
Myocardial Damage by SARS-CoV-2: Emerging Mechanisms and Therapies
by
 , and
, and
Huyen Tran Ho
1,* ,
,
Stefan Peischard
1,
Nathalie Strutz-Seebohm
1,
Karin Klingel
2 and
Guiscard Seebohm
1,* 1
Cellular Electrophysiology and Molecular Biology, Institute for Genetics of Heart Diseases (IfGH), University Hospital Münster, 48149 Münster, Germany
2
Cardiopathology, Institute for Pathology and Neuropathology, University Hospital Tübingen, 72076 Tübingen, Germany
*
Authors to whom correspondence should be addressed.
Viruses 2021, 13(9), 1880; https://0-doi-org.brum.beds.ac.uk/10.3390/v13091880
Submission received: 3 August 2021
/
Revised: 6 September 2021
/
Accepted: 18 September 2021
/
Published: 21 September 2021
(This article belongs to the Special Issue COVID-19-Associated Myocarditis and Cardiac Pathology)
Abstract
:Evidence is emerging that severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) can infect various organs of the body, including cardiomyocytes and cardiac endothelial cells in the heart. This review focuses on the effects of SARS-CoV-2 in the heart after direct infection that can lead to myocarditis and an outline of potential treatment options. The main points are: (1) Viral entry: SARS-CoV-2 uses specific receptors and proteases for docking and priming in cardiac cells. Thus, different receptors or protease inhibitors might be effective in SARS-CoV-2-infected cardiac cells. (2) Viral replication: SARS-CoV-2 uses RNA-dependent RNA polymerase for replication. Drugs acting against ssRNA(+) viral replication for cardiac cells can be effective. (3) Autophagy and double-membrane vesicles: SARS-CoV-2 manipulates autophagy to inhibit viral clearance and promote SARS-CoV-2 replication by creating double-membrane vesicles as replication sites. (4) Immune response: Host immune response is manipulated to evade host cell attacks against SARS-CoV-2 and increased inflammation by dysregulating immune cells. Efficiency of immunosuppressive therapy must be elucidated. (5) Programmed cell death: SARS-CoV-2 inhibits programmed cell death in early stages and induces apoptosis, necroptosis, and pyroptosis in later stages. (6) Energy metabolism: SARS-CoV-2 infection leads to disturbed energy metabolism that in turn leads to a decrease in ATP production and ROS production. (7) Viroporins: SARS-CoV-2 creates viroporins that lead to an imbalance of ion homeostasis. This causes apoptosis, altered action potential, and arrhythmia.
1. Introduction
Currently, severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), which causes coronavirus disease 2019 (COVID-19), is threatening the entire world in one of the worst global pandemics, with over 200 million cases and an estimated 4.4 million deaths as of August 2021, and with new, more-threatening virus variants emerging according to the World Health Organization (WHO) [1].
The virus mainly causes respiratory infection, which in severe cases causes viral pneumonia and acute respiratory distress syndrome (ARDS). Severe COVID-19 cases have been associated with myocardial injury, arrhythmia, acute coronary syndrome, venous thromboembolism, and micro- and macro-thromboses, which are associated with high mortality [2,3,4,5]. Myocarditis is defined as the inflammation in the myocardium. It is usually identified by immunohistochemical analysis of endomyocardial biopsies (EMBs). Characteristics include flulike symptoms, left ventricle systolic dysfunction, enhanced numbers of interstitial immune cells, and an increase in creatine kinase biomarker and cardiac troponin levels in the blood [6]. There have been cases of myocarditis in COVID-19 patients: Different studies have reported rates between 7.2% to 27.8% of COVID-19 patients developing cardiac issues. However, these studies did not elucidate whether the disease was caused by direct infection of the heart or indirectly (see also Section 2) [7,8,9,10,11,12,13,14]. There are studies that analyzed and detected SARS-CoV-2 presence in the heart. Interestingly, in some of these cases, no infection was found in nasopharyngeal swabs of patients. These findings were interpreted as indication of a primary cardiac infection [15,16,17,18,19]. Later, another study based on autopsies of deceased COVID-19 patients demonstrated that SARS-Cov-2 infection in the myocardium was common in these patients: 30 out of 41 patients were identified with SARS-CoV-2 in the myocardium. However, the number of infected cells in the myocardium was particularly low, and only four of the patients developed myocarditis. However, those patients with SARS-CoV-2 in the myocardium correlated with higher numbers of myocardial macrophages and lymphocytes, as well as cardiac inflammation and changes in the electrocardiographs [20]. Additionally, direct infection appeared to cause myofibrillar fragmentation in cardiomyocytes [21,22]. SARS-CoV-2 can infect endothelial cells and cause endotheliitis as well [23,24]. Electron microscopic imaging of a 64-year-old male COVID-19 patient showed viral particles in an endothelial cell of the heart (see Figure 1).
In conclusion, there are indications that SARS-CoV-2 can directly infect cardiomyocytes and cardiac endothelial cells. This poses a possible risk of causing myocarditis or other complex heart diseases. To this date, data for SARS-CoV-2 causing viral myocarditis is scarce and seems uncommon [25]. There were COVID-19 patients with myocarditis, but without SARS-CoV-2 directly present in the myocardium. This means that through other indirect mechanisms, SARS-CoV-2 can cause myocarditis as well [26]. In these cases, possible causes of myocarditis were a cytokine storm and a systemic inflammation in COVID-19 patients that causes autoimmune myocarditis [25,27].
SARS-CoV-2 mutant variants are emerging that have increased transmissibility and lead to higher mortality [28]. This is mainly due to mutations in the spike S protein, most commonly D614G; this leads to higher affinity of the spike protein to the host cell receptor angiotensin-converting enzyme 2 (ACE2), therefore facilitating viral entry. In addition, evasion of HLA-A24-mediated cellular immunity and potentially increased viral replication have been reported for recent mutants [29]. A study analyzing the effect of the variants B.1.1.7, B.1.351, and P.2 on different endothelial cell types in vitro showed higher viral uptake with these variants, especially in coronary artery endothelial cells. Furthermore, the variants had cytotoxic effects, in contrast to the wildtype SARS-CoV-2 [30].
It is important to evaluate how SARS-CoV-2 could affect the heart. This is the topic of this review. Other viruses frequently causing myocarditis, such as enteroviruses like coxsackievirus B3 (CVB3) that infect cardiomyocytes and parvovirus B19 (PVB19) that infects cardiac endothelial cells, are shown to hijack the biological processes of the host cell to their advantage while damaging the heart [31,32,33]. These include, among others, the immune system, viral replication, the autophagosomal pathway, cell death, energy metabolism, and ion homeostasis, which all play a significant role in myocardial function [31]. Hence, by recapitulating our current knowledge about the effects of well-known viruses like enteroviruses, which infect cardiomyocytes, or parvovirus B19, which infects endothelial cells, and with our understanding of similar coronaviruses during viral myocarditis progression, we can suggest potential actions of the new SARS-CoV-2 on the human heart. Several drug candidates targeting some of these mechanisms are in clinical trials, and are elucidated as possible treatment options against SARS-CoV-2 infection in the heart in this review. We included a glossary of key terms addressed in this review for easier understanding (see Table 1).
SARS-CoV-2 is an ssRNA(+) virus that belongs to the order Nidovirales, with a large genome size of approximately 30 kb [34]. It encodes structural proteins that include the spike (S protein), envelope (E), membrane (M), and nucleocapsid proteins (N). It has 14 open reading frames and polyproteins that are processed to 16 nonstructural proteins termed Nsp1 to Nsp16 [35,36]. SARS-CoV-1 shares around 77.1% of the protein sequence with SARS-CoV-2. Our understanding of SARS-CoV-1 can help in understanding analogous actions of SARS-CoV-2, as data for the new SARS-CoV-2 are still incomplete [37].
2. SARS-CoV-2 Causing Myocardial Damage Unrelated to Myocarditis
Myocarditis-like symptoms, including troponin rise and cardiac dysfunction, have also been observed without the presence myocardial inflammation in COVID-19 patients, namely in Takotsubo cardiomyopathy and type I and type II myocardial infarction. Takotsubo cardiomyopathy, also known as broken-heart syndrome, is triggered by a sudden extreme release of catecholamines under physical and emotional stress. High concentrations of catecholamines lead to β-adrenergic toxicity and calcium dysregulation in cardiomyocytes, which is essential for the excitation–contraction coupling. A hypothesis for the increase of Takotsubo cases in COVID-19 pneumonia patients is the interplay between catecholamine release and cytokine release from a dysregulated immune system (see also Section 6), which amplify each other in a positive feedback loop [38,39].
Type 1 myocardial infarction has also been reported in some patients [40]. Type 1 is an atherothrombotic coronary artery disease characterized by plaque rupture and thrombosis. Potential mechanisms include the activation of pathogen-associated molecular pattern (PAMP) receptors in immune cells in atherosclerotic plaques, leading to a higher risk for plaque rupture. PAMP recognition by the innate immune system can also result in coronary endothelial dysfunction. This in turn leads to vasoconstriction and thrombosis [41].
The cytokine storm leads to increase in oxygen consumption in the heart while also causing thrombosis and coronary heart spasm. This leads to a decrease in blood supply and to an imbalance in oxygen demand and supply in the heart. Consequently, this acutely damages the heart, also known as type 2 myocardial infarction [42].
3. Viral Entry via Spike S Protein
SARS-CoV-2 binds to host cell surface proteins via the spike S protein. The host cell receptor is commonly formed by the angiotensin-converting enzyme 2 (ACE2) and by priming either the serine protease TMPRSS2, which mediates membrane fusion, or endosomal cathepsin B and L, which mediate viral entry via endocytosis [43,44]. In the lung, SARS-CoV-2 mainly targets type II pneumocytes that use ACE2 for binding, and TMPRSS2 and furin for priming [45]. Cardiomyocytes express high levels of ACE2 but extremely low levels of TMPRSS2. In cardiomyocytes, TMPRRS2 is localized in the cytoplasm and nucleus, rather than the plasma membrane where the enzyme is needed for priming [46,47]. However, high levels of cathepsin B and L that can compensate for TMPRSS2 are observed in cardiomyocytes. This would indicate endocytosis as the primary mechanism of SARS-CoV-2 entry [48] (Figure 2). Indeed, in vitro studies with hiPSC-cardiomyocytes and human-engineered heart tissues show that SARS-CoV-2 can directly infect these cells by entering via ACE2 and cathepsins, and that it can replicate in these cells [19,49]. Inhibition of cathepsin B and L via the inhibitor E-64d reduces SARS-CoV-2 infection significantly in iPSC-derived cardiomyocytes [21].
Nafamostat and camostat, both in clinical trials, are serine protease inhibitors that can potentially inhibit TMPRSS2, and thus viral entry. However, as cardiomyocytes do not express TMPRSS2, but instead cathepsin B and L, serine protease inhibitors that inhibit TMPRSS2 would most likely not be effective. Indeed, while cathepsin inhibitors are effective, the two TMPRSS2 inhibitors aprotinin and camostat mesilate did not have a significant effect in SARS-CoV-2-infected, iPSC-derived cardiomyocytes [21]. Hydroxychloroquine, among others, inhibits endosome-mediated viral entry of coronaviruses. It is therefore potentially effective against cathepsin-mediated virus entry [44]. However, there are concerns because it prolongs the QT interval and increases the risk of drug-induced sudden cardiac death. Later, it was dropped as a potential drug against SARS-CoV-2 due to low efficiency and the aforementioned risks [50,51,52]. In human-induced pluripotent stem cell (hiPSC)-derived cardiomyocytes, SARS-CoV-2 infection was successfully inhibited by the cathepsin inhibitor N-acetyl-l-leucyl-l-leucyl-l-methionine. Cathepsin inhibitors thus stand as a promising treatment option against SARS-CoV-2-infected hearts [49].
As previously mentioned, SARS-CoV-2 can infect endothelial cells, and can cause endotheliitis as well. Endothelial cells, especially vascular endothelial cells, abundantly express ACE2. Moreover, precursor endothelial cells express TMPRSS2. Nevertheless, it has been suggested that SARS-CoV-2 can also enter endothelial cells via neuropilin-1 and CD209L. Both are expressed abundantly in endothelial cells [53] (Figure 2). In line with this, in vitro studies showed significant decrease in infectivity using small-molecule inhibitors or antibodies against neuropilin-1, and should be considered as a treatment option that can potentially inhibit SARS-CoV-2 spike protein binding in endothelial cells [54,55].
