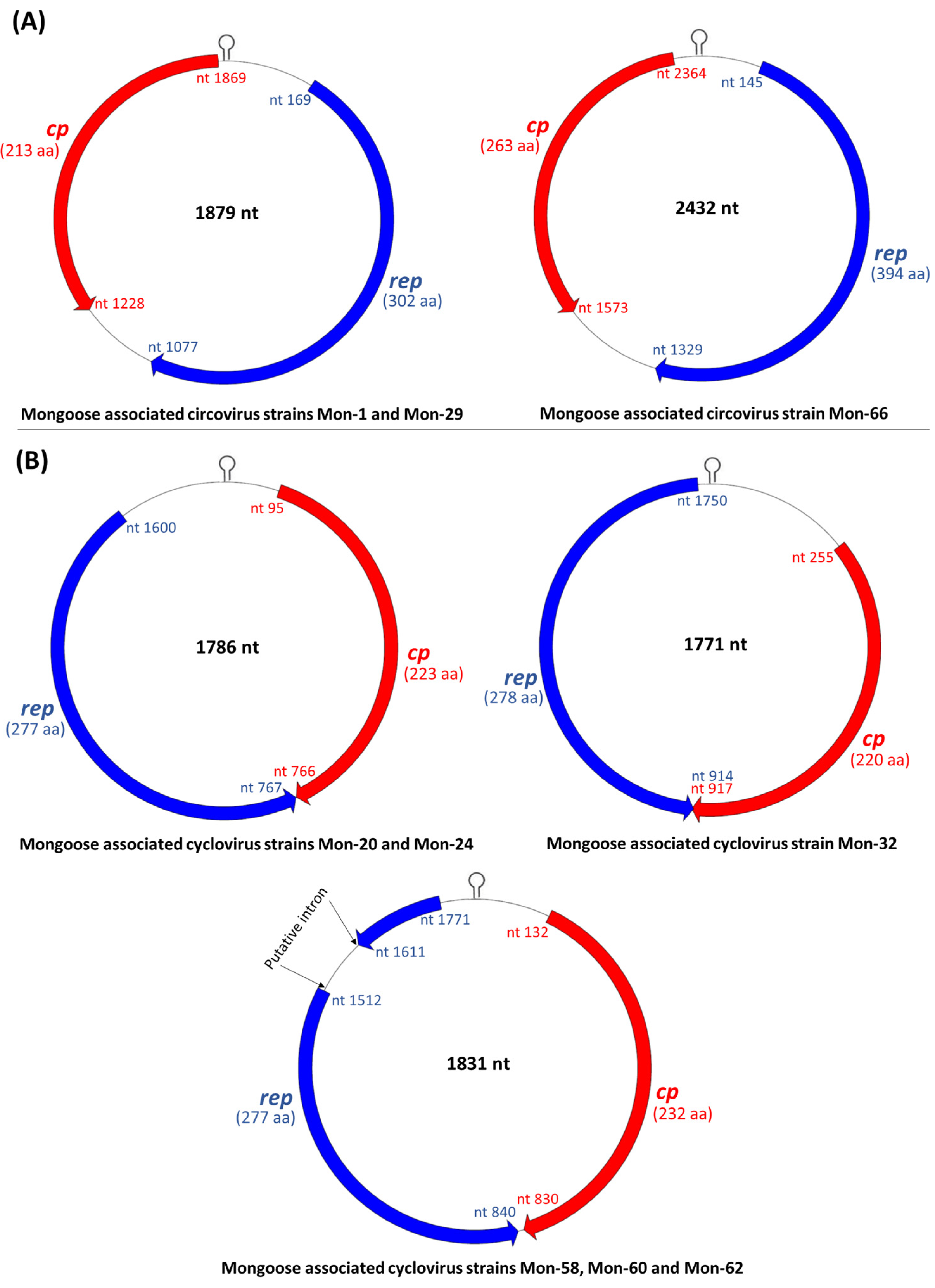
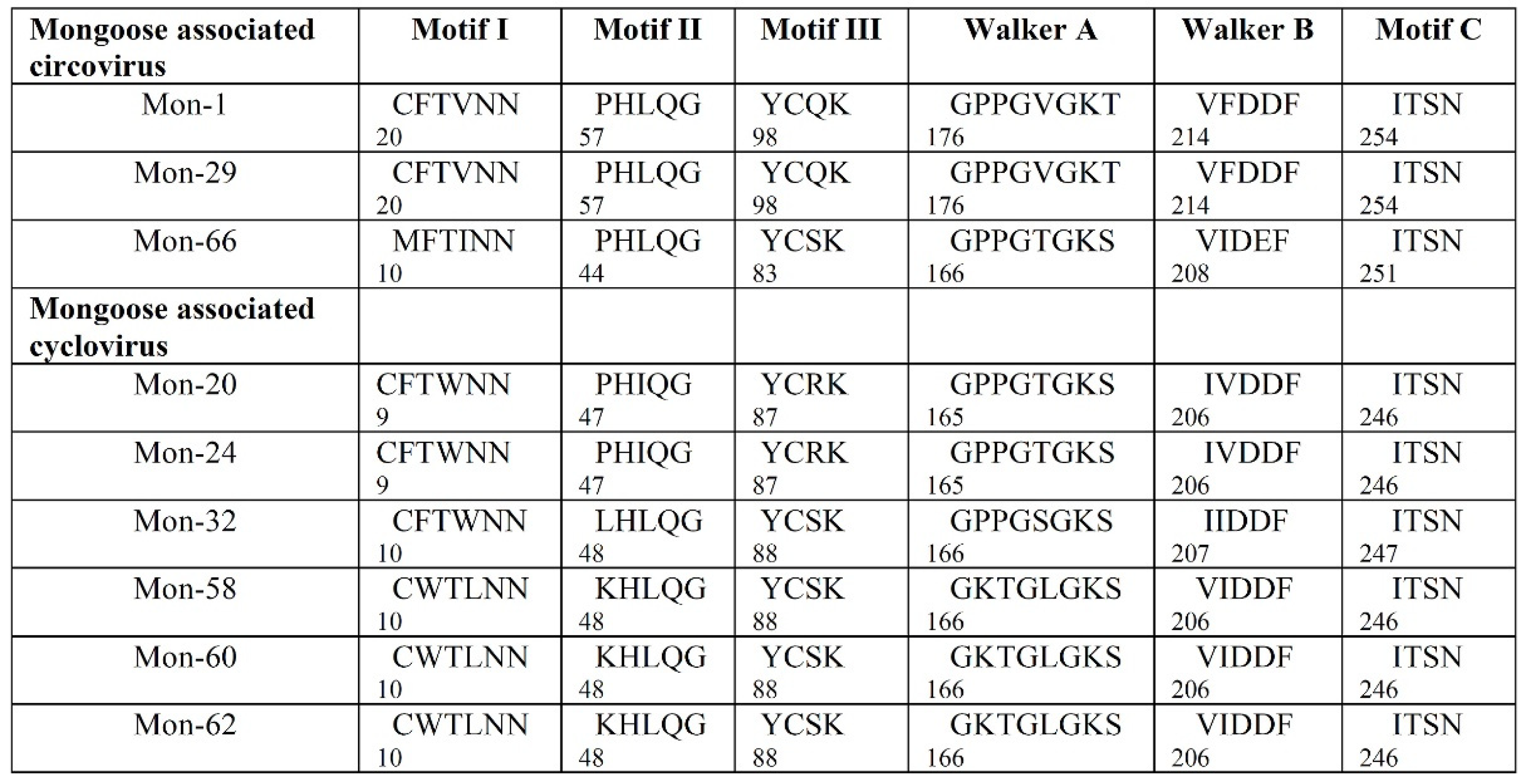
3.2. Analysis of the Complete Genomes of Mongoose Associated Circoviruses and Cycloviruses
3.3. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Breitbart, M.; Delwart, E.; Rosario, K.; Segalés, J.; Varsani, A. ICTV virus taxonomy profile: Circoviridae. J. Gen. Virol. 2017, 98, 1997–1998. [Google Scholar] [CrossRef]
- Rosario, K.; Breitbart, M.; Harrach, B.; Segalés, J.; Delwart, E.; Biagini, P.; Varsani, A. Revisiting the taxonomy of the family Circoviridae: Establishment of the genus Cyclovirus and removal of the genus Gyrovirus. Arch. Virol. 2017, 162, 1447–1463. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Kapoor, A.; Slikas, B.; Bamidele, O.S.; Wang, C.; Shaukat, S.; Masroor, M.A.; Wilson, M.L.; Ndjango, J.-B.N.; Peeters, M.; et al. Multiple Diverse Circoviruses Infect Farm Animals and Are Commonly Found in Human and Chimpanzee Feces. J. Virol. 2010, 84, 1674–1682. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.; Rosario, K.; Breitbart, M.; Duffy, S. Eukaryotic Circular Rep-Encoding Single-Stranded DNA (CRESS DNA) Viruses: Ubiquitous Viruses with Small Genomes and a Diverse Host Range. Adv. Virus Res. 2019, 103, 71–133. [Google Scholar] [CrossRef]
- Delwart, E.; Li, L. Rapidly expanding genetic diversity and host range of the Circoviridae viral family and other Rep encoding small circular ssDNA genomes. Virus Res. 2012, 164, 114–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinfeldt, T.; Finsterbusch, T.; Mankertz, A. Demonstration of Nicking/Joining Activity at the Origin of DNA Replication Associated with the Rep and Rep′ Proteins of Porcine Circovirus Type 1. J. Virol. 2006, 80, 6225–6234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheung, A.K. Identification of an octanucleotide motif sequence essential for viral protein, DNA, and progeny virus biosynthesis at the origin of DNA replication of porcine circovirus type 2. Virology 2004, 324, 28–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alex, C.E.; Fahsbender, E.; Altan, E.; Bildfell, R.; Wolff, P.; Jin, L.; Black, W.; Jackson, K.; Woods, L.; Munk, B.; et al. Viruses in unexplained encephalitis cases in american black bears (ursus americanus). PLoS ONE 2020, 15, e0244056. [Google Scholar] [CrossRef] [PubMed]
- Fisher, M.; Harrison, T.M.R.; Nebroski, M.; Kruczkiewicz, P.; Rothenburger, J.L.; Ambagala, A.; Macbeth, B.; Lung, O. Discovery and comparative genomic analysis of elk circovirus (elkcv), a novel circovirus species and the first reported from a cervid host. Sci. Rep. 2020, 10, 19548. [Google Scholar] [CrossRef]
- Payne, N.; Kraberger, S.; Fontenele, R.S.; Schmidlin, K.; Bergeman, M.H.; Cassaigne, I.; Culver, M.; Varsani, A.; Van Doorslaer, K. Novel circoviruses detected in feces of sonoran felids. Viruses 2020, 12, 1027. [Google Scholar] [CrossRef] [PubMed]
- Tisza, M.J.; Pastrana, D.V.; Welch, N.L.; Stewart, B.; Peretti, A.; Starrett, G.J.; Pang, Y.S.; Krishnamurthy, S.R.; Pesavento, P.A.; McDermott, D.H.; et al. Discovery of several thousand highly diverse circular DNA viruses. Elife 2020, 9, e51971. [Google Scholar] [CrossRef] [PubMed]
