
The Clonal Expansion Dynamics of the HIV-1 Reservoir: Mechanisms of Integration Site-Dependent Proliferation and HIV-1 Persistence

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. The Clonally Expanding HIV-1-Infected Cells Are the Major Barrier to Cure
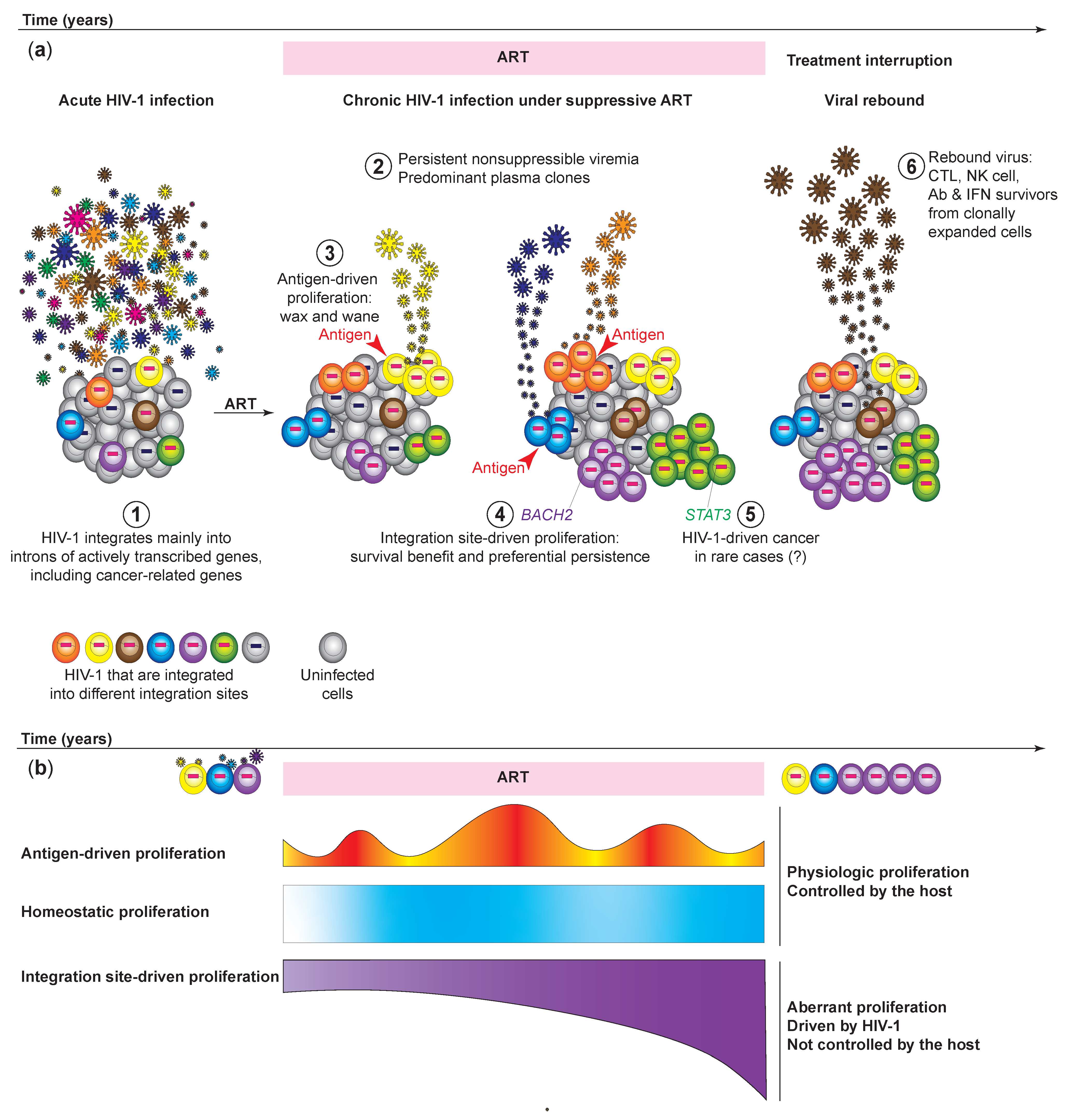
3. HIV-1 Expression from Clonally Expanded HIV-1-Infected Cells Causes Nonsuppressible and Persistent Low-Level Viremia of Predominant HIV-1 Plasma Clones
4. Clonally Expanded HIV-1-Infected Cells Serve as a Source of Viral Rebound after Treatment Interruptions
5. Near Full-Length HIV-1 Proviral Genome Profiling Distinguishes Intact versus Defective HIV-1 Proviruses and Identifies Clonally Expanded HIV-1-Infected Cells
5.1. HIV-1 Proviral Sequencing Provides an Indirect Method for Identifying Clonal Expansion of the Infected Cells
5.2. The Highly Diverse HIV-1 Pol and Env Sequences from Limiting Dilution Viral Outgrowth Culture Serve as Proxies for Measuring Clonally Expanded HIV-1-Infected Cells
6. Integration Site Analysis Provides a Definite Proof of the Clonal Expansion of Infected Cells
6.1. Lessons from HTLV-1: High-Throughput Integration Site Analysis Captures the Clonal Expansion Dynamics of Infected Cells
6.2. The Discovery of Clonally Expanded HIV-1-Infected Cells by Mapping HIV-1 Integration Sites