There seems to be downregulation of ACE2 upon SARS-CoV-2 infection [56]. This could impact the human heart adversely, as ACE2 has a cardioprotective role and is relevant for cardiac contractility. It is part of the renin–angiotensin–aldosterone system (RAAS) that converts angiotensin I to angiotensin (1–7), which in turn promotes vasodilation [57,58,59]. Moreover, there are hints that ACE2 downregulation could lead to increased macrophage activation to promote inflammation [59]. ACE inhibitors (ACEi) and angiotensin-receptor blockers (ARBs) are used as treatment in many older patients with hypertension to regulate RAAS. These compounds enhance ACE2 expression. A meta-analysis of COVID-19 patients showed a tendency towards a decrease in deaths and critical events when using ACEi. Therefore, the use of ACEi and ARBs is evaluated to be safe in hypertensive patients during the pandemic, and might even be preferred in COVID-19 patients [60,61]. The S-mediated membrane fusion is inhibited by the pan-coronavirus fusion inhibitor EK1C4, which could represent a potent therapeutic option [62].
4. Viral Replication
Infected cardiomyocytes in COVID-19 patients are demonstrated to possess transcriptional activity [22]. Nsp1 in coronaviruses, including SARS-CoV-1, suppresses host gene translation by binding to the ribosome and promoting host mRNA degradation [63,64,65]. The other nonstructural proteins Nsp2 to Nsp16 comprise the viral replication and transcription complex (RTC), which include the RNA-processing and RNA-modifying enzymes and proteins for proofreading of the genome [66]. The reviews by V’kovski et al. and Romano et al. provide a good overview about the roles of the proteins in the SARS-CoV-2-RNA replication mechanism [36,67]. In brief, the positive-sense RNA can be immediately used for translating viral proteins (Figure 3). For RNA synthesis, the nascent positive strand is used to generate a negative-strand genomic RNA. This negative-stranded RNA is the template to generate the positive-stranded genomic RNA for the new virus particle, and to generate mRNA for translation of viral proteins. RNA replication is accomplished via discontinuous transcription, in which the RTC pauses at so-called transcription regulatory sequences (TRS). Transcription is performed until reaching a so-called body TRS (TRS-B) and can either continue to the next body TRS or jump to the end, where the leader TRS (TRS-L) is located and the last piece of mRNA is added. This leads to the creation of different-sized subgenomic mRNAs that translate into a plethora of different viral proteins and polyproteins. Further processing of the polyproteins is performed by viral proteases.
Viral RNA polymerase inhibitors, including remdesivir and favipiravir, have been evaluated in clinical trials as potential SARS-CoV-2 treatments. Remdesivir acts as a nucleoside analogue and is incorporated into the viral RNA product by the viral RNA-dependent RNA polymerase [68]. Remdesivir showed no significant effect during clinical trials, but is still FDA-approved as a drug against SARS-CoV-2 [52,69]. However, when remdesivir was used in hiPS-cell-derived cardiomyocytes and EHTs, SARS-CoV-2 infection was successfully inhibited [19,49]. The 3C-like protease encoded by nsp5, along with papain-like proteinase encoded by nsp3, cleave polyprotein precursors to form the replication complex for viral replication. Lopinavir/ritonavir are protease inhibitors and can potentially inhibit viral 3C-like protease, but clinical trials so far have been discouraging [70].
5. Autophagy and Double-Membrane Vesicles
Autophagy is an intracellular process to recycle misfolded proteins and organelles, and eliminate intracellular pathogens in autophagosomes as an innate immune response [71]. However, several positive-stranded RNA viruses such as CVB3 can manipulate autophagosomes by inhibiting lysosome–autophagosome fusion, which prevents virus elimination [72,73,74]. The autophagosmal pathway appear to be relevant for viral replication: Enteroviruses misuse autophagosomes as replication organelles, or use parts of the autophagosomal pathway for replication [75,76,77].
Autophagosomes are manipulated in SARS-CoV-2 infection as well (Figure 4). Analogous to CVB3, Nsp3a of SARS-CoV-2 has been shown to inhibit lysosome–autophagosome fusion by disrupting Rab7–HOPS complex formation, which is needed for lysosome–autophagosome fusion. This leads to blockage of autophagy and accumulation of autophagosomes [78]. The use of autophagy-inducing compounds has been shown to inhibit SARS-CoV-2 propagation in vitro, making autophagy induction an interesting treatment option [79].
Accumulation of nonlytic autophagosomes can be beneficial for SARS-CoV-2 by using them as autophagosome-like double-membrane vesicles (DMVs). Indeed, these DMVs are induced in SARS-CoV-2-infected cells, described as inhabiting double-stranded RNA, and originate from the ER [80]. Coronaviruses can abuse components that are used for formation of vesicles for ER degradation called EDEMosomes to promote DMV formation [81]. DMVs of coronaviruses are in a closed conformation and connected to the ER [82,83,84]. Nsp3, -4, and -6 appear to be relevant for DMV formation in SARS-CoV-1-infected cells [85]. To transport the newly synthesized viral RNA from the vesicle to the cytosol, coronaviruses create molecular pores in the closed DMVs. The pore is potentially composed of nsp3 and possibly -4 and -6, all of which are transmembrane proteins [86].
Hence, targeting DMV formation could be a promising approach against SARS-CoV-2 infection and should be further studied. For example, the inhibition of cytosolic phospholipase A2 and early secretory protein GBF1 reduced DMV numbers and virus replication [87,88]. Other DMV inhibitors that are found for analogous viruses could be studied as potential SARS-CoV-2 treatment [74]. After viral RNA export from the DMVs, the viral RNA, along with viral structural proteins, are assembled in single-membrane compartments made from the ER-to-Golgi intermediate compartment (ERGIC), and the newly assembled virions are excreted via the secretory pathway into the extracellular space [86,89,90].
6. Immune Response
There are indications that the immune system contributes to the pathology of SARS-CoV-2 myocarditis and myocardial injury. Cytotoxic T-lymphocytes and macrophages are reported to infiltrate the myocardium in SARS-CoV-2-induced myocarditis patients [16,17,18]. As exemplarily shown in Figure 5, EMBs from patients with COVID-19 revealed more CD68+ macrophages and a few CD3+ T cells. Patients with severe COVID-19 disease progression have increased IL-17 secreting T-helper 17 (TH17) cells [91]. Cytotoxic T cells and macrophages in CVB3-induced viral myocarditis are described to promote cardiac damage. Studies indicate that there is a major role for TH17 in promoting progression towards dilated cardiomyopathy and heart failure [92,93,94]. Thus, a SARS-CoV-2 infection in the heart could cause analogous adverse immune responses leading to myocarditis, DCM, or heart failure. Humoral and cellular immune signatures that can predict a risk for progression towards heart diseases, including further knowledge of heart-reactive autoantibodies present during SARS-CoV-2 infection, should be further elucidated [95].
Two stages during COVID-19 disease progression have been described: the early stage, which is more focused on immune protection, viral replication, and apoptosis inhibition; and the later severe stage, in which the cytokine storm is described to damage the tissue due to inflammation [96]. In the myocardium, Lindner et al. reported an increase of cytokine expression, including TNF-α; IFN-γ; chemokine ligand 5; and IL-1β, -6, -7, -8, and -18 [97]. Inflammasomes contain pattern-recognition receptors, including NLR family pyrin domain containing 3 (NLRP3), and inflammatory procaspases, including procaspase-1. They contribute to the maturation of proinflammatory cytokines such as IL-1β and induction of pyroptosis [98]. NLRP3 inflammasomes are shown to be elevated by the SARS-CoV-1 nsp3a, -8b, and E proteins, which promote inflammation reactions [99,100,101]. The structural proteins of SARS-CoV-1, as well as nsp1, -3a, and -7a, appear to be able to enhance nuclear factor-κB (NF-κB) signaling. The increase in NF-κB signaling could be responsible for the cytokine storm seen in the later severe stages of COVID-19 [96,102]. Inhibition of NF-κB in SARS-CoV-1-infected mice did not affect virus titers, but decreased inflammatory cytokines, namely TNF, CCL2, and CXCL2 in combination with higher observed survival rates [103].
Coronaviruses interfere with interferon type I (IFN I) induction and signaling in early stages. In agreement with this, SARS-CoV-2 patients do not show high levels of IFN-I response. Impaired or delayed IFN-I response promotes progression towards COVID-19 with accumulation of monocytes–macrophages, increased viral replication, hypercytokinemia, or cytokine storm with a dysregulated T-cell response (see Figure 6) [104,105]. Thus, treatment with IFNs can be beneficial against SARS-CoV-2, but should be applied in early stages. Late administration of IFN I is shown to be ineffective or have adverse effects in animals with SARS-CoV-1 or MERS-CoV infection [104]. In an in vitro study with SARS-CoV-2-infected, hPSC-derived cardiomyocytes and EHTs, elevation of cytokines and chemokines, including IFN I signaling and TNF expression, was observed. In addition, macrophages accumulated in the sites of SARS-CoV-2 infection in the EHT model, indicating that cardiomyocyte infection leads to macrophage activation [19]. It must be further elucidated what role IFN I signaling has in coronavirus induced myocarditis, as IFN I response varies between different cell types and within different microenvironments [104,106].
In general, suppression of certain transcription factors or proinflammatory cytokines such as NF-κB; e.g., with proteasome inhibitors in later-stage infections, and IFN I administration during early infection stages, could be beneficial in COVID-19 [107,108]. Immune modulation via interferons such as IFN-β1a, IFN-α, PegIFN-α2β, IFN-α1β, and IFN-β1β have been evaluated in clinical trials as adjuvants against SARS-CoV-2 [69]. However, interferon IFN-β1a was already dropped from trials because the drug did not show significant effects in patients [52]. In this study, patients that were hospitalized with COVID-19 were treated with IFN-β1a, therefore similar to SARS-CoV-1 and MERS-CoV, it could be ineffective due to late administration. A triple combination of IFN-β1β with lopinavir–ritonavir and ribavirin was shown to have a beneficial effect on COVID-19 patients in a phase 2 study [109]. IFN-β administration in patients with enteroviral or parvovirus-B19-induced myocarditis in clinical studies showed to be safe and have ameliorating effects, so interferon treatment in myocardium SARS-CoV-2 myocarditis may be promising [110,111,112].
7. Programmed Cell Death
To efficiently replicate, cardiogenic viruses such as enteroviruses inhibit apoptosis signaling at the beginning of viral infection. In later stages, the focus shifts to viral release, so enteroviruses manipulate the host cell towards apoptosis [113]. PVB19 utilizes its nonstructural protein NS1 to trigger cell-cycle arrest to induce apoptosis in endothelial cells in the late viral cycle [33,114]. Similar to enteroviruses, SARS-CoV-2 also blocks apoptosis in earlier stages and induces cell death in later stages [115] (Figure 7). An in vitro study with EHT showed cell death as a result of SARS-CoV-2 infection and not inflammation [19].
In early stages, NF-κB signaling is upregulated, which is an important antiapoptotic pathway. Activated NF-κB upregulates apoptosis inhibitors such as cellular FLICE (FADD-like IL-1β-converting enzyme)-inhibitory protein (c-FLIP), Bcl-2 family members, and the X-linked inhibitor of apoptosis proteins (XIAPs) [115].
In later stages, in which the cytokine storm occurs, apoptosis, necroptosis, and pyroptosis are initiated via different pathways. For instance, in contrast to earlier stages, in later stages NF-κB is suppressed by the SARS-CoV-1 M protein, which leads to decreased apoptosis inhibition [116]. In the lungs of a transgenic mouse model, apoptosis and necrosis was induced via caspase-8 activation [117]. The Nsp3, -6, -7, and -8 proteins also appear to play a role in inducing apoptosis: The 3a protein of SARS-CoV-2 was shown to induce apoptosis in HEK293T, HepG2, and Vero E6 cells. Nsp3a cleaves and activates caspase-8. The active caspase-8 then activates the extrinsic apoptotic pathway. Further, SARS-CoV-2 increases and cleaves Bid to tBid, which activates Bax/Bak, cytochrome c, and caspase-9, which in turn activates the mitochondrial intrinsic apoptotic pathway [118]. Thus, it seems likely that caspase-8 localizes to the mitochondrial membrane and cleaves Bid to initiate the intrinsic apoptotic pathway [119]. The Nsp7a protein appears to inhibit host-cell protein synthesis, possibly as a host-cell stress response, and elevates p38 mitogen-activated protein-kinase (MAPK) signaling, which induces apoptosis [120]. SARS-CoV-1 Nsp6 induces caspase-3-mediated, ER-stress-induced, and JNK-dependent apoptosis [121]. Nsp8b promotes cell death by forming intracellular aggregates. The aggregates causes ER stress in a caspase-independent way, and activates NLRP3 inflammasomes, which then activate inflammation reactions and pyroptosis, another form of cell death [100]. There are indications that the structural proteins E, S, and M regulate apoptosis as well: The E protein of SARS-CoV-1 induces T-cell-mediated apoptosis, which in turn can be inhibited by overexpressing Bcl-xL [122]. The S protein alone is able to induce apoptosis, but its mechanism remains elusive [123]. The M protein activates apoptosis by interfering with the PDK1-PKB/Akt signaling, thus activating caspase-8 and -9 [124]. The death domain superfamily also plays a complex role in regulating pyroptosis, necroptosis, and apoptosis in SARS-CoV-2 infection; this has been thoroughly described by Ivanisenko et al. [115].