- Nishizawa, T.; Sugimoto, Y.; Takeda, T.; Kodera, Y.; Hatano, Y.; Takahashi, M.; Okamoto, H. Identification and full-genome characterization of novel circoviruses in masked palm civets (Paguma larvata). Virus Res. 2018, 258, 50–54. [Google Scholar] [CrossRef] [PubMed]
- Nebbak, A.; Monteil-Bouchard, S.; Berenger, J.M.; Almeras, L.; Parola, P.; Desnues, C. Virome diversity among mosquito populations in a sub-urban region of Marseille, France. Viruses 2021, 13, 768. [Google Scholar] [CrossRef]
- Wu, Z.; Lu, L.; Du, J.; Yang, L.; Ren, X.; Liu, B.; Jiang, J.; Yang, J.; Dong, J.; Sun, L.; et al. Comparative analysis of rodent and small mammal viromes to better understand the wildlife origin of emerging infectious diseases. Microbiome 2018, 6, 178. [Google Scholar] [CrossRef]
- Hui, A.; Altan, E.; Slovis, N.; Fletcher, C.; Deng, X.; Delwart, E. Circovirus in blood of a febrile horse with hepatitis. Viruses 2021, 13, 944. [Google Scholar] [CrossRef]
- Zhang, H.H.; Hu, W.Q.; Li, J.Y.; Liu, T.N.; Zhou, J.Y.; Opriessnig, T.; Xiao, C.T. Novel circovirus species identified in farmed pigs designated as porcine circovirus 4, hunan province, china. Transbound. Emerg. Dis. 2020, 67, 1057–1061. [Google Scholar] [CrossRef] [PubMed]
- Kleymann, A.; Soto, E.; Illanes, O.; Malik, Y.S.; Fuentealba, C.; Ghosh, S. High rates of detection and complete genomic analysis of porcine circovirus 2 (PCV2) in the Lesser Antilles island of St. Kitts: Identification of PCV2b-PCV2d recombinants. Transbound. Emerg. Dis. 2020, 67, 2282–2289. [Google Scholar] [CrossRef]
- Bandoo, R.A.; Bautista, J.; Lund, M.; Newkirk, E.; Squires, J.; Varsani, A.; Kraberger, S. Identification of novel circovirus and anelloviruses from wolverines using a non-invasive faecal sampling approach. Infect. Genet. Evol. 2021, 93, 104914. [Google Scholar] [CrossRef]
- Prades, Y.; Pizarro, R.; Ruiz, M.; Moreno, C.; Avendaño, L.F.; Luchsinger, V. Cyclovirus detection in Chilean adults with and without community-acquired pneumonia. J. Med. Virol. 2021, 93, 4786–4793. [Google Scholar] [CrossRef]
- Sasaki, M.; Orba, Y.; Ueno, K.; Ishii, A.; Moonga, L.; Hangombe, B.M.; Mweene, A.S.; Ito, K.; Sawa, H. Metagenomic analysis of the shrew enteric virome reveals novel viruses related to human stool-associated viruses. J. Gen. Virol. 2015, 96, 440–452. [Google Scholar] [CrossRef]
- Rosario, K.; Mettel, K.A.; Benner, B.E.; Johnson, R.; Scott, C.; Yusseff-Vanegas, S.Z.; Baker, C.C.M.; Cassill, D.L.; Storer, C.; Varsani, A.; et al. Virus discovery in all three major lineages of terrestrial arthropods highlights the diversity of single-stranded DNA viruses associated with invertebrates. PeerJ 2018, 6, e5761. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Wang, H.; Ling, Y.; Yang, S.X.; Wang, X.C.; Zhou, R.; Xiao, Y.Q.; Chen, X.; Yang, J.; Fu, W.G.; et al. Viral metagenomics revealed diverse CRESS-DNA virus genomes in faeces of forest musk deer. Virol. J. 2020, 17, 61. [Google Scholar] [CrossRef] [Green Version]
