
Wastewater-Based Epidemiology and Long-Read Sequencing to Identify Enterovirus Circulation in Three Municipalities in Maricopa County, Arizona, Southwest United States between June and October 2020


 , ,
, ,  , , ,
, , , 

Abstract
:1. Introduction
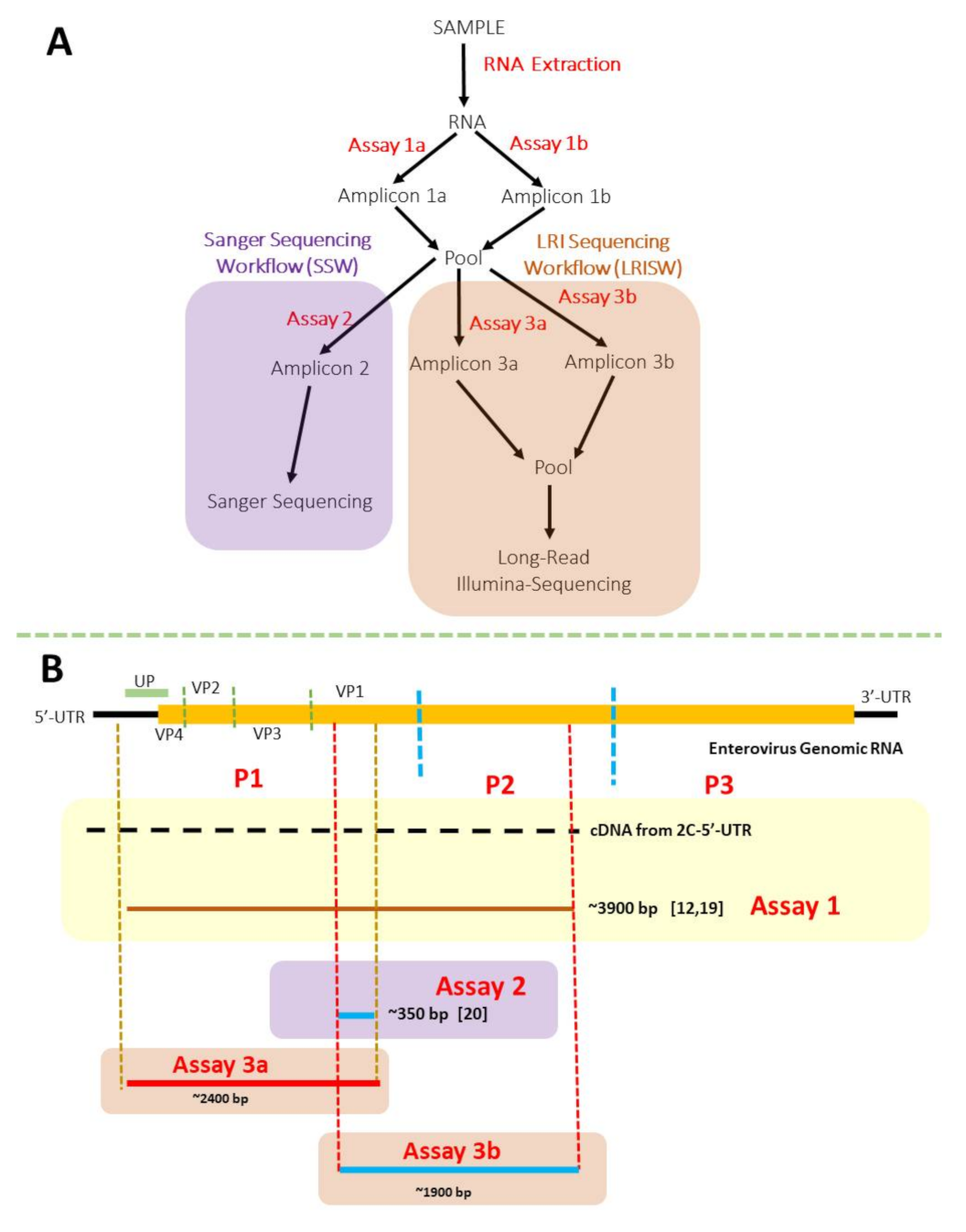
2. Materials and Methods
3. Results
3.1. Nested PCR (SSW and LRISW)
3.1.1. SSW
3.1.2. LRISW
3.2. Sequencing and EV Type Determination
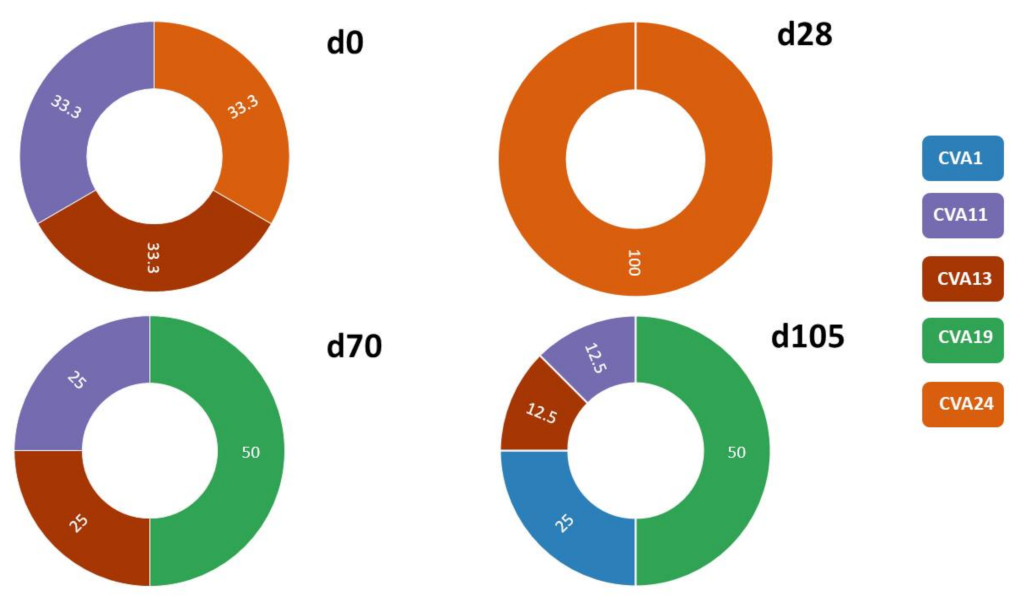
3.2.1. SSW and EV Type Determination
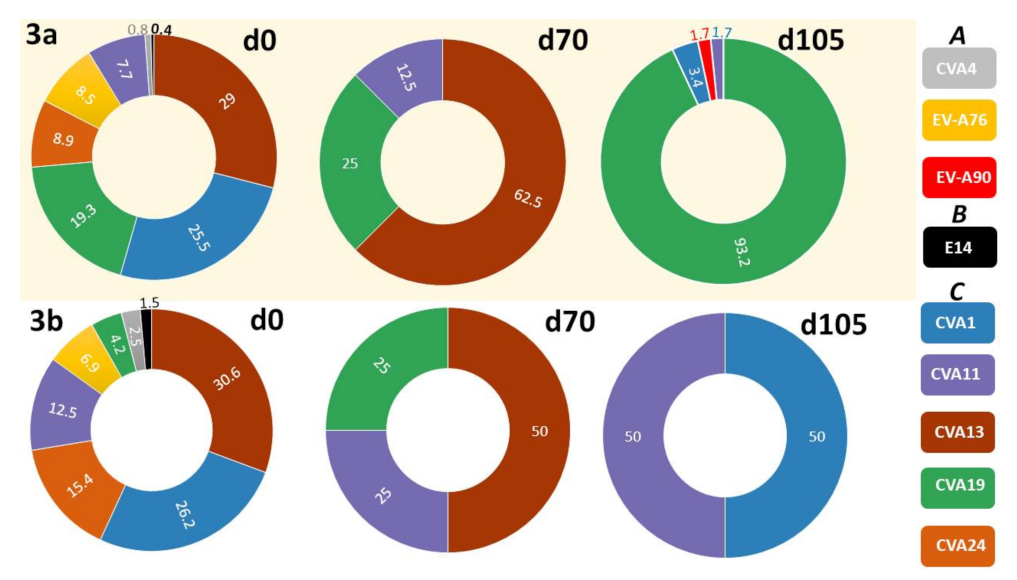
3.2.2. LRISW and EV Type Determination
3.2.3. Comparison of EV Types Identified Using SSW and LRISW
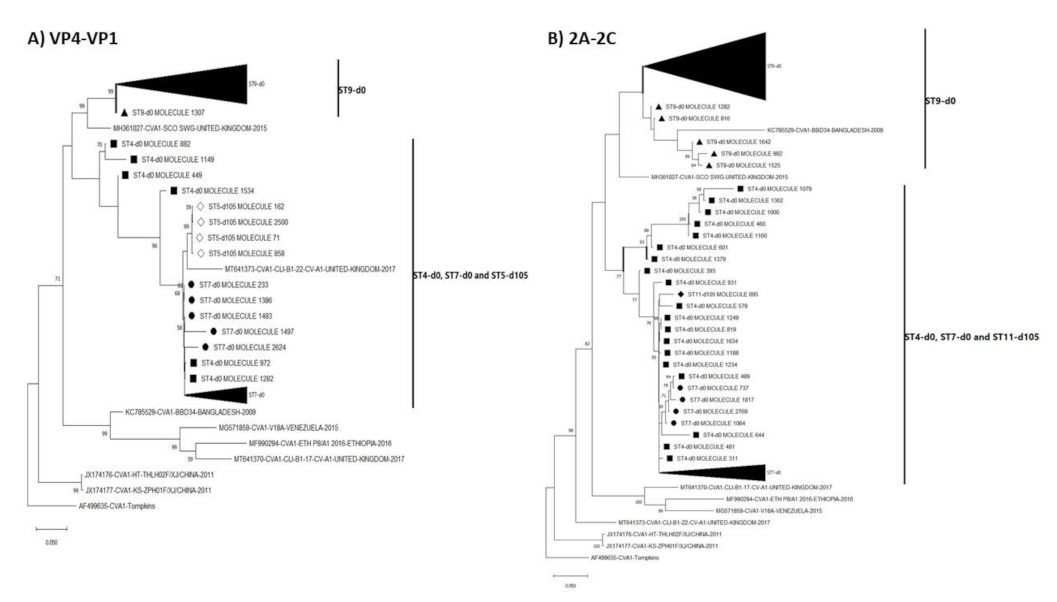
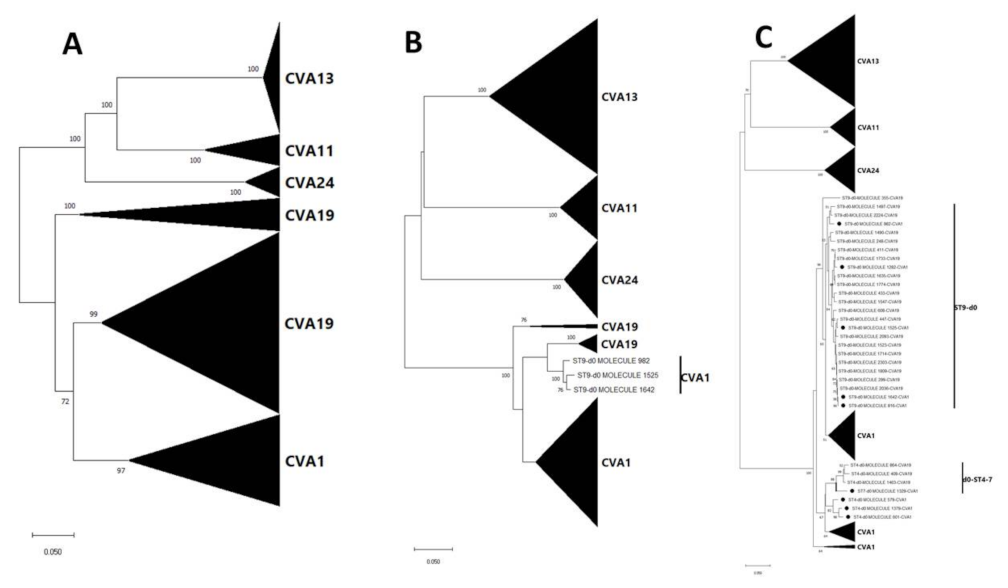
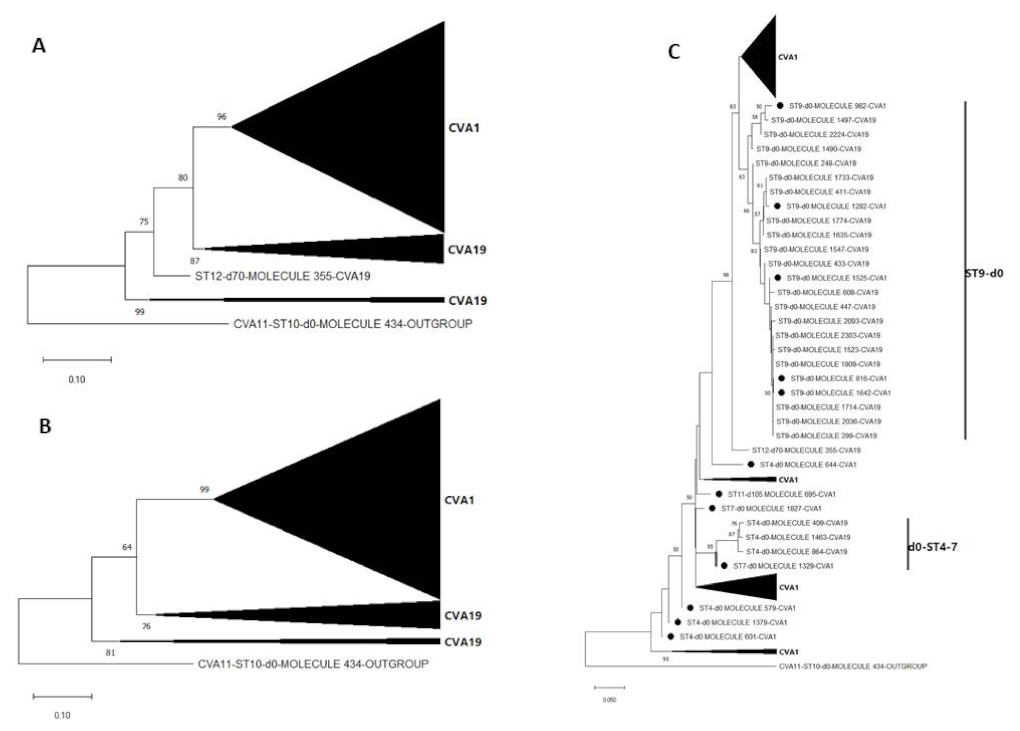
3.3. Pairwise Distance and Phylogenetic Analysis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Enterovirus. Available online: https://www.picornaviridae.com/sg3/enterovirus/enterovirus.htm (accessed on 20 July 2021).
- Oberste, M.S.; Maher, K.; Kilpatrick, D.R.; Pallansch, M.A. Molecular evolution of the human enteroviruses: Correlation of serotype with VP1 sequence and application to picornavirus classification. J. Virol. 1999, 73, 1941–1948. [Google Scholar] [CrossRef] [Green Version]
- Harvala, H.; Broberg, E.; Benschop, K.; Berginc, N.; Ladhani, S.; Susi, P.; Christiansen, C.; McKenna, J.; Allen, D.; Makiello, P.; et al. Recommendations for enterovirus diagnostics and characterisation within and beyond Europe. J. Clin. Virol. 2018, 101, 11–17. [Google Scholar] [CrossRef]
- CDC. Non-Polio Enterovirus. Available online: https://www.cdc.gov/non-polio-enterovirus/index.html (accessed on 20 July 2021).
- Tapparel, C.; Siegrist, F.; Petty, T.J.; Kaiser, L. Picornavirus and enterovirus diversity with associated human diseases. Infect. Genet. Evol. 2013, 14, 282–293. [Google Scholar] [CrossRef]