In summary, SARS-CoV-2 infection leads to suppression and induction of cell death by several viral proteins. There seems to be a pattern of time-dependent manipulation of apoptosis, in which extrinsic apoptosis is inhibited at early stages and apoptosis and pyroptosis are prevalent in later, more severe stages that promote inflammation. However, inhibition of caspase-mediated apoptosis using Bcl-2 in SARS-CoV-1-infected Vero cells showed no significant change in susceptibility to infection, kinetics, viral replication, or release, which poses a question regarding what role apoptosis plays in SARS-CoV-2 pathogenesis [125]. The use of anti-inflammatory treatment strategies and targeting components of the death domain superfamily are promising [115]. Moreover, the regulation of apoptosis seems to be dependent on the cell type, thus cell death in SARS-CoV-2-infected hearts must be further elucidated [126].
8. Energy Metabolism
During viral myocarditis, energy metabolism is disturbed, which can lead to adverse effects on myocardial performance and cell-damaging ROS production. Consistently, disturbed energy metabolism indicates severe disease progression. Mice with CVB3-induced myocarditis have reduced mitochondrial ATP/ADP ratios, and the activity of the respiratory chain (RC) in mitochondria has been shown to be decreased or imbalanced [127]. Interestingly, imbalanced RC activity that leads to high ROS production and apoptosis in mice correlates with efficient viral elimination, while overall decrease of RC activity correlates with persistent infection [128]. High ROS production is also observed in human-iPSC-derived cardiomyocytes that express CVB3 [129].
CVB3 in murine atrial cardiomyocytes can stimulate dynamin-related protein 1 (Drp1) by localizing itself to mitochondria, which leads to mitochondrial fragmentation, producing extracellular membrane vesicles. CVB3 can utilize these vesicles to release viral particles [130]. Inhibition of Drp1 has been shown to prevent mitochondrial damage and myocardial injury in CVB3-induced myocarditis [131].
Mitochondria and energy metabolism are affected by SARS-CoV-2 as well (Figure 8). Computational modeling predicts SARS-CoV-2 RNA to localize to the mitochondria, which may impact mitochondrial function [132]. Indeed, mitochondrial accumulation has been observed using integrative imaging around the viral replication sites where dsRNA is present. Furthermore, the mitochondria appear smaller in size, and intracristal space and matrix density are increased in infected cells. In addition, a decrease was reported in ATP synthase subunit 5B associated with a decrease in ATP production [84]. SARS-CoV-2 also appears to downregulate mitochondrial proteins such as NDUFA10, a component of the mitochondrial complex I, as a master regulator [133]. Moreover, interactions of viral proteins with other components of complex I and complex IV have been observed, which could lead to imbalanced RC complex activity and ROS production [134,135,136,137].
A recent study by Ajaz et al. analyzed mitochondrial respiration in peripheral blood mononuclear cells from COVID-19 patients and described reduced ATP-linked respiration, reserve capacity, and maximal respiration, indicating compromised mitochondrial respiration or mitochondrial dysfunction. In addition, increased glycolysis was observed, which could compensate for the decreased respiration [138]. Therefore, the virus seems to depend more on glycolysis for energy production.
Altogether, cardiogenic viruses such as CVB3 and SARS-CoV-2 alter mitochondrial structures and mitochondrial respiration. SARS-CoV-2 likely manipulates mitochondrial function to suppress antiviral response by the host immune system [139]. The resulting impairment of energy metabolism and ATP production can cause reduction of cardiac function [140]. Furthermore, it is assumed that ROS are increasingly produced, especially in severe cases of COVID-19 due to hypoxia, the cytokine storm, and dysfunction of mitochondria. This would lead to oxidative stress and promote inflammation [141]. Higher ROS levels may lead to more apoptosis, and thus loss of functional cardiomyocytes and the spread of viral particles. On the other hand, more apoptosis can also lead to more efficient viral clearing and prevention of chronic disease development: High apoptosis rates correlated with functional recovery in myocarditis patients, while inhibition of apoptosis correlated with chronic myocarditis [142,143].
9. Viroporins
Viroporins are viral proteins that are similar to ion channels; they both integrate into the membrane of host cells and conduct ions. Most animal RNA viruses form these viroporins [144,145,146]. The main functions of viroporins are to aid virion assembly and release from the host cell. By altering the ion concentration gradient, viroporins can cause depolarization that supports the budding of the virus. Furthermore, viroporins localize at the budding neck, the part of the cellular membrane that constricts and forms the budding enveloped virion. At this location, the viroporins can oligomerize to facilitate virion scission [147]. Further supported functions include cell entry, genome replication, and manipulation of programmed cell death. As a consequence, the host-cell ion concentration; the membrane permeability of ions; and the activity of ion channels that conduct ions such as Na+, K+, and Ca2+ are altered. As a counteraction, the host immune system can sense the intracellular K+ and Ca2+ imbalances caused by viroporins and activate NLRP3 inflammasomes in several respiratory RNA virus infections [146].
Enteroviruses, including CVB3, form a Ca2+-conducting viroporin that alters Ca2+ homeostasis. This viroporin inserts itself into the plasma membrane, as well as the membranes of mitochondria, the Golgi apparatus, the ER, and the SR. This leads to apoptosis suppression and induction in a time-dependent manner [148]. PVB19 does not seem to possess a viroporin, but the virus causes endothelial dysfunction by inducing Ca2+ entry via host ion channels, which disturbs the ion homeostasis [33].
Interestingly, SARS-CoV-1 also creates viroporins from the structural proteins E, nsp3a, and -8a (Figure 9). Proteins E and nsp3a share over 85% similarity with the analogous proteins of SARS-CoV-2 [149,150]. The E protein is shown to be a cationic viroporin for Na+, K+, and Ca2+, being more selective for Na+ and Ca2+, which localize to the ER, Golgi apparatus, and the ER–Golgi compartment (ERGIC) [101,151,152,153]. Nsp3a forms a cationic viroporin at the plasma membrane, ER, and Golgi that conducts Na+, K+, and Ca2+, with selectivity for K+ and Ca2+ [149,154,155,156]. Thus, there is a possibility that the E protein and Nsp3a increase cytosolic Ca2+ concentration by releasing Ca2+ from the intracellular stores, leading to Ca2+ overload, Ca2+ uptake in mitochondria, and thus apoptosis. Both viroporins appear to be relevant for virus viability and replication. Moreover, it was shown that both E protein and Nsp3a cause NLRP3 inflammasome activation by causing Ca2+ or K+ efflux, respectively [101,157]. Nsp8a, on the other hand, forms a weak cation-conducting viroporin, and does not appear to play a major role in virulence, unlike the E protein and Nsp3a [149,158].
A computational model of human ventricular myocytes indicated that SARS-CoV-2 viroporin activity can dysregulate action potential and Ca2+ handling, as the E protein and Nsp3a have similar channel properties as the outward K+ current (Ito) in cardiomyocytes [150]. A SARS-CoV-2 infection in the heart could hence lead to arrhythmia. Suppressing SARS-CoV-2 viroporin activity thus poses an attractive treatment option against SARS-CoV-2 myocarditis and other diseases caused by viroporin-forming viruses. Adamantanes are able to inhibit several viroporins. They are shown to be effective against the SARS-CoV-1 E protein, and a case report on SARS-CoV-2 patients indicated that the use of adamantanes could be protective against progression to COVID-19 [159,160]. Hexamethylene amiloride is also able to inhibit the E protein, resulting in reduced viral replication in several coronaviruses [161]. In patients with viral diseases including severe COVID-19, hypocalcemia, a decrease in serum Ca2+, has been observed. Hypocalcemia is assumed to be a host defense mechanism against increasing intracellular Ca2+ concentrations caused by viruses [162]. Decreasing intracellular Ca2+ can thus be effective against SARS-CoV-2: calcium chelators or calcium inhibitors can be used in reducing inflammasome activation. Whether this can reduce viral replication in SARS-CoV-2 infection has yet to be elucidated, but it is known to have no effect on replication of rhinovirus, which expresses viroporin 2B [163]. Calcium channel blockers can also be considered as a treatment against SARS-CoV-2, as they can suppress viral entry and have anti-inflammatory effects in SARS-CoV-1 or Mers-CoV infections [162].
10. Summary
In this review, we explained SARS-CoV-2 entry in cardiac cells, the replication mechanism and manipulation of autophagy and DMVs for viral replication, SARS-CoV-2 interaction with the immune system, manipulation of programmed cell death, effects on host energy metabolism, and myocardial effects by viroporins expressed by SARS-CoV-2 (see Figure 10). Several of these virus–host interactions for SARS-CoV-2 are analogous to well-known cardiogenic viruses like the ssRNA(+) virus CVB3 and ssDNA virus PVB19 (see Table 2).
To date, the incidence rate of SARS-CoV-2-induced myocarditis has not been determined due to incomplete data. Still, there is a possibility that SARS-CoV-2 has cardiovascular effects, and a high possibility that direct myocardial infection can occur. Myocardial damage caused by SARS-CoV-2 also can become an emerging issue in regard to new mutations of SARS-CoV-2 that were shown to have increased virulence. SARS-CoV-1 and -2 appear to have similar effects on the heart as other myocarditis-causing viruses, but further studies of SARS-CoV-2 in the heart have to be performed. Several antiviral drugs against SARS-CoV-2 infections are in clinical trials, but several have already been found to be ineffective [52,69]. The efficacy of these drugs on SARS-CoV-2 infections in the heart still must be further elucidated. Some of these viral therapies are considered potentially cardiotoxic, and drug-induced myocarditis could occur [25].
Author Contributions
Conceptualization, H.T.H.; writing—original draft preparation, H.T.H.; writing—review and editing, H.T.H., S.P., N.S.-S., and G.S.; visualization, H.T.H.; supervision, G.S.; data curation, K.K.; funding acquisition, G.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Deutsche Forschungsgemeinschaft (DFG) under grant number DFG Se1077/13-1 to G.S.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Patient consent was waived, as histological images are just examples and are not related to any other clinical data for these anonymous patients.
Data Availability Statement
Data sharing not applicable.
Acknowledgments
We thank Royce Anthony Harner and Matthew Cabell Greenwood for language editing.
Conflicts of Interest
The authors declare no conflict of interest.