- Dennis, T.P.W.; Flynn, P.J.; de Souza, W.M.; Singer, J.B.; Moreau, C.S.; Wilson, S.J.; Gifford, R.J. Insights into Circovirus Host Range from the Genomic Fossil Record. J. Virol. 2018, 92, e00145-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altan, E.; Kubiski, S.V.; Burchell, J.; Bicknese, E.; Deng, X.; Delwart, E. The first reptilian circovirus identified infects gut and liver tissues of black-headed pythons. Vet. Res. 2019, 50, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, W.S.; Li, C.X.; Hall, J.; Eden, J.S.; Hyndman, T.H.; Holmes, E.C.; Rose, K. Meta-transcriptomic discovery of a divergent circovirus and a chaphamaparvovirus in captive reptiles with proliferative respiratory syndrome. Viruses 2020, 12, 1073. [Google Scholar] [CrossRef] [PubMed]
- Lorincz, M.; Cságola, A.; Farkas, S.L.; Székely, C.; Tuboly, T. First detection and analysis of a fish circovirus. J. Gen. Virol. 2011, 92, 1817–1821. [Google Scholar] [CrossRef] [PubMed]
- Lorincz, M.; Dan, A.; Lang, M.; Csaba, G.; Toth, A.G.; Szekely, C.; Csagola, A.; Tuboly, T. Novel circovirus in european catfish (Silurus glanis). Arch. Virol. 2012, 157, 1173–1176. [Google Scholar] [CrossRef] [Green Version]
- Doszpoly, A.; Tarján, Z.L.; Glávits, R.; Müller, T.; Benko, M. Full genome sequence of a novel circo-like virus detected in an adult European eel Anguilla anguilla showing signs of cauliflower disease. Dis. Aquat. Organ. 2014, 109, 107–115. [Google Scholar] [CrossRef] [Green Version]
- Opriessnig, T.; Karuppannan, A.K.; Castro, A.M.M.G.; Xiao, C.T. Porcine circoviruses: Current status, knowledge gaps and challenges. Virus Res. 2020, 286, 198044. [Google Scholar] [CrossRef]
- Fogell, D.J.; Martin, R.O.; Groombridge, J.J. Beak and feather disease virus in wild and captive parrots: An analysis of geographic and taxonomic distribution and methodological trends. Arch. Virol. 2016, 161, 2059–2074. [Google Scholar] [CrossRef] [Green Version]
- Soike, D.; Köhler, B.; Albrecht, K. A circovirus-like infection in geese related to a runting syndrome. Avian Pathol. 1999, 28, 199–202. [Google Scholar] [CrossRef]
- Stenzel, T.; Koncicki, A. The epidemiology, molecular characterization and clinical pathology of circovirus infections in pigeons—Current knowledge. Vet. Q. 2017, 37, 166–174. [Google Scholar] [CrossRef]
- Li, L.; McGraw, S.; Zhu, K.; Leutenegger, C.M.; Marks, S.L.; Kubiski, S.; Gaffney, P.; Cruz, F.N.D.; Wang, C.; Delwart, E.; et al. Circovirus in tissues of dogs with vasculitis and hemorrhage. Emerg. Infect. Dis. 2013, 19, 534–541. [Google Scholar] [CrossRef] [PubMed]
- Van Kruiningen, H.J.; Heishima, M.; Kerr, K.M.; Garmendia, A.E.; Helal, Z.; Smyth, J.A. Canine circoviral hemorrhagic enteritis in a dog in Connecticut. J. Vet. Diagn. Investig. 2019, 31, 732–736. [Google Scholar] [CrossRef] [PubMed]
- Lian, H.; Liu, Y.; Li, N.; Wang, Y.; Zhang, S.; Hu, R. Novel circovirus from mink, China. Emerg. Infect. Dis. 2014, 20, 1548–1550. [Google Scholar] [CrossRef]
- Smits, S.L.; Zijlstra, E.E.; van Hellemond, J.J.; Schapendonk, C.M.E.; Bodewes, R.; Schürch, A.C.; Haagmans, B.L.; Osterhaus, A.D.M.E. Novel cyclovirus in human cerebrospinal fluid, Malawi, 2010–2011. Emerg. Infect. Dis. 2013, 19, 1511–1513. [Google Scholar] [CrossRef] [PubMed]