- Khediri, Z.; Vauloup-Fellous, C.; Benachi, A.; Ayoubi, J.M.; Mandelbrot, L.; Picone, O. Adverse effects of maternal enterovirus infection on the pregnancy outcome: A prospective and retrospective pilot study. Virol. J. 2018, 15, 70. [Google Scholar] [CrossRef]
- WHO. Guidelines for Environmental Surveillance of Poliovirus Circulation. Available online: https://apps.who.int/iris/bitstream/handle/10665/67854/WHO_V-B_03.03_eng.pdf?sequence=1&isAllowed=y (accessed on 20 July 2021).
- Manor, Y.; Handsher, R.; Halmut, T.; Neuman, M.; Bobrov, A.; Rudich, H.; Vonsover, A.; Shulman, L.; Kew, O.; Mendelson, E. Detection of poliovirus circulation by environmental surveillance in the absence of clinical cases in Israel and the Palestinian authority. J. Clin. Microbiol. 1999, 37, 1670–1675. [Google Scholar] [CrossRef] [Green Version]
- Anis, E.; Kopel, E.; Singer, S.R.; Kaliner, E.; Moerman, L.; Moran-Gilad, J.; Sofer, D.; Manor, Y.; Shulman, L.M.; Mendelson, E.; et al. Insidious reintroduction of wild poliovirus into Israel, 2013. Euro Surveill. 2013, 18, 20586. [Google Scholar] [CrossRef] [Green Version]
- Brinkman, N.E.; Fout, G.S.; Keely, S.P. Retrospective Surveillance of Wastewater To Examine Seasonal Dynamics of Enterovirus Infections. mSphere 2017, 2, e00099-17. [Google Scholar] [CrossRef] [Green Version]
- Majumdar, M.; Sharif, S.; Klapsa, D.; Wilton, T.; Alam, M.M.; Fernandez-Garcia, M.D.; Rehman, L.; Mujtaba, G.; McAllister, G.; Harvala, H.; et al. Environmental Surveillance Reveals Complex Enterovirus Circulation Patterns in Human Populations. Open Forum Infect. Dis. 2018, 5, ofy250. [Google Scholar] [CrossRef]
- Majumdar, M.; Martin, J. Detection by Direct Next Generation Sequencing Analysis of Emerging Enterovirus D68 and C109 Strains in an Environmental Sample From Scotland. Front. Microbiol. 2018, 9, 1956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bisseux, M.; Debroas, D.; Mirand, A.; Archimbaud, C.; Peigue-Lafeuille, H.; Bailly, J.L.; Henquell, C. Monitoring of enterovirus diversity in wastewater by ultra-deep sequencing: An effective complementary tool for clinical enterovirus surveillance. Water Res. 2020, 169, 115246. [Google Scholar] [CrossRef]