6.3. Simultaneous Integration Site Profiling and Proviral Sequencing Captures HIV-1 Clonal Expansion Dynamics of Intact versus Defective Proviruses
6.4. The Enrichment of HIV-1 Integration into Heterochromatin Reflects Immune Selection Pressure against Actively Transcribed HIV-1
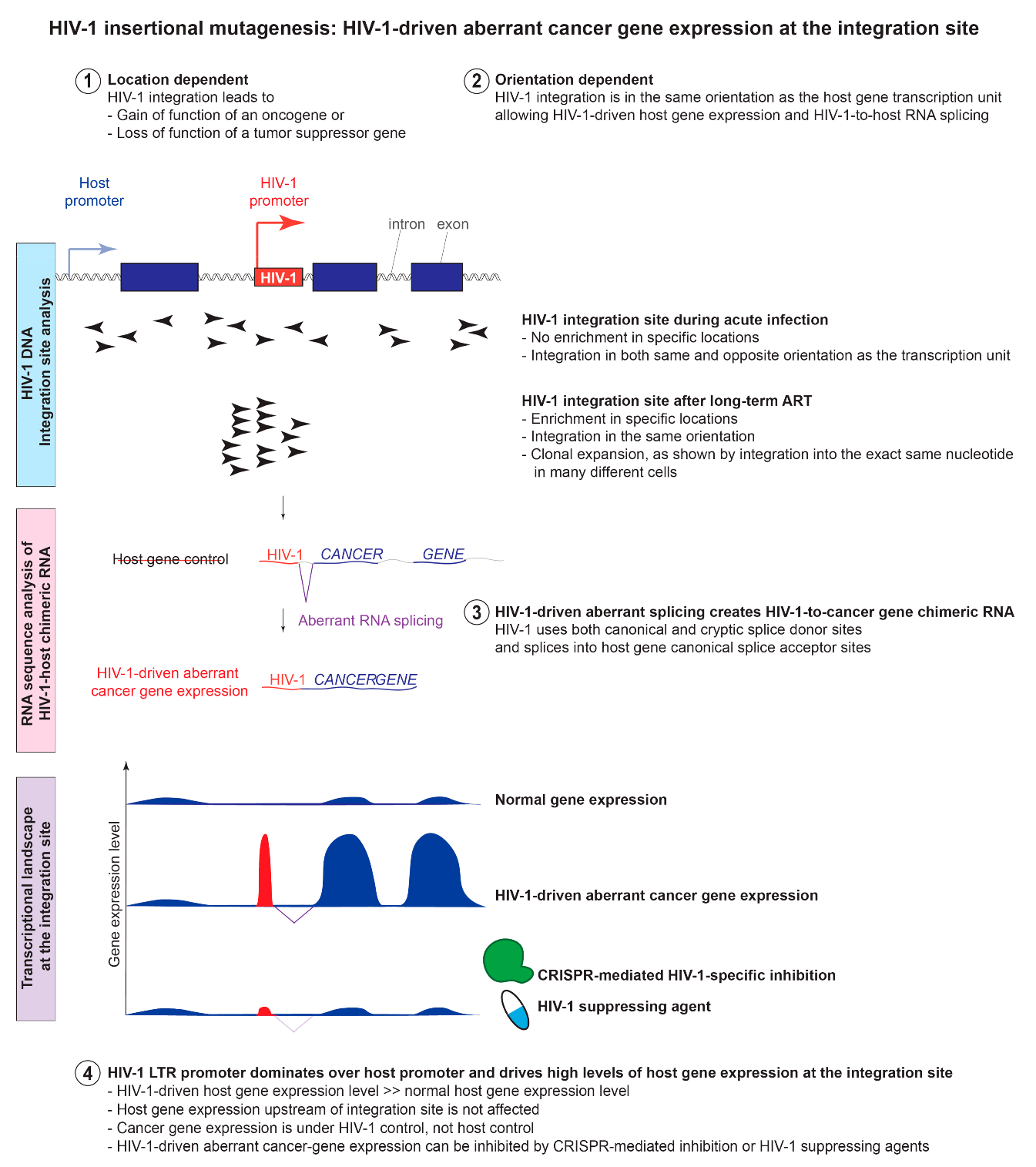
7. HIV-1 Insertional Mutagenesis: The Location-and Orientation-Dependent Impact of HIV-1 Integration into Cancer-Related Genes on HIV-1 Persistence
7.1. Lessons from Acute versus Chronic Infections: Clonal Expansion of HIV-1-Infected Cells Established during Acute Infection Persists after Viral Suppression
7.2. Lessons from Pediatric versus Adult Infections: Clonal Expansion of HIV-1-Infected Cells in Children Is Comparable to That in Adults
7.3. Lessons from Primary Cell Models Support the Location-and Orientation-Dependent Impact of HIV-1 Integration into Cancer-Related Genes on HIV-1 Persistence
7.4. Lessons from Animal Models Support the Location-and Orientation-Dependent Impact of HIV-1 Integration into Cancer-Related Genes on HIV-1 Persistence
8. HIV-1 Insertional Mutagenesis Can Cause Aberrant HIV-1-Driven Proliferation That is out of Control from the Host
8.1. HIV-1 Integration Changes the Host Gene Transcriptional Landscape at the Integration Site through Aberrant Splicing and Read-through Transcription
8.2. HIV-1 Promoter Dominates over the Host Promoter and Drives High Levels of Cancer Gene Expression at the Integration Site