- Barun, A.; Hanson, C.C.; Campell, K.J.; Simberlo, D. A review of small Indian mongoose management and eradications on islands. In Island Invasives: Eradication and Management; Veitch, C.R., Clout, M.N., Towns, D.R., Eds.; IUCN: Gland, Switzerland, 2011; pp. 17–25. [Google Scholar]
- Veron, G.; Jennings, A.P. Javan mongoose or small Indian mongoose—Who is where? Mamm. Biol. 2017, 87, 62–70. [Google Scholar] [CrossRef]
- Sauvé, C.C.; Rees, E.E.; Gilbert, A.T.; Berentsen, A.R.; Allibert, A.; Leighton, P.A. Modeling mongoose rabies in the caribbean: A model—Guided fieldwork approach to identify research priorities. Viruses 2021, 13, 323. [Google Scholar] [CrossRef]
- Nidaira, M.; Takahashi, K.; Ogura, G.; Taira, K.; Okano, S.; Kudaka, J.; Itokazu, K.; Mishiro, S.; Nakamura, M. Detection and phylogenetic analysis of Hepatitis E Viruses from mongooses in Okinawa, Japan. J. Vet. Med. Sci. 2012, 74, 1665–1668. [Google Scholar] [CrossRef] [Green Version]
- Chang, A.M.; Chen, C.C. Molecular characteristics of Carnivore protoparvovirus 1 with high sequence similarity between wild and domestic carnivores in Taiwan. Pathogens 2021, 10, 671. [Google Scholar] [CrossRef]
- Schmiedeknecht, G.; Eickmann, M.; Köhler, K.; Herden, C.E.; Kolesnikova, L.; Förster, C.; Burkhardt, E.H.; König, M.; Thiel, M.; Reinacher, M. Fatal cowpox virus infection in captive Banded mongooses (Mungos mungo). Vet. Pathol. 2010, 47, 547–552. [Google Scholar] [CrossRef] [Green Version]
- Duarte, M.D.; Henriques, A.M.; Barros, S.C.; Fagulha, T.; Mendonça, P.; Carvalho, P.; Monteiro, M.; Fevereiro, M.; Basto, M.P.; Rosalino, L.M.; et al. Snapshot of Viral Infections in Wild Carnivores Reveals Ubiquity of Parvovirus and Susceptibility of Egyptian Mongoose to Feline Panleukopenia Virus. PLoS ONE 2013, 8, e59399. [Google Scholar] [CrossRef]
- Kleymann, A.; Becker, A.A.M.J.; Malik, Y.S.; Kobayashi, N.; Ghosh, S. Detection and molecular characterization of picobirnaviruses (PBVs) in the mongoose: Identification of a novel PBV using an alternative genetic code. Viruses 2020, 12, 99. [Google Scholar] [CrossRef] [Green Version]
- Ogen-Odoi, A.; Miller, B.R.; Happ, C.M.; Maupin, G.O.; Burkot, T.R. Isolation of thogoto virus (Orthomyxoviridae) from the banded mongoose, Mongos mungo (herpestidae), in Uganda. Am. J. Trop. Med. Hyg. 1999, 60, 439–440. [Google Scholar] [CrossRef] [Green Version]
- Conceição-Neto, N.; Zeller, M.; Heylen, E.; Lefrère, H.; Mesquita, J.R.; Matthijnssens, J. Fecal virome analysis of three carnivores reveals a novel nodavirus and multiple gemycircularviruses. Virol. J. 2015, 12, 79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becker, A.A.M.J.; Hill, K.C.; Butaye, P. Unraveling the gut microbiome of the invasive small Indian mongoose (Urva auropunctata) in the Caribbean. Microorganisms 2021, 9, 465. [Google Scholar] [CrossRef] [PubMed]
- Muhire, B.M.; Varsani, A.; Martin, D.P. SDT: A virus classification tool based on pairwise sequence alignment and identity calculation. PLoS ONE 2014, 9, e108277. [Google Scholar] [CrossRef]
- Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003, 31, 3406–3415. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015, 1, vev003. [Google Scholar] [CrossRef] [Green Version]