- Zomahoun, D.J.; Burman, A.L.; Snider, C.J.; Chauvin, C.; Gardner, T.; Lickness, J.S.; Ahmed, J.A.; Diop, O.; Gerber, S.; Anand, A. Impact of COVID-19 Pandemic on Global Poliovirus Surveillance. MMWR Morb. Mortal. Wkly. Rep. 2021, 69, 1648–1652. [Google Scholar] [CrossRef]
- Kies, K.D.; Thomas, A.S.; Binnicker, M.J.; Bashynski, K.L.; Patel, R. Decrease in Enteroviral Meningitis-An Unexpected Benefit of COVID-19 Mitigation? Clin. Infect. Dis. 2020. [Google Scholar] [CrossRef]
- Kuo, S.C.; Tsou, H.H.; Wu, H.Y.; Hsu, Y.T.; Lee, F.J.; Shih, S.M.; Hsiung, C.A.; Chen, W.J. Nonpolio Enterovirus Activity during the COVID-19 Pandemic, Taiwan, 2020. Emerg. Infect. Dis. 2021, 27, 306. [Google Scholar] [CrossRef]
- Pons-Salort, M.; Oberste, M.S.; Pallansch, M.A.; Abedi, G.R.; Takahashi, S.; Grenfell, B.T.; Grassly, N.C. The seasonality of nonpolio enteroviruses in the United States: Patterns and drivers. Proc. Natl. Acad. Sci. USA 2018, 115, 3078–3083. [Google Scholar] [CrossRef] [Green Version]
- US Census Data. Available online: https://www.census.gov/data.html (accessed on 21 July 2021).
- Arita, M.; Kilpatrick, D.R.; Nakamura, T.; Burns, C.C.; Bukbuk, D.; Oderinde, S.B.; Oberste, M.S.; Kew, O.M.; Pallansch, M.A.; Shimizu, H. Development of an efficient entire-capsid-coding-region amplification method for direct detection of poliovirus from stool extracts. J. Clin. Microbiol. 2015, 53, 73–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nix, W.A.; Oberste, M.S.; Pallansch, M.A. Sensitive, seminested PCR amplification of VP1 sequences for direct identification of all enterovirus serotypes from original clinical specimens. J. Clin. Microbiol. 2006, 44, 2698–2704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [Green Version]