8.3. HIV-1 Insertional Mutagenesis May Cause Cancer Transformation in the Infected Cells in Rare Cases
8.4. Why Does HIV-1 Infection Cause Cancer in the Infected Cells Only in Extremely Rare Cases?
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chun, T.W.; Stuyver, L.; Mizell, S.B.; Ehler, L.A.; Mican, J.A.; Baseler, M.; Lloyd, A.L.; Nowak, M.A.; Fauci, A.S. Presence of an inducible HIV-1 latent reservoir during highly active antiretroviral therapy. Proc. Natl. Acad. Sci. USA 1997, 94, 13193–13197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, J.K.; Hezareh, M.; Gunthard, H.F.; Havlir, D.V.; Ignacio, C.C.; Spina, C.A.; Richman, D.D. Recovery of replication-competent HIV despite prolonged suppression of plasma viremia. Science 1997, 278, 1291–1295. [Google Scholar] [CrossRef]
- Finzi, D.; Hermankova, M.; Pierson, T.; Carruth, L.M.; Buck, C.; Chaisson, R.E.; Quinn, T.C.; Chadwick, K.; Margolick, J.; Brookmeyer, R.; et al. Identification of a reservoir for HIV-1 in patients on highly active antiretroviral therapy. Science 1997, 278, 1295–1300. [Google Scholar] [CrossRef] [PubMed]
- Estes, J.D.; Kityo, C.; Ssali, F.; Swainson, L.; Makamdop, K.N.; Del Prete, G.Q.; Deeks, S.G.; Luciw, P.A.; Chipman, J.G.; Beilman, G.J.; et al. Defining total-body AIDS-virus burden with implications for curative strategies. Nat. Med. 2017, 23, 1271–1276. [Google Scholar] [CrossRef] [Green Version]
- Chun, T.W.; Carruth, L.; Finzi, D.; Shen, X.; DiGiuseppe, J.A.; Taylor, H.; Hermankova, M.; Chadwick, K.; Margolick, J.; Quinn, T.C.; et al. Quantification of latent tissue reservoirs and total body viral load in HIV-1 infection. Nature 1997, 387, 183–188. [Google Scholar] [CrossRef]
- Siliciano, J.D.; Kajdas, J.; Finzi, D.; Quinn, T.C.; Chadwick, K.; Margolick, J.B.; Kovacs, C.; Gange, S.J.; Siliciano, R.F. Long-term follow-up studies confirm the stability of the latent reservoir for HIV-1 in resting CD4+ T cells. Nat. Med. 2003, 9, 727–728. [Google Scholar] [CrossRef]
- Crooks, A.M.; Bateson, R.; Cope, A.B.; Dahl, N.P.; Griggs, M.K.; Kuruc, J.D.; Gay, C.L.; Eron, J.J.; Margolis, D.M.; Bosch, R.J.; et al. Precise Quantitation of the Latent HIV-1 Reservoir: Implications for Eradication Strategies. J. Infect. Dis. 2015, 212, 1361–1365. [Google Scholar] [CrossRef]
- Walker, B.D.; Chakrabarti, S.; Moss, B.; Paradis, T.J.; Flynn, T.; Durno, A.G.; Blumberg, R.S.; Kaplan, J.C.; Hirsch, M.S.; Schooley, R.T. HIV-specific cytotoxic T lymphocytes in seropositive individuals. Nature 1987, 328, 345–348. [Google Scholar] [CrossRef]
- Plata, F.; Autran, B.; Martins, L.P.; Wain-Hobson, S.; Raphael, M.; Mayaud, C.; Denis, M.; Guillon, J.M.; Debre, P. AIDS virus-specific cytotoxic T lymphocytes in lung disorders. Nature 1987, 328, 348–351. [Google Scholar] [CrossRef]
- Nixon, D.F.; Townsend, A.R.; Elvin, J.G.; Rizza, C.R.; Gallwey, J.; McMichael, A.J. HIV-1 gag-specific cytotoxic T lymphocytes defined with recombinant vaccinia virus and synthetic peptides. Nature 1988, 336, 484–487. [Google Scholar] [CrossRef] [PubMed]
- Koenig, S.; Earl, P.; Powell, D.; Pantaleo, G.; Merli, S.; Moss, B.; Fauci, A.S. Group-specific, major histocompatibility complex class I-restricted cytotoxic responses to human immunodeficiency virus 1 (HIV-1) envelope proteins by cloned peripheral blood T cells from an HIV-1-infected individual. Proc. Natl. Acad. Sci. USA 1988, 85, 8638–8642. [Google Scholar] [CrossRef] [Green Version]
- Borrow, P.; Lewicki, H.; Wei, X.; Horwitz, M.S.; Peffer, N.; Meyers, H.; Nelson, J.A.; Gairin, J.E.; Hahn, B.H.; Oldstone, M.B.; et al. Antiviral pressure exerted by HIV-1-specific cytotoxic T lymphocytes (CTLs) during primary infection demonstrated by rapid selection of CTL escape virus. Nat. Med. 1997, 3, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Price, D.A.; Goulder, P.J.; Klenerman, P.; Sewell, A.K.; Easterbrook, P.J.; Troop, M.; Bangham, C.R.; Phillips, R.E. Positive selection of HIV-1 cytotoxic T lymphocyte escape variants during primary infection. Proc. Natl. Acad. Sci. USA 1997, 94, 1890–1895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goulder, P.J.; Phillips, R.E.; Colbert, R.A.; McAdam, S.; Ogg, G.; Nowak, M.A.; Giangrande, P.; Luzzi, G.; Morgan, B.; Edwards, A.; et al. Late escape from an immunodominant cytotoxic T-lymphocyte response associated with progression to AIDS. Nat. Med. 1997, 3, 212–217. [Google Scholar] [CrossRef] [PubMed]
- Petrovas, C.; Casazza, J.P.; Brenchley, J.M.; Price, D.A.; Gostick, E.; Adams, W.C.; Precopio, M.L.; Schacker, T.; Roederer, M.; Douek, D.C.; et al. PD-1 is a regulator of virus-specific CD8+ T cell survival in HIV infection. J. Exp. Med. 2006, 203, 2281–2292. [Google Scholar] [CrossRef] [PubMed]
- Trautmann, L.; Janbazian, L.; Chomont, N.; Said, E.A.; Gimmig, S.; Bessette, B.; Boulassel, M.R.; Delwart, E.; Sepulveda, H.; Balderas, R.S.; et al. Upregulation of PD-1 expression on HIV-specific CD8+ T cells leads to reversible immune dysfunction. Nat. Med. 2006, 12, 1198–1202. [Google Scholar] [CrossRef]