References
- World Health Organization. COVID-19 Weekly Epidemiological Update, Edition 53, 17 August 2021; World Health Organization: Geneva, Switzerland, 2021. [Google Scholar]
- Shi, S.; Qin, M.; Shen, B.; Cai, Y.; Liu, T.; Yang, F.; Gong, W.; Liu, X.; Liang, J.; Zhao, Q.; et al. Association of Cardiac Injury with Mortality in Hospitalized Patients with COVID-19 in Wuhan, China. JAMA Cardiol. 2020, 5, 802–810. [Google Scholar] [CrossRef] [Green Version]
- Guo, T.; Fan, Y.; Chen, M.; Wu, X.; Zhang, L.; He, T.; Wang, H.; Wan, J.; Wang, X.; Lu, Z. Cardiovascular Implications of Fatal Outcomes of Patients with Coronavirus Disease 2019 (COVID-19). JAMA Cardiol. 2020, 5, 811–818. [Google Scholar] [CrossRef] [Green Version]
- Shi, S.; Qin, M.; Cai, Y.; Liu, T.; Shen, B.; Yang, F.; Cao, S.; Liu, X.; Xiang, Y.; Zhao, Q.; et al. Characteristics and clinical significance of myocardial injury in patients with severe coronavirus disease 2019. Eur. Heart J. 2020, 41, 2070–2079. [Google Scholar] [CrossRef]
- Tiwari, N.R.; Phatak, S.; Sharma, V.R.; Agarwal, S.K. COVID-19 and thrombotic microangiopathies. Thromb. Res. 2021, 202, 191–198. [Google Scholar] [CrossRef]
- Fung, G.; Luo, H.; Qiu, Y.; Yang, D.; McManus, B. Myocarditis. Circ. Res. 2016, 118, 496–514. [Google Scholar] [CrossRef]
- Knowlton, K.U. Pathogenesis of SARS-CoV-2 induced cardiac injury from the perspective of the virus. J. Mol. Cell Cardiol. 2020, 147, 12–17. [Google Scholar] [CrossRef]
- Basso, C.; Leone, O.; Rizzo, S.; De Gaspari, M.; van der Wal, A.C.; Aubry, M.C.; Bois, M.C.; Lin, P.T.; Maleszewski, J.J.; Stone, J.R. Pathological features of COVID-19-associated myocardial injury: A multicentre cardiovascular pathology study. Eur. Heart J. 2020, 41, 3827–3835. [Google Scholar] [CrossRef] [PubMed]
- Lara, D.; Young, T.; Del Toro, K.; Chan, V.; Ianiro, C.; Hunt, K.; Kleinmahon, J. Acute Fulminant Myocarditis in a Pediatric Patient with COVID-19 Infection. Pediatrics 2020, 146, e20201509. [Google Scholar] [CrossRef] [PubMed]
- Bernal-Torres, W.; Herrera-Escandón, Á.; Hurtado-Rivera, M.; Plata-Mosquera, C.A. COVID-19 fulminant myocarditis: A case report. Eur. Heart J. Case Rep. 2020, 4, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J.H.; Liu, Y.X.; Yuan, J.; Wang, F.X.; Wu, W.B.; Li, J.X.; Wang, L.F.; Gao, H.; Wang, Y.; Dong, C.F.; et al. First case of COVID-19 complicated with fulminant myocarditis: A case report and insights. Infection 2020, 48, 773–777. [Google Scholar] [CrossRef] [Green Version]
- Kesici, S.; Aykan, H.H.; Orhan, D.; Bayrakci, B. Fulminant COVID-19-related myocarditis in an infant. Eur. Heart J. 2020, 41, 3021. [Google Scholar] [CrossRef]
- Naneishvili, T.; Khalil, A.; O’Leary, R.; Prasad, N. Fulminant myocarditis as an early presentation of SARS-CoV-2. BMJ Case Rep. 2020, 13, e237553. [Google Scholar] [CrossRef]
- Garot, J.; Amour, J.; Pezel, T.; Dermoch, F.; Messadaa, K.; Felten, M.L.; Raymond, V.; Baubillier, E.; Sanguineti, F.; Garot, P. SARS-CoV-2 Fulminant Myocarditis. JACC Case Rep. 2020, 2, 1342–1346. [Google Scholar] [CrossRef] [PubMed]
- Albert, C.L.; Carmona-Rubio, A.E.; Weiss, A.J.; Procop, G.G.; Starling, R.C.; Rodriguez, E.R. The Enemy within: Sudden-Onset Reversible Cardiogenic Shock with Biopsy-Proven Cardiac Myocyte Infection by Severe Acute Respiratory Syndrome Coronavirus 2. Circulation 2020, 142, 1865–1870. [Google Scholar] [CrossRef]
- Gauchotte, G.; Venard, V.; Segondy, M.; Cadoz, C.; Esposito-Fava, A.; Barraud, D.; Louis, G. SARS-Cov-2 fulminant myocarditis: An autopsy and histopathological case study. Int. J. Legal Med. 2021, 135, 577–581. [Google Scholar] [CrossRef]
- Wenzel, P.; Kopp, S.; Göbel, S.; Jansen, T.; Geyer, M.; Hahn, F.; Kreitner, K.F.; Escher, F.; Schultheiss, H.P.; Münzel, T. Evidence of SARS-CoV-2 mRNA in endomyocardial biopsies of patients with clinically suspected myocarditis tested negative for COVID-19 in nasopharyngeal swab. Cardiovasc. Res. 2020, 116, 1661–1663. [Google Scholar] [CrossRef] [PubMed]
- Escher, F.; Pietsch, H.; Aleshcheva, G.; Bock, T.; Baumeier, C.; Elsaesser, A.; Wenzel, P.; Hamm, C.; Westenfeld, R.; Schultheiss, M.; et al. Detection of viral SARS-CoV-2 genomes and histopathological changes in endomyocardial biopsies. ESC Heart Fail. 2020, 7, 2440–2447. [Google Scholar] [CrossRef] [PubMed]
- Bailey, A.L.; Dmytrenko, O.; Greenberg, L.; Bredemeyer, A.L.; Ma, P.; Liu, J.; Penna, V.; Winkler, E.S.; Sviben, S.; Brooks, E.; et al. SARS-CoV-2 Infects Human Engineered Heart Tissues and Models COVID-19 Myocarditis. JACC Basic Transl. Sci. 2021, 6, 331–345. [Google Scholar] [CrossRef] [PubMed]
- Bearse, M.; Hung, Y.P.; Krauson, A.J.; Bonanno, L.; Boyraz, B.; Harris, C.K.; Helland, T.L.; Hilburn, C.F.; Hutchison, B.; Jobbagy, S.; et al. Factors associated with myocardial SARS-CoV-2 infection, myocarditis, and cardiac inflammation in patients with COVID-19. Mod. Pathol. 2021, 34, 1345–1357. [Google Scholar] [CrossRef] [PubMed]
- Perez-Bermejo, J.A.; Kang, S.; Rockwood, S.J.; Simoneau, C.R.; Joy, D.A.; Silva, A.C.; Ramadoss, G.N.; Flanigan, W.R.; Fozouni, P.; Li, H.; et al. SARS-CoV-2 infection of human iPSC-derived cardiac cells reflects cytopathic features in hearts of patients with COVID-19. Sci. Transl. Med. 2021, 13. [Google Scholar] [CrossRef] [PubMed]
- Bulfamante, G.P.; Perrucci, G.L.; Falleni, M.; Sommariva, E.; Tosi, D.; Martinelli, C.; Songia, P.; Poggio, P.; Carugo, S.; Pompilio, G. Evidence of SARS-CoV-2 Transcriptional Activity in Cardiomyocytes of COVID-19 Patients without Clinical Signs of Cardiac Involvement. Biomedicines 2020, 8, 626. [Google Scholar] [CrossRef]
- Varga, Z.; Flammer, A.J.; Steiger, P.; Haberecker, M.; Andermatt, R.; Zinkernagel, A.S.; Mehra, M.R.; Schuepbach, R.A.; Ruschitzka, F.; Moch, H. Endothelial cell infection and endotheliitis in COVID-19. Lancet 2020, 395, 1417–1418. [Google Scholar] [CrossRef]
- Maccio, U.; Zinkernagel, A.S.; Shambat, S.M.; Zeng, X.; Cathomas, G.; Ruschitzka, F.; Schuepbach, R.A.; Moch, H.; Varga, Z. SARS-CoV-2 leads to a small vessel endotheliitis in the heart. EBioMedicine 2021, 63, 103182. [Google Scholar] [CrossRef]
- Ozieranski, K.; Tyminska, A.; Jonik, S.; Marcolongo, R.; Baritussio, A.; Grabowski, M.; Filipiak, K.J.; Opolski, G.; Caforio, A.L.P. Clinically Suspected Myocarditis in the Course of Severe Acute Respiratory Syndrome Novel Coronavirus-2 Infection: Fact or Fiction? J. Card. Fail. 2021, 27, 92–96. [Google Scholar] [CrossRef]
- Sala, S.; Peretto, G.; Gramegna, M.; Palmisano, A.; Villatore, A.; Vignale, D.; De Cobelli, F.; Tresoldi, M.; Cappelletti, A.M.; Basso, C.; et al. Acute myocarditis presenting as a reverse Tako-Tsubo syndrome in a patient with SARS-CoV-2 respiratory infection. Eur. Heart J. 2020, 41, 1861–1862. [Google Scholar] [CrossRef]
- Raghavan, S.; Gayathri, R.; Kancharla, S.; Kolli, P.; Ranjitha, J.; Shankar, V. Cardiovascular Impacts on COVID-19 Infected Patients. Front. Cardiovasc. Med. 2021, 8, 670659. [Google Scholar] [CrossRef]
- Davies, N.G.; Jarvis, C.I.; Edmunds, W.J.; Jewell, N.P.; Diaz-Ordaz, K.; Keogh, R.H.; Group, C.C.-W. Increased mortality in community-tested cases of SARS-CoV-2 lineage B.1.1.7. Nature 2021, 593, 270–274. [Google Scholar] [CrossRef] [PubMed]
- Motozono, C.; Toyoda, M.; Zahradnik, J.; Ikeda, T.; Saito, A.; Tan, T.S.; Ngare, I.; Nasser, H.; Kimura, I.; Uriu, K.; et al. An emerging SARS-CoV-2 mutant evading cellular immunity and increasing viral infectivity. bioRxiv 2021. [Google Scholar] [CrossRef]
- Wagner, J.U.G.; Bojkova, D.; Shumliakivska, M.; Luxán, G.; Nicin, L.; Aslan, G.S.; Milting, H.; Kandler, J.D.; Dendorfer, A.; Heumueller, A.W.; et al. Increased susceptibility of human endothelial cells to infections by SARS-CoV-2 variants. Basic Res. Cardiol. 2021, 116, 42. [Google Scholar] [CrossRef] [PubMed]
- Peischard, S.; Ho, H.T.; Theiss, C.; Strutz-Seebohm, N.; Seebohm, G. A Kidnapping Story: How Coxsackievirus B3 and Its Host Cell Interact. Cell Physiol. Biochem. 2019, 53, 121–140. [Google Scholar] [CrossRef]
- Luo, Y.; Qiu, J. Human parvovirus B19: A mechanistic overview of infection and DNA replication. Future Virol. 2015, 10, 155–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verdonschot, J.; Hazebroek, M.; Merken, J.; Debing, Y.; Dennert, R.; Brunner-La Rocca, H.P.; Heymans, S. Relevance of cardiac parvovirus B19 in myocarditis and dilated cardiomyopathy: Review of the literature. Eur. J. Heart Fail. 2016, 18, 1430–1441. [Google Scholar] [CrossRef] [PubMed]
- Khailany, R.A.; Safdar, M.; Ozaslan, M. Genomic characterization of a novel SARS-CoV-2. Gene Rep. 2020, 19, 100682. [Google Scholar] [CrossRef] [PubMed]
- Nishiga, M.; Wang, D.W.; Han, Y.; Lewis, D.B.; Wu, J.C. COVID-19 and cardiovascular disease: From basic mechanisms to clinical perspectives. Nat. Rev. Cardiol. 2020, 17, 543–558. [Google Scholar] [CrossRef]
- Romano, M.; Ruggiero, A.; Squeglia, F.; Maga, G.; Berisio, R. A Structural View of SARS-CoV-2 RNA Replication Machinery: RNA Synthesis, Proofreading and Final Capping. Cells 2020, 9, 1267. [Google Scholar] [CrossRef] [PubMed]
- Yoshimoto, F.K. The Proteins of Severe Acute Respiratory Syndrome Coronavirus-2 (SARS CoV-2 or n-COV19), the Cause of COVID-19. Protein J. 2020, 39, 198–216. [Google Scholar] [CrossRef] [PubMed]
- Esposito, A.; Palmisano, A.; Natale, L.; Ligabue, G.; Peretto, G.; Lovato, L.; Vignale, D.; Fiocchi, F.; Marano, R.; Russo, V. Cardiac Magnetic Resonance Characterization of Myocarditis-Like Acute Cardiac Syndrome in COVID-19. JACC Cardiovasc. Imaging 2020, 13, 2462–2465. [Google Scholar] [CrossRef]
- Ortuno, S.; Jozwiak, M.; Mira, J.P.; Nguyen, L.S. Case Report: Takotsubo Syndrome Associated with Novel Coronavirus Disease 2019. Front. Cardiovasc. Med. 2021, 8, 614562. [Google Scholar] [CrossRef]
- Stefanini, G.G.; Montorfano, M.; Trabattoni, D.; Andreini, D.; Ferrante, G.; Ancona, M.; Metra, M.; Curello, S.; Maffeo, D.; Pero, G.; et al. ST-Elevation Myocardial Infarction in Patients with COVID-19: Clinical and Angiographic Outcomes. Circulation 2020, 141, 2113–2116. [Google Scholar] [CrossRef]
- Giustino, G.; Pinney, S.P.; Lala, A.; Reddy, V.Y.; Johnston-Cox, H.A.; Mechanick, J.I.; Halperin, J.L.; Fuster, V. Coronavirus and Cardiovascular Disease, Myocardial Injury, and Arrhythmia: JACC Focus Seminar. J. Am. Coll. Cardiol. 2020, 76, 2011–2023. [Google Scholar] [CrossRef] [PubMed]
- Dou, Q.; Wei, X.; Zhou, K.; Yang, S.; Jia, P. Cardiovascular Manifestations and Mechanisms in Patients with COVID-19. Trends Endocrinol. Metab. 2020, 31, 893–904. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef]