- Kroneman, A.; Vennema, H.; Deforche, K.; Avoort, H.V.D.; Penaranda, S.; Oberste, M.S.; Vinje, J.; Koopmans, M. An automated genotyping tool for enteroviruses and noroviruses. J. Clin. Virol. 2011, 51, 121–125. [Google Scholar] [CrossRef]
- Callahan, B.J.; Grinevich, D.; Thakur, S.; Balamotis, M.A.; Yehezkel, T.B. Ultra-accurate microbial amplicon sequencing with synthetic long reads. Microbiome 2021, 9, 130. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Sayers, E.W.; Cavanaugh, M.; Clark, K.; Pruitt, K.D.; Schoch, C.L.; Sherry, S.T.; Karsch-Mizrachi, I. GenBank. Nucleic Acids Res. 2021, 49, D92–D96. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Pons-Salort, M.; Grassly, N.C. Serotype-specific immunity explains the incidence of diseases caused by human enteroviruses. Science 2018, 361, 800–803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, C.C.; Wang, C.Y.; Hsueh, P.R. Co-infections among patients with COVID-19: The need for combination therapy with non-anti-SARS-CoV-2 agents? J. Microbiol. Immunol. Infect. 2020, 53, 505–512. [Google Scholar] [CrossRef]
- Lansbury, L.; Lim, B.; Baskaran, V.; Lim, W.S. Co-infections in people with COVID-19: A systematic review and meta-analysis. J. Infect. 2020, 81, 266–275. [Google Scholar] [CrossRef]
- Lange, S.J.; Ritchey, M.D.; Goodman, A.B.; Dias, T.; Twentyman, E.; Fuld, J.; Schieve, L.A.; Imperatore, G.; Benoit, S.R.; Kite-Powell, A.; et al. Potential Indirect Effects of the COVID-19 Pandemic on Use of Emergency Departments for Acute Life-Threatening Conditions-United States, January-May 2020. MMWR Morb. Mortal. Wkly. Rep. 2020, 69, 795–800. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Garcia, M.D.; Kebe, O.; Fall, A.D.; Ndiaye, K. Identification and molecular characterization of non-polio enteroviruses from children with acute flaccid paralysis in West Africa, 2013–2014. Sci. Rep. 2017, 7, 3808. [Google Scholar] [CrossRef]