- Cheng, T.; Halper, B.; Siebert, J.; Cruz-Martinez, L.; Chapwanya, A.; Kelly, P.; Ketzis, J.K.; Vessell, J.; Köster, L.; Yao, C. Parasites of small Indian mongoose, Herpestes auropunctatus, on St. Kitts, West Indies. Parasitol. Res. 2018, 117, 989–994. [Google Scholar] [CrossRef] [Green Version]
- Louppe, V.; Leroy, B.; Herrel, A.; Veron, G. The globally invasive small Indian mongoose Urva auropunctata is likely to spread with climate change. Sci. Rep. 2020, 10, 7461. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Shan, T.; Soji, O.B.; Alam, M.M.; Kunz, T.H.; Zaidi, S.Z.; Delwart, E. Possible cross-species transmission of circoviruses and cycloviruses among farm animals. J. Gen. Virol. 2011, 92, 768–772. [Google Scholar] [CrossRef]
- Reeves, W.K.; Beck, J.; Orlova, M.V.; Daly, J.L.; Pippin, K.; Revan, F.; Loftis, A.D. Ecology of Bats, Their Ectoparasites, and Associated Pathogens on Saint Kitts Island. J. Med. Entomol. 2016, 53, 1218–1225. [Google Scholar] [CrossRef]
- Dayaram, A.; Potter, K.A.; Moline, A.B.; Rosenstein, D.D.; Marinov, M.; Thomas, J.E.; Breitbart, M.; Rosario, K.; Argüello-Astorga, G.R.; Varsani, A. High global diversity of cycloviruses amongst dragonflies. J. Gen. Virol. 2013, 94, 1827–1840. [Google Scholar] [CrossRef] [Green Version]
- Heath, L.; Martin, D.P.; Warburton, L.; Perrin, M.; Horsfield, W.; Kingsley, C.; Rybicki, E.P.; Williamson, A.-L. Evidence of Unique Genotypes of Beak and Feather Disease Virus in Southern Africa. J. Virol. 2004, 78, 9277–9284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaszab, E.; Lengyel, G.; Marton, S.; Dán, Á.; Bányai, K.; Fehér, E. Occurrence and genetic diversity of CRESS DNA viruses in wild birds: A Hungarian study. Sci. Rep. 2020, 10, 7036. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Yang, L.; Ren, X.; He, G.; Zhang, J.; Yang, J.; Qian, Z.; Dong, J.; Sun, L.; Zhu, Y.; et al. Deciphering the bat virome catalog to better understand the ecological diversity of bat viruses and the bat origin of emerging infectious diseases. ISME J. 2016, 10, 609–620. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, T.; Sato, M.; Nishizono, A.; Ahmed, K. A novel bat-associated circovirus identified in northern Hokkaido, Japan. Arch. Virol. 2019, 164, 2179–2182. [Google Scholar] [CrossRef]
- Yinda, C.K.; Ghogomu, S.M.; Conceição-Neto, N.; Beller, L.; Deboutte, W.; Vanhulle, E.; Maes, P.; Van Ranst, M.; Matthijnssens, J. Cameroonian fruit bats harbor divergent viruses, including rotavirus H, bastroviruses, and picobirnaviruses using an alternative genetic code. Virus Evol. 2018, 4, vey008. [Google Scholar] [CrossRef] [Green Version]
- Male, M.F.; Kraberger, S.; Stainton, D.; Kami, V.; Varsani, A. Cycloviruses, gemycircularviruses and other novel replication-associated protein encoding circular viruses in Pacific flying fox (Pteropus tonganus) faeces. Infect. Genet. Evol. 2016, 39, 279–292. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CRESS DNA Viral Sequence from the Small Indian Mongoose | Putative Group | Length of High Quality nt Sequence Analyzed 1 | GenBank Accession Number | Maximum Pairwise nt Sequence (%) Identity with Cognate CRESS Viral DNA Sequence (Virus Name/Detected in Animal, or Environment/Country/Year of Detection/GenBank Accession Number) from Other Animal Species, or Environment 2 |