- Day, C.L.; Kaufmann, D.E.; Kiepiela, P.; Brown, J.A.; Moodley, E.S.; Reddy, S.; Mackey, E.W.; Miller, J.D.; Leslie, A.J.; DePierres, C.; et al. PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature 2006, 443, 350–354. [Google Scholar] [CrossRef]
- Schwartz, O.; Maréchal, V.; Gall, S.L.; Lemonnier, F.; Heard, J.-M. Endocytosis of major histocompatibility complex class I molecules is induced by the HIV–1 Nef protein. Nat. Med. 1996, 2, 338–342. [Google Scholar] [CrossRef]
- Pollack, R.A.; Jones, R.B.; Pertea, M.; Bruner, K.M.; Martin, A.R.; Thomas, A.S.; Capoferri, A.A.; Beg, S.A.; Huang, S.H.; Karandish, S.; et al. Defective HIV-1 Proviruses Are Expressed and Can Be Recognized by Cytotoxic T Lymphocytes, which Shape the Proviral Landscape. Cell Host Microbe 2017, 21, 494–506.e4. [Google Scholar] [CrossRef] [Green Version]
- Bui, J.K.; Sobolewski, M.D.; Keele, B.F.; Spindler, J.; Musick, A.; Wiegand, A.; Luke, B.T.; Shao, W.; Hughes, S.H.; Coffin, J.M.; et al. Proviruses with identical sequences comprise a large fraction of the replication-competent HIV reservoir. PLoS Pathog. 2017, 13, e1006283. [Google Scholar] [CrossRef]
- Lorenzi, J.C.; Cohen, Y.Z.; Cohn, L.B.; Kreider, E.F.; Barton, J.P.; Learn, G.H.; Oliveira, T.; Lavine, C.L.; Horwitz, J.A.; Settler, A.; et al. Paired quantitative and qualitative assessment of the replication-competent HIV-1 reservoir and comparison with integrated proviral DNA. Proc. Natl. Acad. Sci. USA 2016, 113, E7908–E7916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hosmane, N.N.; Kwon, K.J.; Bruner, K.M.; Capoferri, A.A.; Beg, S.; Rosenbloom, D.I.; Keele, B.F.; Ho, Y.C.; Siliciano, J.D.; Siliciano, R.F. Proliferation of latently infected CD4(+) T cells carrying replication-competent HIV-1: Potential role in latent reservoir dynamics. J. Exp. Med. 2017, 214, 959–972. [Google Scholar] [CrossRef] [PubMed]
- Wagner, T.A.; McLaughlin, S.; Garg, K.; Cheung, C.Y.; Larsen, B.B.; Styrchak, S.; Huang, H.C.; Edlefsen, P.T.; Mullins, J.I.; Frenkel, L.M. HIV latency. Proliferation of cells with HIV integrated into cancer genes contributes to persistent infection. Science 2014, 345, 570–573. [Google Scholar] [CrossRef] [Green Version]
- Simonetti, F.R.; Zhang, H.; Soroosh, G.P.; Duan, J.; Rhodehouse, K.; Hill, A.L.; Beg, S.A.; McCormick, K.; Raymond, H.E.; Nobles, C.L.; et al. Antigen-driven clonal selection shapes the persistence of HIV-1 infected CD4+ T cells in vivo. J. Clin. Investig. 2020, 131, e145254. [Google Scholar] [CrossRef]
- Simonetti, F.R.; Sobolewski, M.D.; Fyne, E.; Shao, W.; Spindler, J.; Hattori, J.; Anderson, E.M.; Watters, S.A.; Hill, S.; Wu, X.; et al. Clonally expanded CD4+ T cells can produce infectious HIV-1 in vivo. Proc. Natl. Acad. Sci. USA 2016, 113, 1883–1888. [Google Scholar] [CrossRef] [Green Version]
- Mendoza, P.; Jackson, J.R.; Oliveira, T.Y.; Gaebler, C.; Ramos, V.; Caskey, M.; Jankovic, M.; Nussenzweig, M.C.; Cohn, L.B. Antigen-responsive CD4+ T cell clones contribute to the HIV-1 latent reservoir. J. Exp. Med. 2020, 217, e20200051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Douek, D.C.; Brenchley, J.M.; Betts, M.R.; Ambrozak, D.R.; Hill, B.J.; Okamoto, Y.; Casazza, J.P.; Kuruppu, J.; Kunstman, K.; Wolinsky, S.; et al. HIV preferentially infects HIV-specific CD4+ T cells. Nature 2002, 417, 95–98. [Google Scholar] [CrossRef]
- Chomont, N.; El-Far, M.; Ancuta, P.; Trautmann, L.; Procopio, F.A.; Yassine-Diab, B.; Boucher, G.; Boulassel, M.R.; Ghattas, G.; Brenchley, J.M.; et al. HIV reservoir size and persistence are driven by T cell survival and homeostatic proliferation. Nat. Med. 2009, 15, 893–900. [Google Scholar] [CrossRef]
- Macallan, D.C.; Busch, R.; Asquith, B. Current estimates of T cell kinetics in humans. Curr. Opin. Syst. Biol. 2019, 18, 77–86. [Google Scholar] [CrossRef]
- Macallan, D.C.; Wallace, D.; Zhang, Y.; De Lara, C.; Worth, A.T.; Ghattas, H.; Griffin, G.E.; Beverley, P.C.; Tough, D.F. Rapid turnover of effector-memory CD4(+) T cells in healthy humans. J. Exp. Med. 2004, 200, 255–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venanzi Rullo, E.; Pinzone, M.R.; Cannon, L.; Weissman, S.; Ceccarelli, M.; Zurakowski, R.; Nunnari, G.; O’Doherty, U. Persistence of an intact HIV reservoir in phenotypically naive T cells. JCI Insight 2020, 5, e133157. [Google Scholar] [CrossRef]
- Bosque, A.; Famiglietti, M.; Weyrich, A.S.; Goulston, C.; Planelles, V. Homeostatic proliferation fails to efficiently reactivate HIV-1 latently infected central memory CD4+ T cells. PLoS Pathog. 2011, 7, e1002288. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Gurule, E.E.; Brennan, T.P.; Gerold, J.M.; Kwon, K.J.; Hosmane, N.N.; Kumar, M.R.; Beg, S.A.; Capoferri, A.A.; Ray, S.C.; et al. Expanded cellular clones carrying replication-competent HIV-1 persist, wax, and wane. Proc. Natl. Acad. Sci. USA 2018, 115, E2575–E2584. [Google Scholar] [CrossRef] [Green Version]