- Faleye, T.O.C.; Driver, E.; Bowes, D.; Adhikari, S.; Adams, D.; Varsani, A.; Halden, R.U.; Scotch, M. Pan-Enterovirus Amplicon-Based High-Throughput Sequencing Detects the Complete Capsid of a EVA71 Genotype C1 Variant via Wastewater-Based Epidemiology in Arizona. Viruses 2021, 13, 74. [Google Scholar] [CrossRef]
- Sadeuh-Mba, S.A.; Bessaud, M.; Massenet, D.; Joffret, M.L.; Endegue, M.C.; Njouom, R.; Reynes, J.M.; Rousset, D.; Delpeyroux, F. High frequency and diversity of species C enteroviruses in Cameroon and neighboring countries. J. Clin. Microbiol. 2013, 51, 759–770. [Google Scholar] [CrossRef] [Green Version]
- Brouwer, L.; Moreni, G.; Wolthers, K.C.; Pajkrt, D. World-Wide Prevalence and Genotype Distribution of Enteroviruses. Viruses 2021, 13, 434. [Google Scholar] [CrossRef] [PubMed]
- Jorba, J.; Campagnoli, R.; De, L.; Kew, O. Calibration of multiple poliovirus molecular clocks covering an extended evolutionary range. J. Virol. 2008, 82, 4429–4440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- WHO. Enterovirus Surveillance Guidelines. Available online: https://www.euro.who.int/__data/assets/pdf_file/0020/272810/EnterovirusSurveillanceGuidelines.pdf (accessed on 21 July 2021).
- Domingo, E.; Perales, C. Viral quasispecies. PLoS Genet. 2019, 15, e1008271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bessaud, M.; Joffret, M.L.; Holmblat, B.; Razafindratsimandresy, R.; Delpeyroux, F. Genetic relationship between cocirculating Human enteroviruses species C. PLoS ONE 2011, 6, e24823. [Google Scholar] [CrossRef]
- Combelas, N.; Holmblat, B.; Joffret, M.L.; Colbere-Garapin, F.; Delpeyroux, F. Recombination between poliovirus and coxsackie A viruses of species C: A model of viral genetic plasticity and emergence. Viruses 2011, 3, 1460–1484. [Google Scholar] [CrossRef] [Green Version]