|---|---|---|---|---|
| Mon-1 3 | I | 358 nt | MZ382570 | 77.65% with Bat circovirus isolate C072/Bat/China/2015/KX834490 3 |
| Mon-2 | II | 463 nt | MZ382579 | 97.62% with Pacific flying fox associated cyclovirus-3/Bat/Tonga/2015/KT732789 |
| Mon-3 | III | 364 nt | MZ382580 | 94.25% with Cockroach-associated cyclovirus/Palmetto bug/USA/2011/JX569794 |
| Mon-6 | IV | 363 nt | MZ382581 | 96.69% with Cyclovirus TN2/Human/Tunisia/2007/GQ404904 |
| Mon-10 | IV | 390 nt | MZ382582 | 97.18% with Cyclovirus TN2/Human/Tunisia/2005/GQ404904 |
| Mon-14 | IV | 381 nt | MZ382583 | 97.38% with Cyclovirus TN2/Human/Tunisia/2005/GQ404904 |
| Mon-16 | V | 363 nt | MZ382584 | 93.11% with Cyclovirus NG_sheep50/Sheep/Nigeria/2009/GQ404982 |
| Mon-18 | IV | 324 nt | MZ382585 | 95.37% with Cyclovirus TN2/Human/Tunisia/2005/GQ404904 |
| Mon-20 3 | VI | 364 nt | MZ382573 | 77.47% with Bat cyclovirus isolate CyV-LimbeP14/Bat/Cameroon/2013/MG693173 3 |
| Mon-22 | IV | 343 nt | MZ382586 | 96.79% with Cyclovirus TN2/Human/Tunisia/2005/GQ404904 |
| Mon-24 3 | VI | 364 nt | MZ382574 | 77.47% with Bat cyclovirus isolate CyV-LimbeP14/Bat/Cameroon/2013/MG693173 3 |
| Mon-25 | IV | 372 nt | MZ382587 | 97.04% with Cyclovirus TN2/Human/Tunisia/2005/GQ404904 |
| Mon-29 3 | I | 358 nt | MZ382571 | 77.65% with Bat circovirus isolate C072/Bat/China/2015/KX834490 3 |
| Mon-32 3 | IV | 363 nt | MZ382572 | 97.25% with Cyclovirus TN2/Human/Tunisia/2005/GQ404904 3 |
| Mon-33 | I | 327 nt | MZ382588 | 79.20% with Bat circovirus isolate C072/Bat/China/2015/KX834490 |
| Mon-36 | VI | 281 nt | MZ382589 | 82.21% with Bat cyclovirus isolate CyV-LimbeP14/Bat/Cameroon/2013/MG693173 |
| Mon-37 | IV | 374 nt | MZ382590 | 96.79% with Cyclovirus TN2/Human/Tunisia/2005/GQ404904 |
| Mon-39 | IV | 350 nt | MZ382591 | 96.86% with Cyclovirus TN2/Human/Tunisia/2005/GQ404904 |
| Mon-41 | IV | 354 nt | MZ382592 | 97.18% with Cyclovirus TN2/Human/Tunisia/2005/GQ404904 |
| Mon-44 | IV | 357 nt | MZ382593 | 97.48% with Cyclovirus TN2/Human/Tunisia/2005/GQ404904 |
| Mon-45 | VI | 275 nt | MZ382594 | 81.45% with Bat cyclovirus isolate CyV-LimbeP14/Bat/Cameroon/2013/MG693173 |
| Mon-56 | IV | 366 nt | MZ382595 | 97.27% with Cyclovirus TN2/Human/Tunisia/2005/GQ404904 |
| Mon-58 3 | VII | 428 nt | MZ382575 | 91.84% with Human cyclovirus VS5700009/Human/Malawi/2010-2011/KC771281 3 |
| Mon-59 | II | 472 nt | MZ382596 | 98.31% with Pacific flying fox associated cyclovirus-3/Bat/Tonga/2015/KT732789 |
| Mon-60 3 | VII | 428 nt | MZ382576 | 91.84% with Human cyclovirus VS5700009/Human/Malawi/2010-2011/KC771281 3 |
| Mon-61 | IV | 354 nt | MZ382597 | 96.61% with Cyclovirus TN2/Human/Tunisia/2005/GQ404904 |
| Mon-62 3 | VII | 428 nt | MZ382577 | 92.07% with Human cyclovirus VS5700009/Human/Malawi/2010-2011/KC771281 3 |
| Mon-66 3 | VIII | 370 nt | MZ382578 | 63.37% with Uncultured virus clone CG263/Environmental sample/USA/2015/KY487932 3 |