- Nettles, R.E.; Kieffer, T.L.; Kwon, P.; Monie, D.; Han, Y.; Parsons, T.; Cofrancesco, J.; Gallant, J.E.; Quinn, T.C.; Jackson, B.; et al. Intermittent HIV-1 Viremia (Blips) and Drug Resistance in Patients Receiving HAART. JAMA 2005, 293, 817–829. [Google Scholar] [CrossRef] [Green Version]
- Havlir, D.V.; Bassett, R.; Levitan, D.; Gilbert, P.; Tebas, P.; Collier, A.C.; Hirsch, M.S.; Ignacio, C.; Condra, J.; Günthard, H.F.; et al. Prevalence and Predictive Value of Intermittent Viremia With Combination HIV Therapy. JAMA 2001, 286, 171–179. [Google Scholar] [CrossRef] [Green Version]
- Bailey, J.R.; Sedaghat, A.R.; Kieffer, T.; Brennan, T.; Lee, P.K.; Wind-Rotolo, M.; Haggerty, C.M.; Kamireddi, A.R.; Liu, Y.; Lee, J.; et al. Residual human immunodeficiency virus type 1 viremia in some patients on Antiretroviral therapy is dominated by a small number of invariant clones rarely found in circulating CD4(+) T cells. J. Virol. 2006, 80, 6441–6457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halvas, E.K.; Joseph, K.W.; Brandt, L.D.; Guo, S.; Sobolewski, M.D.; Jacobs, J.L.; Tumiotto, C.; Bui, J.K.; Cyktor, J.C.; Keele, B.F.; et al. HIV-1 viremia not suppressible by antiretroviral therapy can originate from large T cell clones producing infectious virus. J. Clin. Investig. 2020, 130, 5847–5857. [Google Scholar] [CrossRef] [PubMed]
- Cohn, L.B.; da Silva, I.T.; Valieris, R.; Huang, A.S.; Lorenzi, J.C.C.; Cohen, Y.Z.; Pai, J.A.; Butler, A.L.; Caskey, M.; Jankovic, M.; et al. Clonal CD4(+) T cells in the HIV-1 latent reservoir display a distinct gene profile upon reactivation. Nat. Med. 2018, 24, 604–609. [Google Scholar] [CrossRef]
- Liu, R.; Yeh, Y.J.; Varabyou, A.; Collora, J.A.; Sherrill-Mix, S.; Talbot, C.C., Jr.; Mehta, S.; Albrecht, K.; Hao, H.; Zhang, H.; et al. Single-cell transcriptional landscapes reveal HIV-1-driven aberrant host gene transcription as a potential therapeutic target. Sci. Transl. Med. 2020, 12, eaaz0802. [Google Scholar] [CrossRef] [PubMed]
- Cole, B.; Lambrechts, L.; Gantner, P.; Noppe, Y.; Bonine, N.; Witkowski, W.; Chen, L.; Palmer, S.; Mullins, J.I.; Chomont, N.; et al. In-depth single-cell analysis of translation-competent HIV-1 reservoirs identifies cellular sources of plasma viremia. Nat. Commun. 2021, 12, 3727. [Google Scholar] [CrossRef] [PubMed]
- Salantes, D.B.; Zheng, Y.; Mampe, F.; Srivastava, T.; Beg, S.; Lai, J.; Li, J.Z.; Tressler, R.L.; Koup, R.A.; Hoxie, J.; et al. HIV-1 latent reservoir size and diversity are stable following brief treatment interruption. J. Clin. Investig. 2018, 128, 3102–3115. [Google Scholar] [CrossRef] [Green Version]
- Lu, C.-L.; Pai, J.A.; Nogueira, L.; Mendoza, P.; Gruell, H.; Oliveira, T.Y.; Barton, J.; Lorenzi, J.C.C.; Cohen, Y.Z.; Cohn, L.B.; et al. Relationship between intact HIV-1 proviruses in circulating CD4+ T cells and rebound viruses emerging during treatment interruption. Proc. Natl. Acad. Sci. USA 2018, 115, E11341–E11348. [Google Scholar] [CrossRef] [Green Version]
- Bertagnolli, L.N.; Varriale, J.; Sweet, S.; Brockhurst, J.; Simonetti, F.R.; White, J.; Beg, S.; Lynn, K.; Mounzer, K.; Frank, I.; et al. Autologous IgG antibodies block outgrowth of a substantial but variable fraction of viruses in the latent reservoir for HIV-1. Proc. Natl. Acad. Sci. USA 2020, 117, 32066–32077. [Google Scholar] [CrossRef] [PubMed]
- Gondim, M.V.P.; Sherrill-Mix, S.; Bibollet-Ruche, F.; Russell, R.M.; Trimboli, S.; Smith, A.G.; Li, Y.; Liu, W.; Avitto, A.N.; DeVoto, J.C.; et al. Heightened resistance to host type 1 interferons characterizes HIV-1 at transmission and after antiretroviral therapy interruption. Sci. Transl. Med. 2021, 13, eabd8179. [Google Scholar] [CrossRef] [PubMed]
- Bagasra, O.; Lavi, E.; Bobroski, L.; Khalili, K.; Pestaner, J.P.; Tawadros, R.; Pomerantz, R.J. Cellular reservoirs of HIV-1 in the central nervous system of infected individuals: Identification by the combination of in situ polymerase chain reaction and immunohistochemistry. AIDS 1996, 10, 573–585. [Google Scholar] [CrossRef]
- Fukazawa, Y.; Lum, R.; Okoye, A.A.; Park, H.; Matsuda, K.; Bae, J.Y.; Hagen, S.I.; Shoemaker, R.; Deleage, C.; Lucero, C.; et al. B cell follicle sanctuary permits persistent productive simian immunodeficiency virus infection in elite controllers. Nat. Med. 2015, 21, 132–139. [Google Scholar] [CrossRef] [Green Version]
- Kuo, H.H.; Banga, R.; Lee, G.Q.; Gao, C.; Cavassini, M.; Corpataux, J.M.; Blackmer, J.E.; Zur Wiesch, S.; Yu, X.G.; Pantaleo, G.; et al. Blood and Lymph Node Dissemination of Clonal Genome-Intact Human Immunodeficiency Virus 1 DNA Sequences During Suppressive Antiretroviral Therapy. J. Infect. Dis. 2020, 222, 655–660. [Google Scholar] [CrossRef]