- Ou, T.; Mou, H.; Zhang, L.; Ojha, A.; Choe, H.; Farzan, M. Hydroxychloroquine-mediated inhibition of SARS-CoV-2 entry is attenuated by TMPRSS2. PLoS Pathog. 2021, 17, e1009212. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Pöhlmann, S. A Multibasic Cleavage Site in the Spike Protein of SARS-CoV-2 Is Essential for Infection of Human Lung Cells. Mol. Cell 2020, 78, 779–784.e5. [Google Scholar] [CrossRef]
- Liu, H.; Gai, S.; Wang, X.; Zeng, J.; Sun, C.; Zhao, Y.; Zheng, Z. Single-cell analysis of SARS-CoV-2 receptor ACE2 and spike protein priming expression of proteases in the human heart. Cardiovasc. Res. 2020, 116, 1733–1741. [Google Scholar] [CrossRef]
- Sakamoto, A.; Kawakami, R.; Kawai, K.; Gianatti, A.; Pellegrini, D.; Kutys, R.; Guo, L.; Mori, M.; Cornelissen, A.; Sato, Y.; et al. ACE2 (Angiotensin-Converting Enzyme 2) and TMPRSS2 (Transmembrane Serine Protease 2) Expression and Localization of SARS-CoV-2 Infection in the Human Heart. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 542–544. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Chen, T.; Zhou, Y. Mediators of SARS-CoV-2 entry are preferentially enriched in cardiomyocytes. Hereditas 2021, 158, 4. [Google Scholar] [CrossRef] [PubMed]
- Bojkova, D.; Wagner, J.U.G.; Shumliakivska, M.; Aslan, G.S.; Saleem, U.; Hansen, A.; Luxán, G.; Günther, S.; Pham, M.D.; Krishnan, J.; et al. SARS-CoV-2 infects and induces cytotoxic effects in human cardiomyocytes. Cardiovasc. Res. 2020, 116, 2207–2215. [Google Scholar] [CrossRef]
- Pers, Y.M.; Padern, G. Revisiting the cardiovascular risk of hydroxychloroquine in RA. Nat. Rev. Rheumatol. 2020, 16, 671–672. [Google Scholar] [CrossRef]
- Mercuro, N.J.; Yen, C.F.; Shim, D.J.; Maher, T.R.; McCoy, C.M.; Zimetbaum, P.J.; Gold, H.S. Risk of QT Interval Prolongation Associated with Use of Hydroxychloroquine with or without Concomitant Azithromycin Among Hospitalized Patients Testing Positive for Coronavirus Disease 2019 (COVID-19). JAMA Cardiol. 2020, 5, 1036–1041. [Google Scholar] [CrossRef]
- Pan, H.; Peto, R.; Henao-Restrepo, A.M.; Preziosi, M.P.; Sathiyamoorthy, V.; Abdool Karim, Q.; Alejandria, M.M.; Hernández García, C.; Kieny, M.P.; Malekzadeh, R.; et al. Repurposed Antiviral Drugs for COVID-19—Interim WHO Solidarity Trial Results. N. Engl. J. Med. 2021, 384, 497–511. [Google Scholar] [CrossRef]
- Giordo, R.; Paliogiannis, P.; Mangoni, A.A.; Pintus, G. SARS-CoV-2 and endothelial cell interaction in COVID-19: Molecular perspectives. Vasc. Biol. 2021, 3, R15–R23. [Google Scholar] [CrossRef] [PubMed]
- Perez-Miller, S.; Patek, M.; Moutal, A.; Cabel, C.R.; Thorne, C.A.; Campos, S.K.; Khanna, R. In silico identification and validation of inhibitors of the interaction between neuropilin receptor 1 and SARS-CoV-2 Spike protein. bioRxiv 2020. [Google Scholar] [CrossRef]
- Cantuti-Castelvetri, L.; Ojha, R.; Pedro, L.D.; Djannatian, M.; Franz, J.; Kuivanen, S.; van der Meer, F.; Kallio, K.; Kaya, T.; Anastasina, M.; et al. Neuropilin-1 facilitates SARS-CoV-2 cell entry and infectivity. Science 2020, 370, 856–860. [Google Scholar] [CrossRef]
- Lei, Y.; Zhang, J.; Schiavon, C.R.; He, M.; Chen, L.; Shen, H.; Zhang, Y.; Yin, Q.; Cho, Y.; Andrade, L.; et al. SARS-CoV-2 Spike Protein Impairs Endothelial Function via Downregulation of ACE 2. Circ. Res. 2021, 128, 1323–1326. [Google Scholar] [CrossRef] [PubMed]
- Jiang, F.; Yang, J.; Zhang, Y.; Dong, M.; Wang, S.; Zhang, Q.; Liu, F.F.; Zhang, K.; Zhang, C. Angiotensin-converting enzyme 2 and angiotensin 1-7: Novel therapeutic targets. Nat. Rev. Cardiol. 2014, 11, 413–426. [Google Scholar] [CrossRef] [PubMed]
- Crackower, M.A.; Sarao, R.; Oudit, G.Y.; Yagil, C.; Kozieradzki, I.; Scanga, S.E.; Oliveira-dos-Santos, A.J.; da Costa, J.; Zhang, L.; Pei, Y.; et al. Angiotensin-converting enzyme 2 is an essential regulator of heart function. Nature 2002, 417, 822–828. [Google Scholar] [CrossRef]
- Banu, N.; Panikar, S.S.; Leal, L.R.; Leal, A.R. Protective role of ACE2 and its downregulation in SARS-CoV-2 infection leading to Macrophage Activation Syndrome: Therapeutic implications. Life Sci. 2020, 256, 117905. [Google Scholar] [CrossRef] [PubMed]
- Baral, R.; White, M.; Vassiliou, V.S. Effect of Renin-Angiotensin-Aldosterone System Inhibitors in Patients with COVID-19: A Systematic Review and Meta-analysis of 28,872 Patients. Curr. Atheroscler. Rep. 2020, 22, 61. [Google Scholar] [CrossRef]
- Lopes, R.D.; Macedo, A.V.S.; de Barros e Silva, P.G.M.; Moll-Bernardes, R.J.; Feldman, A.; D’Andréa Saba Arruda, G.; de Souza, A.S.; de Albuquerque, D.C.; Mazza, L.; Santos, M.F.; et al. Continuing versus suspending angiotensin-converting enzyme inhibitors and angiotensin receptor blockers: Impact on adverse outcomes in hospitalized patients with severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2)—The BRACE CORONA Trial. Am. Heart J. 2020, 226, 49–59. [Google Scholar] [CrossRef]
- Xia, S.; Liu, M.; Wang, C.; Xu, W.; Lan, Q.; Feng, S.; Qi, F.; Bao, L.; Du, L.; Liu, S.; et al. Inhibition of SARS-CoV-2 (previously 2019-nCoV) infection by a highly potent pan-coronavirus fusion inhibitor targeting its spike protein that harbors a high capacity to mediate membrane fusion. Cell Res. 2020, 30, 343–355. [Google Scholar] [CrossRef] [Green Version]
- Kamitani, W.; Narayanan, K.; Huang, C.; Lokugamage, K.; Ikegami, T.; Ito, N.; Kubo, H.; Makino, S. Severe acute respiratory syndrome coronavirus nsp1 protein suppresses host gene expression by promoting host mRNA degradation. Proc. Natl. Acad. Sci. USA 2006, 103, 12885–12890. [Google Scholar] [CrossRef] [Green Version]
- Tohya, Y.; Narayanan, K.; Kamitani, W.; Huang, C.; Lokugamage, K.; Makino, S. Suppression of host gene expression by nsp1 proteins of group 2 bat coronaviruses. J. Virol. 2009, 83, 5282–5288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schubert, K.; Karousis, E.D.; Jomaa, A.; Scaiola, A.; Echeverria, B.; Gurzeler, L.A.; Leibundgut, M.; Thiel, V.; Mühlemann, O.; Ban, N. SARS-CoV-2 Nsp1 binds the ribosomal mRNA channel to inhibit translation. Nat. Struct. Mol. Biol. 2020, 27, 959–966. [Google Scholar] [CrossRef] [PubMed]
- Gorbalenya, A.E.; Enjuanes, L.; Ziebuhr, J.; Snijder, E.J. Nidovirales: Evolving the largest RNA virus genome. Virus Res. 2006, 117, 17–37. [Google Scholar] [CrossRef] [PubMed]
- V’kovski, P.; Kratzel, A.; Steiner, S.; Stalder, H.; Thiel, V. Coronavirus biology and replication: Implications for SARS-CoV-2. Nat. Rev. Microbiol. 2020, 19, 155–170. [Google Scholar] [CrossRef] [PubMed]
- Kokic, G.; Hillen, H.S.; Tegunov, D.; Dienemann, C.; Seitz, F.; Schmitzova, J.; Farnung, L.; Siewert, A.; Höbartner, C.; Cramer, P. Mechanism of SARS-CoV-2 polymerase stalling by remdesivir. Nat. Commun. 2021, 12, 279. [Google Scholar] [CrossRef] [PubMed]
- Zeitlinger, M.; Koch, B.C.P.; Bruggemann, R.; De Cock, P.; Felton, T.; Hites, M.; Le, J.; Luque, S.; MacGowan, A.P.; Marriott, D.J.E.; et al. Pharmacokinetics/Pharmacodynamics of Antiviral Agents Used to Treat SARS-CoV-2 and Their Potential Interaction with Drugs and Other Supportive Measures: A Comprehensive Review by the PK/PD of Anti-Infectives Study Group of the European Society of Antimicrobial Agents. Clin. Pharmacokinet. 2020, 59, 1195–1216. [Google Scholar] [CrossRef]
- Uzunova, K.; Filipova, E.; Pavlova, V.; Vekov, T. Insights into antiviral mechanisms of remdesivir, lopinavir/ritonavir and chloroquine/hydroxychloroquine affecting the new SARS-CoV-2. Biomed. Pharmacother. 2020, 131, 110668. [Google Scholar] [CrossRef]
- Glick, D.; Barth, S.; Macleod, K.F. Autophagy: Cellular and molecular mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar] [CrossRef] [Green Version]
- Mohamud, Y.; Shi, J.; Qu, J.; Poon, T.; Xue, Y.C.; Deng, H.; Zhang, J.; Luo, H. Enteroviral Infection Inhibits Autophagic Flux via Disruption of the SNARE Complex to Enhance Viral Replication. Cell Rep. 2018, 22, 3292–3303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, L.; Yue, J. The interplay of autophagy and enterovirus. Semin. Cell Dev. Biol. 2020, 101, 12–19. [Google Scholar] [CrossRef]
- Wolff, G.; Melia, C.E.; Snijder, E.J.; Bárcena, M. Double-Membrane Vesicles as Platforms for Viral Replication. Trends Microbiol. 2020, 28, 1022–1033. [Google Scholar] [CrossRef]
- Wong, J.; Zhang, J.; Si, X.; Gao, G.; Mao, I.; McManus, B.M.; Luo, H. Autophagosome supports coxsackievirus B3 replication in host cells. J. Virol. 2008, 82, 9143–9153. [Google Scholar] [CrossRef] [Green Version]
- Tabor-Godwin, J.M.; Tsueng, G.; Sayen, M.R.; Gottlieb, R.A.; Feuer, R. The role of autophagy during coxsackievirus infection of neural progenitor and stem cells. Autophagy 2012, 8, 938–953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Schaar, H.M.; Melia, C.E.; van Bruggen, J.A.; Strating, J.R.; van Geenen, M.E.; Koster, A.J.; Bárcena, M.; van Kuppeveld, F.J. Illuminating the Sites of Enterovirus Replication in Living Cells by Using a Split-GFP-Tagged Viral Protein. mSphere 2016, 1. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Sun, H.; Pei, R.; Mao, B.; Zhao, Z.; Li, H.; Lin, Y.; Lu, K. The SARS-CoV-2 protein ORF3a inhibits fusion of autophagosomes with lysosomes. Cell Discov. 2021, 7, 31. [Google Scholar] [CrossRef] [PubMed]
- Gassen, N.C.; Papies, J.; Bajaj, T.; Emanuel, J.; Dethloff, F.; Chua, R.L.; Trimpert, J.; Heinemann, N.; Niemeyer, C.; Weege, F.; et al. SARS-CoV-2-mediated dysregulation of metabolism and autophagy uncovers host-targeting antivirals. Nat. Commun. 2021, 12, 3818. [Google Scholar] [CrossRef]
- Klein, S.; Cortese, M.; Winter, S.L.; Wachsmuth-Melm, M.; Neufeldt, C.J.; Cerikan, B.; Stanifer, M.L.; Boulant, S.; Bartenschlager, R.; Chlanda, P. SARS-CoV-2 structure and replication characterized by in situ cryo-electron tomography. Nat. Commun. 2020, 11, 5885. [Google Scholar] [CrossRef] [PubMed]
- Reggiori, F.; Monastyrska, I.; Verheije, M.H.; Calì, T.; Ulasli, M.; Bianchi, S.; Bernasconi, R.; de Haan, C.A.; Molinari, M. Coronaviruses Hijack the LC3-I-positive EDEMosomes, ER-derived vesicles exporting short-lived ERAD regulators, for replication. Cell Host Microbe 2010, 7, 500–508. [Google Scholar] [CrossRef] [Green Version]
- Knoops, K.; Kikkert, M.; Worm, S.H.; Zevenhoven-Dobbe, J.C.; van der Meer, Y.; Koster, A.J.; Mommaas, A.M.; Snijder, E.J. SARS-coronavirus replication is supported by a reticulovesicular network of modified endoplasmic reticulum. PLoS Biol. 2008, 6, e226. [Google Scholar] [CrossRef] [PubMed]
- Snijder, E.J.; Limpens, R.W.A.L.; de Wilde, A.H.; de Jong, A.W.M.; Zevenhoven-Dobbe, J.C.; Maier, H.J.; Faas, F.F.G.A.; Koster, A.J.; Bárcena, M. A unifying structural and functional model of the coronavirus replication organelle: Tracking down RNA synthesis. PLoS Biol. 2020, 18, e3000715. [Google Scholar] [CrossRef]