| Mon-71 | IV | 284 nt | MZ382598 | 97.18% with Cyclovirus TN2/Human/Tunisia/2005/GQ404904 |
| Mon-76 | IV | 374 nt | MZ382599 | 97.33% with Cyclovirus TN2/Human/Tunisia/2005/GQ404904 |
| Mongoose associated circovirus | GenBank accession number | Maximum/Significant Pairwise nt Sequence (%) Identities 1 | |
| Between mongoose associated circoviruses | With circovirus (Strain Name/Detected in Animal Species/Country/Year/GenBank Accession Number) from other animal species | ||
| Mon-1 | MZ382570 | 99.60% with Mon-29 | 67.40% with Bat circovirus isolate BtPspp.-CV/GD2012/Bat/China/2012/KJ641716 66.90% with Bat associated circovirus 10, isolate HK02976/Bat/Japan/2013/LC456717 |
| Mon-29 | MZ382571 | 99.60% with Mon-1 | 67.20% with Bat circovirus isolate BtPspp.-CV/GD2012/Bat/China/2012/KJ641716 66.60% with Bat associated circovirus 10, isolate HK02976/Bat/Japan/2013/LC456717 |
| Mon-66 | MZ382578 | 58.60% with Mon-29 58.50% with Mon-1 | 60.70% with Porcine circovirus 2 isolate MZ-5/Pig/India/2013/LC004750 60.50% with Porcine circovirus 2 strain MLP-25/Pig/India/2016/MH509731 |
| Mongoose associated cyclovirus | Between mongoose associated cycloviruses | With cyclovirus (Strain name/Detected in animal species/Country/Year/GenBank accession number) from other animal species | |
| Mon-20 | MZ382573 | 100% between Mon-20 and Mon-24 | 72.10% with Bat cyclovirus isolate CyV-LysokaP4/Bat/Cameroon/2013/MG693174 71.50% with Bat cyclovirus isolate CyV-LimbeP14/Bat/Cameroon/2013/MG693173 |
| Mon-24 | MZ382574 | ||
| Mon-32 | MZ382572 | 65.70% with Mon-20 and Mon-24 | 77.30% with Cyclovirus isolate PKgoat11/Goat/Pakistan/2009/HQ738636 75.30% with Cyclovirus isolate PK5006/Human/Pakistan/2007/GQ404844 |
| Mon-58 | MZ382575 | 99.30% with Mon-60 and Mon-62 | 80.20% with Human cyclovirus isolate VS570000/Human/Malawi/2010-2011/KC771281 80.20% with Pacific flying fox associated cyclovirus-3 isolate Tbat_H_103923/Bat/Tonga/2015/KT732788 |
| Mon-60 | MZ382576 | 99.50% with Mon-62 99.30% with Mon-58 | 79.90% with Human cyclovirus isolate VS570000/Human/Malawi/2010-2011/KC771281 79.90% with Pacific flying fox associated cyclovirus-3 isolate Tbat_H_103923/Bat/Tonga/2015/KT732788 |
| Mon-62 | MZ382577 | 99.50% with Mon-60 99.30% with Mon-58 | 80.00% with Pacific flying fox associated cyclovirus-3 isolate Tbat_H_103923/Bat/Tonga/2015/KT732788 79.90% with Human cyclovirus isolate VS570000/Human/Malawi/2010-2011/KC771281 |
| Mongoose associated circovirus | Maximum/Significant Pairwise Deduced aa Sequence (%) Identities 1 | |
| Between mongoose associated circoviruses | With circovirus/CRESS DNA Virus (Strain Name/Detected in Animal Species/Country/Year/GenBank Accession Number) from other animal species | |
| Mon-1 | 99.70% with Mon-29 | 71.50% with Bat associated circovirus 10, isolate HK02976/Bat/Japan/2013/LC456717 |
| Mon-29 | 99.70% with Mon-1 | 71.20% with Bat associated circovirus 10, isolate HK02976/Bat/Japan/2013/LC456717 |
| Mon-66 | 40.00% with Mon-1 39.60% with Mon-29 | 51.20% with Syrmaticus reevesii CRESS-DNA-virus sp. isolate phe68cre9/Wild bird/China/2018/MW182878 |