- McManus, W.R.; Bale, M.J.; Spindler, J.; Wiegand, A.; Musick, A.; Patro, S.C.; Sobolewski, M.D.; Musick, V.K.; Anderson, E.M.; Cyktor, J.C.; et al. HIV-1 in lymph nodes is maintained by cellular proliferation during antiretroviral therapy. J. Clin. Investig. 2019, 129, 4629–4642. [Google Scholar] [CrossRef]
- Wu, V.H.; Nobles, C.L.; Kuri-Cervantes, L.; McCormick, K.; Everett, J.K.; Nguyen, S.; Del Rio Estrada, P.M.; González-Navarro, M.; Torres-Ruiz, F.; Ávila-Ríos, S.; et al. Assessment of HIV-1 integration in tissues and subsets across infection stages. JCI Insight 2020, 5, e139783. [Google Scholar] [CrossRef] [PubMed]
- Cuevas, J.M.; Geller, R.; Garijo, R.; López-Aldeguer, J.; Sanjuán, R. Extremely High Mutation Rate of HIV-1 In Vivo. PLoS Biol. 2015, 13, e1002251. [Google Scholar] [CrossRef] [Green Version]
- Roberts, J.D.; Bebenek, K.; Kunkel, T.A. The accuracy of reverse transcriptase from HIV-1. Science 1988, 242, 1171–1173. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, S.; Graf, E.H.; Dahl, V.; Strain, M.C.; Yukl, S.A.; Lysenko, E.S.; Bosch, R.J.; Lai, J.; Chioma, S.; Emad, F.; et al. Comparative analysis of measures of viral reservoirs in HIV-1 eradication studies. PLoS Pathog. 2013, 9, e1003174. [Google Scholar] [CrossRef] [PubMed]
- Ho, Y.C.; Shan, L.; Hosmane, N.N.; Wang, J.; Laskey, S.B.; Rosenbloom, D.I.; Lai, J.; Blankson, J.N.; Siliciano, J.D.; Siliciano, R.F. Replication-competent noninduced proviruses in the latent reservoir increase barrier to HIV-1 cure. Cell 2013, 155, 540–551. [Google Scholar] [CrossRef] [Green Version]
- Bruner, K.M.; Murray, A.J.; Pollack, R.A.; Soliman, M.G.; Laskey, S.B.; Capoferri, A.A.; Lai, J.; Strain, M.C.; Lada, S.M.; Hoh, R.; et al. Defective proviruses rapidly accumulate during acute HIV-1 infection. Nat. Med. 2016, 22, 1043–1049. [Google Scholar] [CrossRef] [Green Version]
- Hiener, B.; Horsburgh, B.A.; Eden, J.S.; Barton, K.; Schlub, T.E.; Lee, E.; von Stockenstrom, S.; Odevall, L.; Milush, J.M.; Liegler, T.; et al. Identification of Genetically Intact HIV-1 Proviruses in Specific CD4(+) T Cells from Effectively Treated Participants. Cell Rep. 2017, 21, 813–822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, G.Q.; Orlova-Fink, N.; Einkauf, K.; Chowdhury, F.Z.; Sun, X.; Harrington, S.; Kuo, H.H.; Hua, S.; Chen, H.R.; Ouyang, Z.; et al. Clonal expansion of genome-intact HIV-1 in functionally polarized Th1 CD4+ T cells. J. Clin. Investig. 2017, 127, 2689–2696. [Google Scholar] [CrossRef] [PubMed]
- Bruner, K.M.; Wang, Z.; Simonetti, F.R.; Bender, A.M.; Kwon, K.J.; Sengupta, S.; Fray, E.J.; Beg, S.A.; Antar, A.A.R.; Jenike, K.M.; et al. A quantitative approach for measuring the reservoir of latent HIV-1 proviruses. Nature 2019, 566, 120–125. [Google Scholar] [CrossRef]
- Silver, J.; Keerikatte, V. Novel use of polymerase chain reaction to amplify cellular DNA adjacent to an integrated provirus. J. Virol. 1989, 63, 1924–1928. [Google Scholar] [CrossRef] [Green Version]
- Han, Y.F.; Lassen, K.; Monie, D.; Sedaghat, A.R.; Shimoji, S.; Liu, X.; Pierson, T.C.; Margolick, J.B.; Siliciano, R.F.; Siliciano, J.D. Resting CD4(+) T cells from human immunodeficiency virus type I (HIV-1)-infected individuals carry integrated HIV-1 Genomes within actively transcribed host genes. J. Virol. 2004, 78, 6122–6133. [Google Scholar] [CrossRef] [Green Version]
- Chun, T.W.; Finzi, D.; Margolick, J.; Chadwick, K.; Schwartz, D.; Siliciano, R.F. In vivo fate of HIV-1-infected T cells: Quantitative analysis of the transition to stable latency. Nat. Med. 1995, 1, 1284–1290. [Google Scholar] [CrossRef]
- Gillet, N.A.; Malani, N.; Melamed, A.; Gormley, N.; Carter, R.; Bentley, D.; Berry, C.; Bushman, F.D.; Taylor, G.P.; Bangham, C.R. The host genomic environment of the provirus determines the abundance of HTLV-1-infected T-cell clones. Blood 2011, 117, 3113–3122. [Google Scholar] [CrossRef] [PubMed]
- Berry, C.C.; Gillet, N.A.; Melamed, A.; Gormley, N.; Bangham, C.R.; Bushman, F.D. Estimating abundances of retroviral insertion sites from DNA fragment length data. Bioinformatics 2012, 28, 755–762. [Google Scholar] [CrossRef] [Green Version]
- Maldarelli, F.; Wu, X.; Su, L.; Simonetti, F.R.; Shao, W.; Hill, S.; Spindler, J.; Ferris, A.L.; Mellors, J.W.; Kearney, M.F.; et al. HIV latency. Specific HIV integration sites are linked to clonal expansion and persistence of infected cells. Science 2014, 345, 179–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patro, S.C.; Brandt, L.D.; Bale, M.J.; Halvas, E.K.; Joseph, K.W.; Shao, W.; Wu, X.; Guo, S.; Murrell, B.; Wiegand, A.; et al. Combined HIV-1 sequence and integration site analysis informs viral dynamics and allows reconstruction of replicating viral ancestors. Proc. Natl. Acad. Sci. USA 2019, 116, 25891–25899. [Google Scholar] [CrossRef] [PubMed]
- Einkauf, K.B.; Lee, G.Q.; Gao, C.; Sharaf, R.; Sun, X.; Hua, S.; Chen, S.M.; Jiang, C.; Lian, X.; Chowdhury, F.Z.; et al. Intact HIV-1 proviruses accumulate at distinct chromosomal positions during prolonged antiretroviral therapy. J. Clin. Investig. 2019, 129, 988–998. [Google Scholar] [CrossRef] [Green Version]