- Cortese, M.; Lee, J.Y.; Cerikan, B.; Neufeldt, C.J.; Oorschot, V.M.J.; Köhrer, S.; Hennies, J.; Schieber, N.L.; Ronchi, P.; Mizzon, G.; et al. Integrative Imaging Reveals SARS-CoV-2-Induced Reshaping of Subcellular Morphologies. Cell Host Microbe 2020, 28, 853–866.e855. [Google Scholar] [CrossRef]
- Angelini, M.M.; Akhlaghpour, M.; Neuman, B.W.; Buchmeier, M.J. Severe acute respiratory syndrome coronavirus nonstructural proteins 3, 4, and 6 induce double-membrane vesicles. mBio 2013, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolff, G.; Limpens, R.W.A.L.; Zevenhoven-Dobbe, J.C.; Laugks, U.; Zheng, S.; de Jong, A.W.M.; Koning, R.I.; Agard, D.A.; Grünewald, K.; Koster, A.J.; et al. A molecular pore spans the double membrane of the coronavirus replication organelle. Science 2020, 369, 1395–1398. [Google Scholar] [CrossRef] [PubMed]
- Müller, C.; Hardt, M.; Schwudke, D.; Neuman, B.W.; Pleschka, S.; Ziebuhr, J. Inhibition of Cytosolic Phospholipase A. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [Green Version]
- Verheije, M.H.; Raaben, M.; Mari, M.; Te Lintelo, E.G.; Reggiori, F.; van Kuppeveld, F.J.; Rottier, P.J.; de Haan, C.A. Mouse hepatitis coronavirus RNA replication depends on GBF1-mediated ARF1 activation. PLoS Pathog. 2008, 4, e1000088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Haan, C.A.; Rottier, P.J. Molecular interactions in the assembly of coronaviruses. Adv. Virus Res. 2005, 64, 165–230. [Google Scholar] [CrossRef]
- Harris, K.G.; Coyne, C.B. Death waits for no man—Does it wait for a virus? How enteroviruses induce and control cell death. Cytokine Growth Factor Rev. 2014, 25, 587–596. [Google Scholar] [CrossRef] [Green Version]
- Hotez, P.J.; Bottazzi, M.E.; Corry, D.B. The potential role of Th17 immune responses in coronavirus immunopathology and vaccine-induced immune enhancement. Microbes Infect. 2020, 22, 165–167. [Google Scholar] [CrossRef] [PubMed]
- Corsten, M.F.; Schroen, B.; Heymans, S. Inflammation in viral myocarditis: Friend or foe? Trends Mol. Med. 2012, 18, 426–437. [Google Scholar] [CrossRef] [PubMed]
- Baldeviano, G.C.; Barin, J.G.; Talor, M.V.; Srinivasan, S.; Bedja, D.; Zheng, D.; Gabrielson, K.; Iwakura, Y.; Rose, N.R.; Cihakova, D. Interleukin-17A is dispensable for myocarditis but essential for the progression to dilated cardiomyopathy. Circ. Res. 2010, 106, 1646–1655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Myers, J.M.; Cooper, L.T.; Kem, D.C.; Stavrakis, S.; Kosanke, S.D.; Shevach, E.M.; Fairweather, D.; Stoner, J.A.; Cox, C.J.; Cunningham, M.W. Cardiac myosin-Th17 responses promote heart failure in human myocarditis. JCI Insight 2016, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tschöpe, C.; Ammirati, E.; Bozkurt, B.; Caforio, A.L.P.; Cooper, L.T.; Felix, S.B.; Hare, J.M.; Heidecker, B.; Heymans, S.; Hübner, N.; et al. Myocarditis and inflammatory cardiomyopathy: Current evidence and future directions. Nat. Rev. Cardiol. 2021, 18, 169–193. [Google Scholar] [CrossRef]
- Shi, Y.; Wang, Y.; Shao, C.; Huang, J.; Gan, J.; Huang, X.; Bucci, E.; Piacentini, M.; Ippolito, G.; Melino, G. COVID-19 infection: The perspectives on immune responses. Cell Death Differ. 2020, 27, 1451–1454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindner, D.; Fitzek, A.; Bräuninger, H.; Aleshcheva, G.; Edler, C.; Meissner, K.; Scherschel, K.; Kirchhof, P.; Escher, F.; Schultheiss, H.P.; et al. Association of Cardiac Infection with SARS-CoV-2 in Confirmed COVID-19 Autopsy Cases. JAMA Cardiol. 2020, 5, 1281–1285. [Google Scholar] [CrossRef]
- Kepp, O.; Galluzzi, L.; Zitvogel, L.; Kroemer, G. Pyroptosis—A cell death modality of its kind? Eur. J. Immunol. 2010, 40, 627–630. [Google Scholar] [CrossRef]
- Siu, K.L.; Yuen, K.S.; Castaño-Rodriguez, C.; Ye, Z.W.; Yeung, M.L.; Fung, S.Y.; Yuan, S.; Chan, C.P.; Yuen, K.Y.; Enjuanes, L.; et al. Severe acute respiratory syndrome coronavirus ORF3a protein activates the NLRP3 inflammasome by promoting TRAF3-dependent ubiquitination of ASC. FASEB J. 2019, 33, 8865–8877. [Google Scholar] [CrossRef]
- Shi, C.S.; Nabar, N.R.; Huang, N.N.; Kehrl, J.H. SARS-Coronavirus Open Reading Frame-8b triggers intracellular stress pathways and activates NLRP3 inflammasomes. Cell Death Discov. 2019, 5, 101. [Google Scholar] [CrossRef] [Green Version]
- Nieto-Torres, J.L.; Verdiá-Báguena, C.; Jimenez-Guardeño, J.M.; Regla-Nava, J.A.; Castaño-Rodriguez, C.; Fernandez-Delgado, R.; Torres, J.; Aguilella, V.M.; Enjuanes, L. Severe acute respiratory syndrome coronavirus E protein transports calcium ions and activates the NLRP3 inflammasome. Virology 2015, 485, 330–339. [Google Scholar] [CrossRef] [Green Version]
- Lim, Y.X.; Ng, Y.L.; Tam, J.P.; Liu, D.X. Human Coronaviruses: A Review of Virus-Host Interactions. Diseases 2016, 4, 26. [Google Scholar] [CrossRef]
- DeDiego, M.L.; Nieto-Torres, J.L.; Regla-Nava, J.A.; Jimenez-Guardeño, J.M.; Fernandez-Delgado, R.; Fett, C.; Castaño-Rodriguez, C.; Perlman, S.; Enjuanes, L. Inhibition of NF-κB-mediated inflammation in severe acute respiratory syndrome coronavirus-infected mice increases survival. J. Virol. 2014, 88, 913–924. [Google Scholar] [CrossRef] [Green Version]
- Sa Ribero, M.; Jouvenet, N.; Dreux, M.; Nisole, S. Interplay between SARS-CoV-2 and the type I interferon response. PLoS Pathog. 2020, 16, e1008737. [Google Scholar] [CrossRef]
- Chen, Z.; John Wherry, E. T cell responses in patients with COVID-19. Nat. Rev. Immunol. 2020, 20, 529–536. [Google Scholar] [CrossRef] [PubMed]
- Makris, S.; Paulsen, M.; Johansson, C. Type I Interferons as Regulators of Lung Inflammation. Front. Immunol. 2017, 8, 259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costela-Ruiz, V.J.; Illescas-Montes, R.; Puerta-Puerta, J.M.; Ruiz, C.; Melguizo-Rodríguez, L. SARS-CoV-2 infection: The role of cytokines in COVID-19 disease. Cytokine Growth Factor Rev. 2020, 54, 62–75. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.C.; Huang, R.; Sakamuru, S.; Shukla, S.J.; Attene-Ramos, M.S.; Shinn, P.; Van Leer, D.; Leister, W.; Austin, C.P.; Xia, M. Identification of known drugs that act as inhibitors of NF-kappaB signaling and their mechanism of action. Biochem. Pharmacol. 2010, 79, 1272–1280. [Google Scholar] [CrossRef] [Green Version]
- Hung, I.F.; Lung, K.C.; Tso, E.Y.; Liu, R.; Chung, T.W.; Chu, M.Y.; Ng, Y.Y.; Lo, J.; Chan, J.; Tam, A.R.; et al. Triple combination of interferon beta-1b, lopinavir-ritonavir, and ribavirin in the treatment of patients admitted to hospital with COVID-19: An open-label, randomised, phase 2 trial. Lancet 2020, 395, 1695–1704. [Google Scholar] [CrossRef]
- Kühl, U.; Pauschinger, M.; Schwimmbeck, P.L.; Seeberg, B.; Lober, C.; Noutsias, M.; Poller, W.; Schultheiss, H.P. Interferon-beta treatment eliminates cardiotropic viruses and improves left ventricular function in patients with myocardial persistence of viral genomes and left ventricular dysfunction. Circulation 2003, 107, 2793–2798. [Google Scholar] [CrossRef] [Green Version]
- Kuhl, U.; Lassner, D.; von Schlippenbach, J.; Poller, W.; Schultheiss, H.P. Interferon-Beta improves survival in enterovirus-associated cardiomyopathy. J. Am. Coll. Cardiol. 2012, 60, 1295–1296. [Google Scholar] [CrossRef] [Green Version]
- Schmidt-Lucke, C.; Spillmann, F.; Bock, T.; Kühl, U.; Van Linthout, S.; Schultheiss, H.P.; Tschöpe, C. Interferon beta modulates endothelial damage in patients with cardiac persistence of human parvovirus b19 infection. J. Infect. Dis. 2010, 201, 936–945. [Google Scholar] [CrossRef]
- Lai, Y.; Wang, M.; Cheng, A.; Mao, S.; Ou, X.; Yang, Q.; Wu, Y.; Jia, R.; Liu, M.; Zhu, D.; et al. Regulation of Apoptosis by Enteroviruses. Front. Microbiol. 2020, 11, 1145. [Google Scholar] [CrossRef]
- Chen, A.Y.; Qiu, J. Parvovirus infection-induced cell death and cell cycle arrest. Future Virol. 2010, 5, 731–743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivanisenko, N.V.; Seyrek, K.; Kolchanov, N.A.; Ivanisenko, V.A.; Lavrik, I.N. The role of death domain proteins in host response upon SARS-CoV-2 infection: Modulation of programmed cell death and translational applications. Cell Death Discov. 2020, 6, 101. [Google Scholar] [CrossRef]
- Fang, X.; Gao, J.; Zheng, H.; Li, B.; Kong, L.; Zhang, Y.; Wang, W.; Zeng, Y.; Ye, L. The membrane protein of SARS-CoV suppresses NF-kappaB activation. J. Med. Virol. 2007, 79, 1431–1439. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Zhang, Y.; Guan, Z.; Li, H.; Ye, M.; Chen, X.; Shen, J.; Zhou, Y.; Shi, Z.L.; Zhou, P.; et al. SARS-CoV-2 triggers inflammatory responses and cell death through caspase-8 activation. Signal Transduct. Target. Ther. 2020, 5, 235. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Shu, T.; Wu, D.; Mu, J.; Wang, C.; Huang, M.; Han, Y.; Zhang, X.Y.; Zhou, W.; Qiu, Y.; et al. The ORF3a protein of SARS-CoV-2 induces apoptosis in cells. Cell Mol. Immunol. 2020, 17, 881–883. [Google Scholar] [CrossRef] [PubMed]
- Schug, Z.T.; Gonzalvez, F.; Houtkooper, R.H.; Vaz, F.M.; Gottlieb, E. BID is cleaved by caspase-8 within a native complex on the mitochondrial membrane. Cell Death Differ. 2011, 18, 538–548. [Google Scholar] [CrossRef] [PubMed]
- Kopecky-Bromberg, S.A.; Martinez-Sobrido, L.; Palese, P. 7a protein of severe acute respiratory syndrome coronavirus inhibits cellular protein synthesis and activates p38 mitogen-activated protein kinase. J. Virol. 2006, 80, 785–793. [Google Scholar] [CrossRef] [Green Version]
- Ye, Z.; Wong, C.K.; Li, P.; Xie, Y. A SARS-CoV protein, ORF-6, induces caspase-3 mediated, ER stress and JNK-dependent apoptosis. Biochim. Biophys. Acta 2008, 1780, 1383–1387. [Google Scholar] [CrossRef]
- Yang, Y.; Xiong, Z.; Zhang, S.; Yan, Y.; Nguyen, J.; Ng, B.; Lu, H.; Brendese, J.; Yang, F.; Wang, H.; et al. Bcl-xL inhibits T-cell apoptosis induced by expression of SARS coronavirus E protein in the absence of growth factors. Biochem. J. 2005, 392, 135–143. [Google Scholar] [CrossRef] [Green Version]
- Chow, K.Y.; Yeung, Y.S.; Hon, C.C.; Zeng, F.; Law, K.M.; Leung, F.C. Adenovirus-mediated expression of the C-terminal domain of SARS-CoV spike protein is sufficient to induce apoptosis in Vero E6 cells. FEBS Lett. 2005, 579, 6699–6704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsoi, H.; Li, L.; Chen, Z.S.; Lau, K.F.; Tsui, S.K.; Chan, H.Y. The SARS-coronavirus membrane protein induces apoptosis via interfering with PDK1-PKB/Akt signalling. Biochem. J. 2014, 464, 439–447. [Google Scholar] [CrossRef] [Green Version]