| Mongoose associated cyclovirus | Between mongoose associated cycloviruses | With cyclovirus (Strain name/Detected in animal species/Country/Year/GenBank accession number) from other animal species |
| Mon-20 | 100% between Mon-20 and Mon-24 | 78.30% with Bat cyclovirus isolate CyV-LysokaP4/Bat/Cameroon/2013/MG693174 and Bat cyclovirus isolate CyV-LimbeP14/Bat/Cameroon/2013/MG693173 |
| Mon-24 | ||
| Mon-32 | 62.80% with Mon-20 and Mon-24 | 87.10% with Cyclovirus isolate PKgoat11/Goat/Pakistan/2009/HQ738636 |
| Mon-58 | 100% between Mon-58, Mon-60, and Mon-62 | 92.40% with Human cyclovirus isolate VS570000/Human/Malawi/2010-2011/KC771281 88.40% with Pacific flying fox associated cyclovirus-3 isolate Tbat_H_103923/Bat/Tonga/2015/KT732788 |
| Mon-60 | ||
| Mon-62 | ||
| Mongoose associated circovirus | Maximum/Significant Pairwise Deduced aa Sequence (%) Identities 1 | |
| Between mongoose associated circoviruses | With circovirus/CRESS DNA Virus (Strain Name/Detected in Animal Species/Country/Year/GenBank Accession Number) from other animal species, or environmental samples | |
| Mon-1 | 100% between Mon-1 and Mon-29 | 42.90% with Bat associated circovirus 10, isolate HK02976/Bat/Japan/2013/LC456717 |
| Mon-29 | ||
| Mon-66 | 24.40% with Mon-1 and Mon-29 | 44.40% with Uncultured virus clone CG83/Wastewater/USA/2015/KY487977 |
| Mongoose associated cyclovirus | Between mongoose associated cycloviruses | With cyclovirus (Strain name/Detected in animal species/Country/Year/GenBank accession number) from other animal species |
| Mon-20 | 100% between Mon-20 and Mon-24 | 59.30% with Bat cyclovirus isolate CyV-LysokaP4/Bat/Cameroon/2013/MG693174 and Bat cyclovirus isolate CyV-LimbeP14/Bat/Cameroon/2013/MG693173 |
| Mon-24 | ||
| Mon-32 | 35.90% with Mon-20 and Mon-24 | 51.60% with Cyclovirus isolate PK5006/Human/Pakistan/2007/GQ404844 |
| Mon-58 | 100% with Mon-62 | 56.10% with Pacific flying fox associated cyclovirus-3 isolate Tbat_H_103923/Bat/Tonga/2015/KT732788 |
| Mon-60 | 99.6% with Mon-58 and Mon-62 | 56.60% with Pacific flying fox associated cyclovirus-3 isolate Tbat_H_103923/Bat/Tonga/2015/KT732788 |
| Mon-62 | 100% with Mon-58 | 56.10% with Pacific flying fox associated cyclovirus-3 isolate Tbat_H_103923/Bat/Tonga/2015/KT732788 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gainor, K.; Becker, A.A.M.J.; Malik, Y.S.; Ghosh, S. Detection and Complete Genome Analysis of Circoviruses and Cycloviruses in the Small Indian Mongoose (Urva auropunctata): Identification of Novel Species. Viruses 2021, 13, 1700. https://0-doi-org.brum.beds.ac.uk/10.3390/v13091700
Gainor K, Becker AAMJ, Malik YS, Ghosh S. Detection and Complete Genome Analysis of Circoviruses and Cycloviruses in the Small Indian Mongoose (Urva auropunctata): Identification of Novel Species. Viruses. 2021; 13(9):1700. https://0-doi-org.brum.beds.ac.uk/10.3390/v13091700
Chicago/Turabian StyleGainor, Kerry, Anne A. M. J. Becker, Yashpal S. Malik, and Souvik Ghosh. 2021. "Detection and Complete Genome Analysis of Circoviruses and Cycloviruses in the Small Indian Mongoose (Urva auropunctata): Identification of Novel Species" Viruses 13, no. 9: 1700. https://0-doi-org.brum.beds.ac.uk/10.3390/v13091700