- Muslin, C.; Mac Kain, A.; Bessaud, M.; Blondel, B.; Delpeyroux, F. Recombination in Enteroviruses, a Multi-Step Modular Evolutionary Process. Viruses 2019, 11, 859. [Google Scholar] [CrossRef] [Green Version]
- Chard, A.N.; Datta, S.D.; Tallis, G.; Burns, C.C.; Wassilak, S.G.F.; Vertefeuille, J.F.; Zaffran, M. Progress Toward Polio Eradication-Worldwide, January 2018-March 2020. MMWR Morb. Mortal. Wkly. Rep. 2020, 69, 784–789. [Google Scholar] [CrossRef]
- Alleman, M.M.; Jorba, J.; Greene, S.A.; Diop, O.M.; Iber, J.; Tallis, G.; Goel, A.; Wiesen, E.; Wassilak, S.G.F.; Burns, C.C. Update on Vaccine-Derived Poliovirus Outbreaks-Worldwide, July 2019-February 2020. MMWR Morb. Mortal. Wkly. Rep. 2020, 69, 489–495. [Google Scholar] [CrossRef] [Green Version]
- Shulman, L.M.; Gavrilin, E.; Jorba, J.; Martin, J.; Burns, C.C.; Manor, Y.; Moran-Gilad, J.; Sofer, D.; Hindiyeh, M.Y.; Gamzu, R.; et al. Molecular epidemiology of silent introduction and sustained transmission of wild poliovirus type 1, Israel, 2013. Euro Surveill 2014, 19, 20709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexander, J.P.; Ehresmann, K.; Seward, J.; Wax, G.; Harriman, K.; Fuller, S.; Cebelinski, E.A.; Chen, Q.; Jorba, J.; Kew, O.M.; et al. Transmission of imported vaccine-derived poliovirus in an undervaccinated community in Minnesota. J. Infect. Dis. 2009, 199, 391–397. [Google Scholar] [CrossRef] [Green Version]
- Kitajima, M.; Ahmed, W.; Bibby, K.; Carducci, A.; Gerba, C.P.; Hamilton, K.A.; Haramoto, E.; Rose, J.B. SARS-CoV-2 in wastewater: State of the knowledge and research needs. Sci. Total Environ. 2020, 739, 139076. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Ellenberg, R.M.; Graham, K.E.; Wigginton, K.R. Survivability, Partitioning, and Recovery of Enveloped Viruses in Untreated Municipal Wastewater. Environ. Sci. Technol. 2016, 50, 5077–5085. [Google Scholar] [CrossRef] [PubMed]
- Weblogo. Available online: https://weblogo.berkeley.edu/logo.cgi (accessed on 30 August 2021).