- Einkauf, K.B.; Osborn, M.; Gao, C.; Parsons, E.; Jiang, C.; Lian, X.; Sun, X.; Blackmer, J.E.; Rosenberg, E.S.; Yu, X.; et al. Evolutionary dynamics of HIV reservoir cells via a novel single-cell multiomics assay. Presented at the Conferences on Retroviruses and Opportunistic Infections, Virtual. 6 March–10 March 2021. [Google Scholar]
- Jiang, C.; Lian, X.; Gao, C.; Sun, X.; Einkauf, K.B.; Chevalier, J.M.; Chen, S.M.Y.; Hua, S.; Rhee, B.; Chang, K.; et al. Distinct viral reservoirs in individuals with spontaneous control of HIV-1. Nature 2020, 585, 261–267. [Google Scholar] [CrossRef]
- Blankson, J.N.; Bailey, J.R.; Thayil, S.; Yang, H.C.; Lassen, K.; Lai, J.; Gandhi, S.K.; Siliciano, J.D.; Williams, T.M.; Siliciano, R.F. Isolation and characterization of replication-competent human immunodeficiency virus type 1 from a subset of elite suppressors. J. Virol. 2007, 81, 2508–2518. [Google Scholar] [CrossRef] [Green Version]
- Veenhuis, R.T.; Kwaa, A.K.; Garliss, C.C.; Latanich, R.; Salgado, M.; Pohlmeyer, C.W.; Nobles, C.L.; Gregg, J.; Scully, E.P.; Bailey, J.R.; et al. Long-term remission despite clonal expansion of replication-competent HIV-1 isolates. JCI Insight 2018, 3, e122795. [Google Scholar] [CrossRef] [Green Version]
- Schroder, A.R.; Shinn, P.; Chen, H.; Berry, C.; Ecker, J.R.; Bushman, F. HIV-1 integration in the human genome favors active genes and local hotspots. Cell 2002, 110, 521–529. [Google Scholar] [CrossRef] [Green Version]
- Ikeda, T.; Shibata, J.; Yoshimura, K.; Koito, A.; Matsushita, S. Recurrent HIV-1 integration at the BACH2 locus in resting CD4+ T cell populations during effective highly active antiretroviral therapy. J. Infect. Dis. 2007, 195, 716–725. [Google Scholar] [CrossRef]
- Cesana, D.; Santoni de Sio, F.R.; Rudilosso, L.; Gallina, P.; Calabria, A.; Beretta, S.; Merelli, I.; Bruzzesi, E.; Passerini, L.; Nozza, S.; et al. HIV-1-mediated insertional activation of STAT5B and BACH2 trigger viral reservoir in T regulatory cells. Nat. Commun. 2017, 8, 498. [Google Scholar] [CrossRef]
- Wang, G.P.; Ciuffi, A.; Leipzig, J.; Berry, C.C.; Bushman, F.D. HIV integration site selection: Analysis by massively parallel pyrosequencing reveals association with epigenetic modifications. Genome Res. 2007, 17, 1186–1194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vansant, G.; Chen, H.-C.; Zorita, E.; Trejbalová, K.; Miklík, D.; Filion, G.; Debyser, Z. The chromatin landscape at the HIV-1 provirus integration site determines viral expression. Nucleic Acids Res. 2020, 48, 7801–7817. [Google Scholar] [CrossRef]
- Haworth, K.G.; Schefter, L.E.; Norgaard, Z.K.; Ironside, C.; Adair, J.E.; Kiem, H.P. HIV infection results in clonal expansions containing integrations within pathogenesis-related biological pathways. JCI Insight 2018, 3, e99127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coffin, J.M.; Wells, D.W.; Zerbato, J.M.; Kuruc, J.D.; Guo, S.; Luke, B.T.; Eron, J.J.; Bale, M.; Spindler, J.; Simonetti, F.R.; et al. Clones of infected cells arise early in HIV-infected individuals. JCI Insight 2019, 4, e128432. [Google Scholar] [CrossRef] [PubMed]
- von Stockenstrom, S.; Odevall, L.; Lee, E.; Sinclair, E.; Bacchetti, P.; Killian, M.; Epling, L.; Shao, W.; Hoh, R.; Ho, T.; et al. Longitudinal Genetic Characterization Reveals That Cell Proliferation Maintains a Persistent HIV Type 1 DNA Pool During Effective HIV Therapy. J. Infect. Dis. 2015, 212, 596–607. [Google Scholar] [CrossRef] [PubMed]
- Abrahams, M.R.; Joseph, S.B.; Garrett, N.; Tyers, L.; Moeser, M.; Archin, N.; Council, O.D.; Matten, D.; Zhou, S.; Doolabh, D.; et al. The replication-competent HIV-1 latent reservoir is primarily established near the time of therapy initiation. Sci. Transl. Med. 2019, 11, eaaw5589. [Google Scholar] [CrossRef]
- Katusiime, M.G.; Halvas, E.K.; Wright, I.; Joseph, K.; Bale, M.J.; Kirby-McCullough, B.; Engelbrecht, S.; Shao, W.; Hu, W.-S.; Cotton, M.F.; et al. Intact HIV Proviruses Persist in Children Seven to Nine Years after Initiation of Antiretroviral Therapy in the First Year of Life. J. Virol. 2020, 94, e01519-19. [Google Scholar] [CrossRef]
- Bale, M.J.; Katusiime, M.G.; Wells, D.; Wu, X.; Spindler, J.; Halvas, E.K.; Cyktor, J.C.; Wiegand, A.; Shao, W.; Cotton, M.F.; et al. Early Emergence and Long-Term Persistence of HIV-Infected T-Cell Clones in Children. mBio 2021, 12, e00568-21. [Google Scholar] [CrossRef]
- Coffin, J.M.; Bale, M.J.; Wells, D.; Guo, S.; Luke, B.; Zerbato, J.M.; Sobolewski, M.D.; Sia, T.; Shao, W.; Wu, X.; et al. Integration in oncogenes plays only a minor role in determining the in vivo distribution of HIV integration sites before or during suppressive antiretroviral therapy. PLoS Pathog. 2021, 17, e1009141. [Google Scholar] [CrossRef]