- Bordi, L.; Castilletti, C.; Falasca, L.; Ciccosanti, F.; Calcaterra, S.; Rozera, G.; Di Caro, A.; Zaniratti, S.; Rinaldi, A.; Ippolito, G.; et al. Bcl-2 inhibits the caspase-dependent apoptosis induced by SARS-CoV without affecting virus replication kinetics. Arch. Virol 2006, 151, 369–377. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Garron, T.M.; Chang, Q.; Su, Z.; Zhou, C.; Gong, E.C.; Zheng, J.; Yin, Y.; Ksiazek, T.; Brasel, T.; et al. Cell-type apoptosis in lung during SARS-CoV-2 infection. bioRxiv 2020. [Google Scholar] [CrossRef]
- Schulze, K.; Witzenbichler, B.; Christmann, C.; Schultheiss, H.P. Disturbance of myocardial energy metabolism in experimental virus myocarditis by antibodies against the adenine nucleotide translocator. Cardiovasc. Res. 1999, 44, 91–100. [Google Scholar] [CrossRef] [Green Version]
- Ebermann, L.; Wika, S.; Klumpe, I.; Hammer, E.; Klingel, K.; Lassner, D.; Volker, U.; Erben, U.; Zeichhardt, H.; Schultheiss, H.P.; et al. The mitochondrial respiratory chain has a critical role in the antiviral process in Coxsackievirus B3-induced myocarditis. Lab. Investig. 2012, 92, 125–134. [Google Scholar] [CrossRef] [Green Version]
- Peischard, S.; Ho, H.T.; Piccini, I.; Strutz-Seebohm, N.; Röpke, A.; Liashkovich, I.; Gosain, H.; Rieger, B.; Klingel, K.; Eggers, B.; et al. The first versatile human iPSC-based model of ectopic virus induction allows new insights in RNA-virus disease. Sci. Rep. 2020, 10, 16804. [Google Scholar] [CrossRef]
- Sin, J.; McIntyre, L.; Stotland, A.; Feuer, R.; Gottlieb, R.A. Coxsackievirus B Escapes the Infected Cell in Ejected Mitophagosomes. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [Green Version]
- Lin, L.; Zhang, M.; Yan, R.; Shan, H.; Diao, J.; Wei, J. Inhibition of Drp1 attenuates mitochondrial damage and myocardial injury in Coxsackievirus B3 induced myocarditis. Biochem. Biophys. Res. Commun. 2017, 484, 550–556. [Google Scholar] [CrossRef]
- Wu, K.; Zou, J.; Chang, H.Y. RNA-GPS Predicts SARS-CoV-2 RNA Localization to Host Mitochondria and Nucleolus. bioRxiv 2020. [Google Scholar] [CrossRef]
- Guzzi, P.H.; Mercatelli, D.; Ceraolo, C.; Giorgi, F.M. Master Regulator Analysis of the SARS-CoV-2/Human Interactome. J. Clin. Med. 2020, 9, 982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.; Xu, J.; Obernier, K.; O’Meara, M.J.; Guo, J.Z.; Swaney, D.L.; Tummino, T.A.; Hüttenhain, R.; et al. A SARS-CoV-2-Human Protein-Protein Interaction Map Reveals Drug Targets and Potential Drug-Repurposing. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Wang, L.; Dong, C.; Che, Y.; Jiang, L.; Liu, L.; Zhao, H.; Liao, Y.; Sheng, Y.; Dong, S.; et al. The interaction of the SARS coronavirus non-structural protein 10 with the cellular oxido-reductase system causes an extensive cytopathic effect. J. Clin. Virol. 2005, 34, 133–139. [Google Scholar] [CrossRef]
- Miller, B.; Silverstein, A.; Flores, M.; Cao, K.; Kumagai, H.; Mehta, H.H.; Yen, K.; Kim, S.J.; Cohen, P. Host mitochondrial transcriptome response to SARS-CoV-2 in multiple cell models and clinical samples. Sci. Rep. 2021, 11, 3. [Google Scholar] [CrossRef] [PubMed]
- Larosa, V.; Remacle, C. Insights into the respiratory chain and oxidative stress. Biosci Rep. 2018, 38. [Google Scholar] [CrossRef] [Green Version]
- Ajaz, S.; McPhail, M.J.; Singh, K.K.; Mujib, S.; Trovato, F.M.; Napoli, S.; Agarwal, K. Mitochondrial metabolic manipulation by SARS-CoV-2 in peripheral blood mononuclear cells of patients with COVID-19. Am. J. Physiol. Cell Physiol. 2021, 320, C57–C65. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.K.; Chaubey, G.; Chen, J.Y.; Suravajhala, P. Decoding SARS-CoV-2 hijacking of host mitochondria in COVID-19 pathogenesis. Am. J. Physiol. Cell Physiol. 2020, 319, C258–C267. [Google Scholar] [CrossRef]
- Schulze, K.; Becker, B.F.; Schauer, R.; Schultheiss, H.P. Antibodies to ADP-ATP carrier--an autoantigen in myocarditis and dilated cardiomyopathy--impair cardiac function. Circulation 1990, 81, 959–969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cecchini, R.; Cecchini, A.L. SARS-CoV-2 infection pathogenesis is related to oxidative stress as a response to aggression. Med. Hypotheses 2020, 143, 110102. [Google Scholar] [CrossRef]
- Abbate, A.; Sinagra, G.; Bussani, R.; Hoke, N.N.; Merlo, M.; Varma, A.; Toldo, S.; Salloum, F.N.; Biondi-Zoccai, G.G.; Vetrovec, G.W.; et al. Apoptosis in patients with acute myocarditis. Am. J. Cardiol. 2009, 104, 995–1000. [Google Scholar] [CrossRef]
- Alter, P.; Maisch, B. Escape from cardiomyocyte apoptosis by enterovirus persistence due to elevated soluble Fas-receptors. Z. Kardiol. 2004, 93, 524–532. [Google Scholar] [CrossRef]
- Nieva, J.L.; Madan, V.; Carrasco, L. Viroporins: Structure and biological functions. Nat. Rev. Microbiol. 2012, 10, 563–574. [Google Scholar] [CrossRef] [PubMed]
- Hyser, J.M.; Estes, M.K. Pathophysiological Consequences of Calcium-Conducting Viroporins. Annu. Rev. Virol. 2015, 2, 473–496. [Google Scholar] [CrossRef]
- Triantafilou, K.; Triantafilou, M. Ion flux in the lung: Virus-induced inflammasome activation. Trends Microbiol. 2014, 22, 580–588. [Google Scholar] [CrossRef] [PubMed]
- Nieto-Torres, J.L.; Verdiá-Báguena, C.; Castaño-Rodriguez, C.; Aguilella, V.M.; Enjuanes, L. Relevance of Viroporin Ion Channel Activity on Viral Replication and Pathogenesis. Viruses 2015, 7, 3552–3573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Frey, T.K.; Yang, J.J. Viral calciomics: Interplays between Ca2+ and virus. Cell Calcium 2009, 46, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Castaño-Rodriguez, C.; Honrubia, J.M.; Gutiérrez-Álvarez, J.; DeDiego, M.L.; Nieto-Torres, J.L.; Jimenez-Guardeño, J.M.; Regla-Nava, J.A.; Fernandez-Delgado, R.; Verdia-Báguena, C.; Queralt-Martín, M.; et al. Role of Severe Acute Respiratory Syndrome Coronavirus Viroporins E, 3a, and 8a in Replication and Pathogenesis. mBio 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Polina, I.; Sung, J.H.; Jhun, B.S.; O-uchi, J. Abstract 15857: SARS-Cov-2 Genes Encode Ito-like Potassium Channels: Linkage Between Viroporins and High Mortality Rate in COVID-19 Patients with Pre-existing Cardiovascular Diseases. Circulation 2020, 142, A15857. [Google Scholar] [CrossRef]
- Wilson, L.; McKinlay, C.; Gage, P.; Ewart, G. SARS coronavirus E protein forms cation-selective ion channels. Virology 2004, 330, 322–331. [Google Scholar] [CrossRef] [Green Version]
- Nieto-Torres, J.L.; Dediego, M.L.; Alvarez, E.; Jiménez-Guardeño, J.M.; Regla-Nava, J.A.; Llorente, M.; Kremer, L.; Shuo, S.; Enjuanes, L. Subcellular location and topology of severe acute respiratory syndrome coronavirus envelope protein. Virology 2011, 415, 69–82. [Google Scholar] [CrossRef] [Green Version]
- Liao, Y.; Yuan, Q.; Torres, J.; Tam, J.P.; Liu, D.X. Biochemical and functional characterization of the membrane association and membrane permeabilizing activity of the severe acute respiratory syndrome coronavirus envelope protein. Virology 2006, 349, 264–275. [Google Scholar] [CrossRef]
- Lu, W.; Zheng, B.J.; Xu, K.; Schwarz, W.; Du, L.; Wong, C.K.; Chen, J.; Duan, S.; Deubel, V.; Sun, B. Severe acute respiratory syndrome-associated coronavirus 3a protein forms an ion channel and modulates virus release. Proc. Natl. Acad. Sci. USA 2006, 103, 12540–12545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minakshi, R.; Padhan, K. The YXXΦ motif within the severe acute respiratory syndrome coronavirus (SARS-CoV) 3a protein is crucial for its intracellular transport. Virol. J. 2014, 11, 75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chien, T.H.; Chiang, Y.L.; Chen, C.P.; Henklein, P.; Hänel, K.; Hwang, I.S.; Willbold, D.; Fischer, W.B. Assembling an ion channel: ORF 3a from SARS-CoV. Biopolymers 2013, 99, 628–635. [Google Scholar] [CrossRef]
- Chen, I.Y.; Moriyama, M.; Chang, M.F.; Ichinohe, T. Severe Acute Respiratory Syndrome Coronavirus Viroporin 3a Activates the NLRP3 Inflammasome. Front. Microbiol. 2019, 10, 50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.C.; Krüger, J.; Sramala, I.; Hsu, H.J.; Henklein, P.; Chen, Y.M.; Fischer, W.B. ORF8a of SARS-CoV forms an ion channel: Experiments and molecular dynamics simulations. Biochim. Biophys. Acta 2011, 1808, 572–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torres, J.; Maheswari, U.; Parthasarathy, K.; Ng, L.; Liu, D.X.; Gong, X. Conductance and amantadine binding of a pore formed by a lysine-flanked transmembrane domain of SARS coronavirus envelope protein. Protein Sci. 2007, 16, 2065–2071. [Google Scholar] [CrossRef]
- Rejdak, K.; Grieb, P. Adamantanes might be protective from COVID-19 in patients with neurological diseases: Multiple sclerosis, parkinsonism and cognitive impairment. Mult. Scler. Relat. Disord. 2020, 42, 102163. [Google Scholar] [CrossRef]
- Wilson, L.; Gage, P.; Ewart, G. Hexamethylene amiloride blocks E protein ion channels and inhibits coronavirus replication. Virology 2006, 353, 294–306. [Google Scholar] [CrossRef] [Green Version]
- Crespi, B.; Alcock, J. Conflicts over calcium and the treatment of COVID-19. Evol. Med. Public Health 2021, 9, 149–156. [Google Scholar] [CrossRef]
- Triantafilou, K.; Kar, S.; van Kuppeveld, F.J.; Triantafilou, M. Rhinovirus-induced calcium flux triggers NLRP3 and NLRC5 activation in bronchial cells. Am. J. Respir. Cell Mol. Biol. 2013, 49, 923–934. [Google Scholar] [CrossRef]
- Bowles, N.E.; Richardson, P.J.; Olsen, E.G.; Archard, L.C. Detection of Coxsackie-B-virus-specific RNA sequences in myocardial biopsy samples from patients with myocarditis and dilated cardiomyopathy. Lancet 1986, 1, 1120–1123. [Google Scholar] [CrossRef]
- Zhi, N.; Zádori, Z.; Brown, K.E.; Tijssen, P. Construction and sequencing of an infectious clone of the human parvovirus B19. Virology 2004, 318, 142–152. [Google Scholar] [CrossRef] [Green Version]
- Klingel, K.; Rieger, P.; Mall, G.; Selinka, H.C.; Huber, M.; Kandolf, R. Visualization of enteroviral replication in myocardial tissue by ultrastructural in situ hybridization: Identification of target cells and cytopathic effects. Lab. Investig. 1998, 78, 1227–1237. [Google Scholar]
- Bültmann, B.D.; Klingel, K.; Sotlar, K.; Bock, C.T.; Baba, H.A.; Sauter, M.; Kandolf, R. Fatal parvovirus B19-associated myocarditis clinically mimicking ischemic heart disease: An endothelial cell-mediated disease. Hum. Pathol. 2003, 34, 92–95. [Google Scholar] [CrossRef] [PubMed]
- Bergelson, J.M.; Krithivas, A.; Celi, L.; Droguett, G.; Horwitz, M.S.; Wickham, T.; Crowell, R.L.; Finberg, R.W. The murine CAR homolog is a receptor for coxsackie B viruses and adenoviruses. J. Virol. 1998, 72, 415–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shafren, D.R.; Williams, D.T.; Barry, R.D. A decay-accelerating factor-binding strain of coxsackievirus B3 requires the coxsackievirus-adenovirus receptor protein to mediate lytic infection of rhabdomyosarcoma cells. J. Virol. 1997, 71, 9844–9848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, K.E.; Anderson, S.M.; Young, N.S. Erythrocyte P antigen: Cellular receptor for B19 parvovirus. Science 1993, 262, 114–117. [Google Scholar] [CrossRef] [PubMed]