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Assay Name | Primer Name | Annealing Temperatures | Extension Temperature and Time | Amplicon Size (bp) | Reference |
|---|---|---|---|---|---|
| 1a | 5-UTR | 55 °C for 30 s, 32× | 68 °C for 8 min | ~3900 | [19] |
| CRE-R | |||||
| 1b | MM-EV-F2 | 55 °C for 30 s, 32× | 68 °C for 8 min | ~3900 | [12] |
| MM-EV-R1 | |||||
| 2 | AN89 | 45 °C for 30 s, 5× 60 °C for 30 s, 30× | 72 °C for 30 s | ~350 | [20] |
| AN88 | |||||
| 3a | LG-MM-EV-F2 | 45 °C for 30 s, 5× 55 °C for 30 s, 35× | 68 °C for 3 min | ~2400 | This study |
| LG-AN88 | |||||
| 3b | LG-AN89 | 45 °C for 30 s, 5× 55 °C for 30 s, 35× | 68 °C for 3 min | ~1900 | This study |
| LG-CRE-R |
| SSW | LRISW | |||||
|---|---|---|---|---|---|---|
| Site # | Sampling Day | EV Type | Assay 3a EV Type (#) | Assay 3a Divergence (%) | Assay 3b EV Type (#) | Assay 3b Divergence (%) |
| 1 | 105 | CVA13 | EV-A90 (2) | 0.64 | ||
| 2 | 70 | Multiple peaks—EV type undetermined | ||||
| 3 | 0 | Multiple peaks—EV type undetermined | EV-A76 (8) | 0.18–0.55 | EV-A76 (14) | 0.11–0.54 |
| E14 (1) | NA | E14 (8) | 0.05–0.58 | |||
| CVA19 (6) | 0.05–0.41 | |||||
| 105 | CVA19 | CVA19 (20) | 0.05–0.91 | |||
| 4 | 0 | Multiple peaks—EV type undetermined | EV-A76 (2) | 0.32 | EV-A76 (7) | 0.05–0.48 |
| CVA1 (6) | 0.36–16.63 | CVA1 (19) | 0.05–16.70 | |||
| CVA19 (2) | 0.23 | CVA19 (3) | 1.07–1.35 | |||
| CVA13 (9) | 0.05–0.59 | |||||
| 70 | CVA19 | CVA19 (2) | 0.36 | |||
| 105 | CVA19 | CVA19 (52) | 0.09–24.37 | |||
| 5 | 0 | CVA24 | CVA24 (23) | 0.04–1.31 | CVA24 (80) | 0.05–0.96 |
| CVA4 (2) | 0.65 | CVA4 (13) | 0.05–0.48 | |||
| 28 | CVA24 | |||||
| 70 | Multiple peaks—EV type undetermined | |||||
| 105 | CVA1 | CVA1 (4) | 0.23–0.50 | |||
| 6 | 28 | Multiple peaks—EV type undetermined | ||||
| 7 | 0 | Multiple peaks—EV type undetermined | EVA76 (12) | 0.18–0.60 | EVA76 (15) | 0.11–0.54 |
| CVA1(21) | 0.05–5.97 | CVA1 (25) | 0.05–4.48 | |||
| CVA13 (1) | NA | CVA13 (15) | 0.05–28.08 | |||
| CVA11(2) | 15.17 | |||||
| CVA19 (9) | 0.27–0.73 | |||||
| 28 | Multiple peaks—EV type undetermined | |||||
| 105 | CVA19 | CVA19 (16) | 0.27–24.63 | |||
| 8 | 70 | CVA13 | CVA13 (10) | 0.09–0.72 | CVA13 (2) | 0.05 |
| 9 | 0 | Multiple peaks—EV type undetermined | CVA1(39) | 0.05–14.97 | CVA1 (92) | 0.05–10.81 |
| CVA19 (33) | 0.05–5.66 | CVA19 (19) | 0.16–2.79 | |||
| 28 | Multiple peaks—EV type undetermined | |||||
| 105 | CVA19 | CVA19 (20) | 0.14–0.82 | |||
| 10 | 0 | CVA11 | CVA11 (18) | 0.09–0.63 | CVA11 (65) | 0.05–0.64 |
| 105 | CVA11 | CVA11 (2) | 0.72 | CVA11 (1) | NA | |