- Yoon, J.K.; Holloway, J.R.; Wells, D.W.; Kaku, M.; Jetton, D.; Brown, R.; Coffin, J.M. HIV proviral DNA integration can drive T cell growth ex vivo. Proc. Natl. Acad. Sci. USA 2020, 117, 32880–32882. [Google Scholar] [CrossRef]
- Satou, Y.; Katsuya, H.; Fukuda, A.; Misawa, N.; Ito, J.; Uchiyama, Y.; Miyazato, P.; Islam, S.; Fassati, A.; Melamed, A.; et al. Dynamics and mechanisms of clonal expansion of HIV-1-infected cells in a humanized mouse model. Sci. Rep. 2017, 7, 6913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lenasi, T.; Contreras, X.; Peterlin, B.M. Transcriptional interference antagonizes proviral gene expression to promote HIV latency. Cell Host Microbe 2008, 4, 123–133. [Google Scholar] [CrossRef] [Green Version]
- Han, Y.; Lin, Y.B.; An, W.; Xu, J.; Yang, H.C.; O’Connell, K.; Dordai, D.; Boeke, J.D.; Siliciano, J.D.; Siliciano, R.F. Orientation-dependent regulation of integrated HIV-1 expression by host gene transcriptional readthrough. Cell Host Microbe 2008, 4, 134–146. [Google Scholar] [CrossRef] [Green Version]
- Yeh, Y.J.; Jenike, K.M.; Calvi, R.M.; Chiarella, J.; Hoh, R.; Deeks, S.G.; Ho, Y.C. Filgotinib suppresses HIV-1-driven gene transcription by inhibiting HIV-1 splicing and T cell activation. J. Clin. Investig. 2020, 130, 4969–4984. [Google Scholar] [CrossRef]
- Bangham, C.R.M.; Cook, L.B.; Melamed, A. HTLV-1 clonality in adult T-cell leukaemia and non-malignant HTLV-1 infection. Semin. Cancer Biol. 2014, 26, 89–98. [Google Scholar] [CrossRef] [Green Version]
- Katano, H.; Sato, Y.; Hoshino, S.; Tachikawa, N.; Oka, S.; Morishita, Y.; Ishida, T.; Watanabe, T.; Rom, W.N.; Mori, S.; et al. Integration of HIV-1 caused STAT3-associated B cell lymphoma in an AIDS patient. Microbes Infect. 2007, 9, 1581–1589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mellors, J.W.; Guo, S.; Naqvi, A.; Brandt, L.D.; Su, L.; Sun, D.; Joseph, K.W.; Demirov, D.; Halvas, E.K.; Butcher, D.; et al. Insertional activation of STAT3 and LCK by HIV-1 proviruses in T cell lymphomas. Presented at the Virtual. In Proceedings of the Cold Spring Harbor Meetings on Retroviruses, Virtual. 25 May–28 May 2021. [Google Scholar]
- Matsuoka, M.; Jeang, K.-T. Human T-cell leukemia virus type 1 (HTLV-1) and leukemic transformation: Viral infectivity, Tax, HBZ and therapy. Oncogene 2011, 30, 1379–1389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Satou, Y.; Yasunaga, J.-i.; Yoshida, M.; Matsuoka, M. HTLV-I basic leucine zipper factor gene mRNA supports proliferation of adult T cell leukemia cells. Proc. Natl. Acad. Sci. USA 2006, 103, 720–725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Satou, Y.; Miyazato, P.; Ishihara, K.; Yaguchi, H.; Melamed, A.; Miura, M.; Fukuda, A.; Nosaka, K.; Watanabe, T.; Rowan, A.G.; et al. The retrovirus HTLV-1 inserts an ectopic CTCF-binding site into the human genome. Proc. Natl. Acad. Sci. USA 2016, 113, 3054–3059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demarchi, F.; d’Adda di Fagagna, F.; Falaschi, A.; Giacca, M. Activation of transcription factor NF-kappaB by the Tat protein of human immunodeficiency virus type 1. J. Virol. 1996, 70, 4427–4437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinberger, L.S.; Burnett, J.C.; Toettcher, J.E.; Arkin, A.P.; Schaffer, D.V. Stochastic gene expression in a lentiviral positive-feedback loop: HIV-1 Tat fluctuations drive phenotypic diversity. Cell 2005, 122, 169–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knudson, A.G., Jr. Mutation and cancer: Statistical study of retinoblastoma. Proc. Natl. Acad. Sci. USA 1971, 68, 820–823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, F.; Bailes, E.; Robertson, D.L.; Chen, Y.; Rodenburg, C.M.; Michael, S.F.; Cummins, L.B.; Arthur, L.O.; Peeters, M.; Shaw, G.M.; et al. Origin of HIV-1 in the chimpanzee Pan troglodytes troglodytes. Nature 1999, 397, 436–441. [Google Scholar] [CrossRef] [PubMed]
- Pneumocystis Pneumonia—Los Angeles. In Center for Disease Control. Morbidity and Mortality Weekly Report; MMWR: Riga, Latvia, 1981; Volume 30, pp. 1–3.


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yeh, Y.-H.J.; Yang, K.; Razmi, A.; Ho, Y.-C. The Clonal Expansion Dynamics of the HIV-1 Reservoir: Mechanisms of Integration Site-Dependent Proliferation and HIV-1 Persistence. Viruses 2021, 13, 1858. https://0-doi-org.brum.beds.ac.uk/10.3390/v13091858
Yeh Y-HJ, Yang K, Razmi A, Ho Y-C. The Clonal Expansion Dynamics of the HIV-1 Reservoir: Mechanisms of Integration Site-Dependent Proliferation and HIV-1 Persistence. Viruses. 2021; 13(9):1858. https://0-doi-org.brum.beds.ac.uk/10.3390/v13091858
Chicago/Turabian StyleYeh, Yang-Hui Jimmy, Kerui Yang, Anya Razmi, and Ya-Chi Ho. 2021. "The Clonal Expansion Dynamics of the HIV-1 Reservoir: Mechanisms of Integration Site-Dependent Proliferation and HIV-1 Persistence" Viruses 13, no. 9: 1858. https://0-doi-org.brum.beds.ac.uk/10.3390/v13091858