| 11 | 0 | CVA13 | CVA13 (74) | 0.04–0.67 | CVA13 (135) | 0.05–0.85 |
| 105 | CVA1 | CVA19 (2) | 0.41 | |||
| CVA1 (1) | NA | |||||
| 12 | 70 | CVA19 | CVA19 (2) | 0.59 | CVA19 (1) | NA |
| 13 | 70 | CVA11 | CVA11 (2) | 0.40 | CVA11 (1) | NA |
| Total Variants | 16 | 393 | 525 | |||
| Assay 3a | Assay 3b | |||||
|---|---|---|---|---|---|---|
| S/N | Site No. | Sampling Day | Highest # of Small Reads/Contig | Smallest # of Small Reads/Contig | Highest # of Small Reads/Contig | Smallest # of Small Reads/Contig |
| 1 | 1 | 105 | 1740 | 1234 | NA | NA |
| 2 | 3 | 0 | 1528 | 252 | 1950 | 406 |
| 3 | 3 | 105 | 1754 | 244 | NA | NA |
| 4 | 4 | 0 | 1566 | 356 | 2028 | 196 |
| 5 | 4 | 70 | 828 | 788 | NA | NA |
| 6 | 4 | 105 | 1878 | 230 | NA | NA |
| 7 | 5 | 0 | 1710 | 368 | 2258 | 198 |
| 8 | 5 | 105 | 1280 | 364 | NA | NA |
| 9 | 7 | 0 | 1776 | 202 | 2332 | 152 |
| 10 | 7 | 105 | 1724 | 256 | NA | NA |
| 11 | 8 | 70 | 1518 | 178 | 1056 | 476 |
| 12 | 9 | 0 | 1884 | 240 | 2142 | 124 |
| 13 | 9 | 105 | 1785 | 342 | NA | NA |
| 14 | 10 | 0 | 1626 | 264 | 2108 | 100 |
| 15 | 10 | 105 | 1506 | 484 | 1488 | 1488 |
| 16 | 11 | 0 | 1880 | 150 | 2240 | 150 |
| 17 | 11 | 105 | 864 | 168 | 1637 | 1637 |
| 18 | 12 | 70 | 1162 | 260 | 842 | 700 |
| 19 | 13 | 70 | 834 | 400 | 958 | 958 |
| Range # of small reads/contigs | 1884 | 150 | 2332 | 100 | ||
| Coverage range | 235.5× | 18.75× | 368.21× | 15.79× | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Faleye, T.O.C.; Bowes, D.A.; Driver, E.M.; Adhikari, S.; Adams, D.; Varsani, A.; Halden, R.U.; Scotch, M. Wastewater-Based Epidemiology and Long-Read Sequencing to Identify Enterovirus Circulation in Three Municipalities in Maricopa County, Arizona, Southwest United States between June and October 2020. Viruses 2021, 13, 1803. https://0-doi-org.brum.beds.ac.uk/10.3390/v13091803
Faleye TOC, Bowes DA, Driver EM, Adhikari S, Adams D, Varsani A, Halden RU, Scotch M. Wastewater-Based Epidemiology and Long-Read Sequencing to Identify Enterovirus Circulation in Three Municipalities in Maricopa County, Arizona, Southwest United States between June and October 2020. Viruses. 2021; 13(9):1803. https://0-doi-org.brum.beds.ac.uk/10.3390/v13091803
Chicago/Turabian StyleFaleye, Temitope O. C., Devin A. Bowes, Erin M. Driver, Sangeet Adhikari, Deborah Adams, Arvind Varsani, Rolf U. Halden, and Matthew Scotch. 2021. "Wastewater-Based Epidemiology and Long-Read Sequencing to Identify Enterovirus Circulation in Three Municipalities in Maricopa County, Arizona, Southwest United States between June and October 2020" Viruses 13, no. 9: 1803. https://0-doi-org.brum.beds.ac.uk/10.3390/v13091803