- Munakata, Y.; Saito-Ito, T.; Kumura-Ishii, K.; Huang, J.; Kodera, T.; Ishii, T.; Hirabayashi, Y.; Koyanagi, Y.; Sasaki, T. Ku80 autoantigen as a cellular coreceptor for human parvovirus B19 infection. Blood 2005, 106, 3449–3456. [Google Scholar] [CrossRef] [PubMed]
- Weigel-Kelley, K.A.; Yoder, M.C.; Srivastava, A. Alpha5beta1 integrin as a cellular coreceptor for human parvovirus B19: Requirement of functional activation of beta1 integrin for viral entry. Blood 2003, 102, 3927–3933. [Google Scholar] [CrossRef]
- Gruez, A.; Selisko, B.; Roberts, M.; Bricogne, G.; Bussetta, C.; Jabafi, I.; Coutard, B.; De Palma, A.M.; Neyts, J.; Canard, B. The crystal structure of coxsackievirus B3 RNA-dependent RNA polymerase in complex with its protein primer VPg confirms the existence of a second VPg binding site on Picornaviridae polymerases. J. Virol. 2008, 82, 9577–9590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Limpens, R.W.; van der Schaar, H.M.; Kumar, D.; Koster, A.J.; Snijder, E.J.; van Kuppeveld, F.J.; Bárcena, M. The transformation of enterovirus replication structures: A three-dimensional study of single- and double-membrane compartments. mBio 2011, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mann, D.L. The emerging role of innate immunity in the heart and vascular system: For whom the cell tolls. Circ. Res. 2011, 108, 1133–1145. [Google Scholar] [CrossRef] [PubMed]
- Nigro, G.; Bastianon, V.; Colloridi, V.; Ventriglia, F.; Gallo, P.; D’Amati, G.; Koch, W.C.; Adler, S.P. Human parvovirus B19 infection in infancy associated with acute and chronic lymphocytic myocarditis and high cytokine levels: Report of 3 cases and review. Clin. Infect. Dis. 2000, 31, 65–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, J.; Yu, M.; Lin, Q.W.; Cao, A.L.; Yu, X.; Dong, J.H.; Wang, J.P.; Zhang, J.H.; Wang, M.; Guo, H.P.; et al. Th17 cells contribute to viral replication in coxsackievirus B3-induced acute viral myocarditis. J. Immunol. 2010, 185, 4004–4010. [Google Scholar] [CrossRef] [Green Version]
- Streitz, M.; Noutsias, M.; Volkmer, R.; Rohde, M.; Brestrich, G.; Block, A.; Klippert, K.; Kotsch, K.; Ay, B.; Hummel, M.; et al. NS1 specific CD8+ T-cells with effector function and TRBV11 dominance in a patient with parvovirus B19 associated inflammatory cardiomyopathy. PLoS ONE 2008, 3, e2361. [Google Scholar] [CrossRef] [PubMed]
- Moffatt, S.; Yaegashi, N.; Tada, K.; Tanaka, N.; Sugamura, K. Human parvovirus B19 nonstructural (NS1) protein induces apoptosis in erythroid lineage cells. J. Virol. 1998, 72, 3018–3028. [Google Scholar] [CrossRef] [Green Version]
- Kuhl, U.; Lassner, D.; Dorner, A.; Rohde, M.; Escher, F.; Seeberg, B.; Hertel, E.; Tschope, C.; Skurk, C.; Gross, U.M.; et al. A distinct subgroup of cardiomyopathy patients characterized by transcriptionally active cardiotropic erythrovirus and altered cardiac gene expression. Basic Res. Cardiol. 2013, 108, 372. [Google Scholar] [CrossRef]
- De Jong, A.S.; de Mattia, F.; Van Dommelen, M.M.; Lanke, K.; Melchers, W.J.; Willems, P.H.; van Kuppeveld, F.J. Functional analysis of picornavirus 2B proteins: Effects on calcium homeostasis and intracellular protein trafficking. J. Virol. 2008, 82, 3782–3790. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
SARS-CoV-2 viral particles in an endothelial cell of the heart of a 64-year-old male COVID-19 patient. The yellow arrow indicates viral particles.
Figure 1.
SARS-CoV-2 viral particles in an endothelial cell of the heart of a 64-year-old male COVID-19 patient. The yellow arrow indicates viral particles.

Figure 2.
SARS-CoV-2 entry into cells of the heart. severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) can use angiotensin-converting-enzyme 2 (ACE2) as a receptor and different proteases to prime the spike protein. Depending on which protease is used for spike S protein priming, a different path is used for viral entry; i.e., membrane fusion or endocytosis. ACE2, angiotensin-converting-enzyme 2; TMPRSS2, transmembrane protease serine 2; NRP1, neuropilin-1.
Figure 2.
SARS-CoV-2 entry into cells of the heart. severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) can use angiotensin-converting-enzyme 2 (ACE2) as a receptor and different proteases to prime the spike protein. Depending on which protease is used for spike S protein priming, a different path is used for viral entry; i.e., membrane fusion or endocytosis. ACE2, angiotensin-converting-enzyme 2; TMPRSS2, transmembrane protease serine 2; NRP1, neuropilin-1.

Figure 3.
SARS-CoV-2 replication and viral protein translation. SARS-CoV-2 has ssRNA(+) as genetic material that is translated to polyproteins. The polyproteins are processed by viral proteases to generate more functional viral proteins; e.g., RNA-dependent RNA polymerase and replication complex for RNA replication.
Figure 3.
SARS-CoV-2 replication and viral protein translation. SARS-CoV-2 has ssRNA(+) as genetic material that is translated to polyproteins. The polyproteins are processed by viral proteases to generate more functional viral proteins; e.g., RNA-dependent RNA polymerase and replication complex for RNA replication.

Figure 4.
SARS-CoV-2 uses double-membrane vesicles and the autophagosomal pathway for replication. SARS-CoV-2 inhibits the fusion of lysosomes with autophagosomes. SARS-CoV-2 uses DMVs originated from the ER as replication sites and uses parts of the autophagosomal pathway for replication. Viral particles are assembled in the ERGIC and are excreted via the secretory pathway. ER, endoplasmic reticulum; DMV, double-membrane vesicle; ERGIC, ER–Golgi intermediate compartment.
Figure 4.
SARS-CoV-2 uses double-membrane vesicles and the autophagosomal pathway for replication. SARS-CoV-2 inhibits the fusion of lysosomes with autophagosomes. SARS-CoV-2 uses DMVs originated from the ER as replication sites and uses parts of the autophagosomal pathway for replication. Viral particles are assembled in the ERGIC and are excreted via the secretory pathway. ER, endoplasmic reticulum; DMV, double-membrane vesicle; ERGIC, ER–Golgi intermediate compartment.

Figure 5.
Healing myocarditis in a SARS-CoV-2 positive patient (75 years old, male). (A) Masson Trichrome (B) H&E, (C) CD3+ T cells, (D) CD68+ macrophages of the interstitium (bar = 100 μm).
Figure 5.
Healing myocarditis in a SARS-CoV-2 positive patient (75 years old, male). (A) Masson Trichrome (B) H&E, (C) CD3+ T cells, (D) CD68+ macrophages of the interstitium (bar = 100 μm).

Figure 6.
SARS-CoV-2 manipulates and dysregulates immune response. The dotted arrows indicate a decreased effect through viral effect. NF-κB, nuclear factor-κB, NLRP3, NLR family pyrin domain containing 3; IFN-I, interferon type I; TNF, tumor necrosis factor; CCL2, chemokine (C-C motif) ligand 2; CXCL2, chemokine (C-X-C motif) ligand 2.
Figure 6.
SARS-CoV-2 manipulates and dysregulates immune response. The dotted arrows indicate a decreased effect through viral effect. NF-κB, nuclear factor-κB, NLRP3, NLR family pyrin domain containing 3; IFN-I, interferon type I; TNF, tumor necrosis factor; CCL2, chemokine (C-C motif) ligand 2; CXCL2, chemokine (C-X-C motif) ligand 2.

Figure 7.
SARS-CoV-2 manipulates cell death. In early infection stages, cell death is inhibited to promote viral replication, while in later infection stages, cell death is promoted to enhance viral spread. The dotted arrows indicate a decreased effect through viral effect. C-FLIP, cellular FLICE (FADD-like IL-1β-converting enzyme)-inhibitory protein; BCL-2, B-cell lymphoma 2; XIAP, X-linked inhibitor of apoptosis protein, Bax, Bcl-2-associated X protein, Bak, BCL-2 antagonist/killer-1; ER, endoplasmic reticulum; JNK, c-Jun N-terminal kinase; NF-κB, nuclear factor-κB; PDK, 3-phosphoinositide-dependent kinase; PKB/Akt, serine/threonine kinase protein kinase B; tBid, truncated BH3 interacting-domain death agonist; Cyt c, cytochrome c; NLRP3, NLR family pyrin domain containing 3.
Figure 7.
SARS-CoV-2 manipulates cell death. In early infection stages, cell death is inhibited to promote viral replication, while in later infection stages, cell death is promoted to enhance viral spread. The dotted arrows indicate a decreased effect through viral effect. C-FLIP, cellular FLICE (FADD-like IL-1β-converting enzyme)-inhibitory protein; BCL-2, B-cell lymphoma 2; XIAP, X-linked inhibitor of apoptosis protein, Bax, Bcl-2-associated X protein, Bak, BCL-2 antagonist/killer-1; ER, endoplasmic reticulum; JNK, c-Jun N-terminal kinase; NF-κB, nuclear factor-κB; PDK, 3-phosphoinositide-dependent kinase; PKB/Akt, serine/threonine kinase protein kinase B; tBid, truncated BH3 interacting-domain death agonist; Cyt c, cytochrome c; NLRP3, NLR family pyrin domain containing 3.

Figure 8.
SARS-CoV-2 manipulates energy metabolism. SARS-CoV-2 affects the morphology of mitochondria and components of the respiratory chain. A decrease in ATP and an increase in ROS production is observed, which could be the result of altered mitochondria function and respiratory chain activity. ATP, adenosine triphosphate; ROS, reactive oxygen species.
Figure 8.
SARS-CoV-2 manipulates energy metabolism. SARS-CoV-2 affects the morphology of mitochondria and components of the respiratory chain. A decrease in ATP and an increase in ROS production is observed, which could be the result of altered mitochondria function and respiratory chain activity. ATP, adenosine triphosphate; ROS, reactive oxygen species.

Figure 9.
SARS-CoV-2 proteins form cationic viroporins, causing inflammasome activation, altered action potential and Ca2+ handling, and apoptosis. ER, endoplasmic reticulum; ERGIC, ER–Golgi intermediate compartment; AP, action potential; cyt, cytosolic; mito, mitochondrial.
Figure 9.
SARS-CoV-2 proteins form cationic viroporins, causing inflammasome activation, altered action potential and Ca2+ handling, and apoptosis. ER, endoplasmic reticulum; ERGIC, ER–Golgi intermediate compartment; AP, action potential; cyt, cytosolic; mito, mitochondrial.

Figure 10.
SARS-CoV-2 virus–host interactions. ACE2, angiotensin-converting enzyme 2; ER, endoplasmic reticulum; ERGIC, ER–Golgi intermediate compartment; ATP, adenosine triphosphate; ROS, reactive oxygen species.
Figure 10.
SARS-CoV-2 virus–host interactions. ACE2, angiotensin-converting enzyme 2; ER, endoplasmic reticulum; ERGIC, ER–Golgi intermediate compartment; ATP, adenosine triphosphate; ROS, reactive oxygen species.


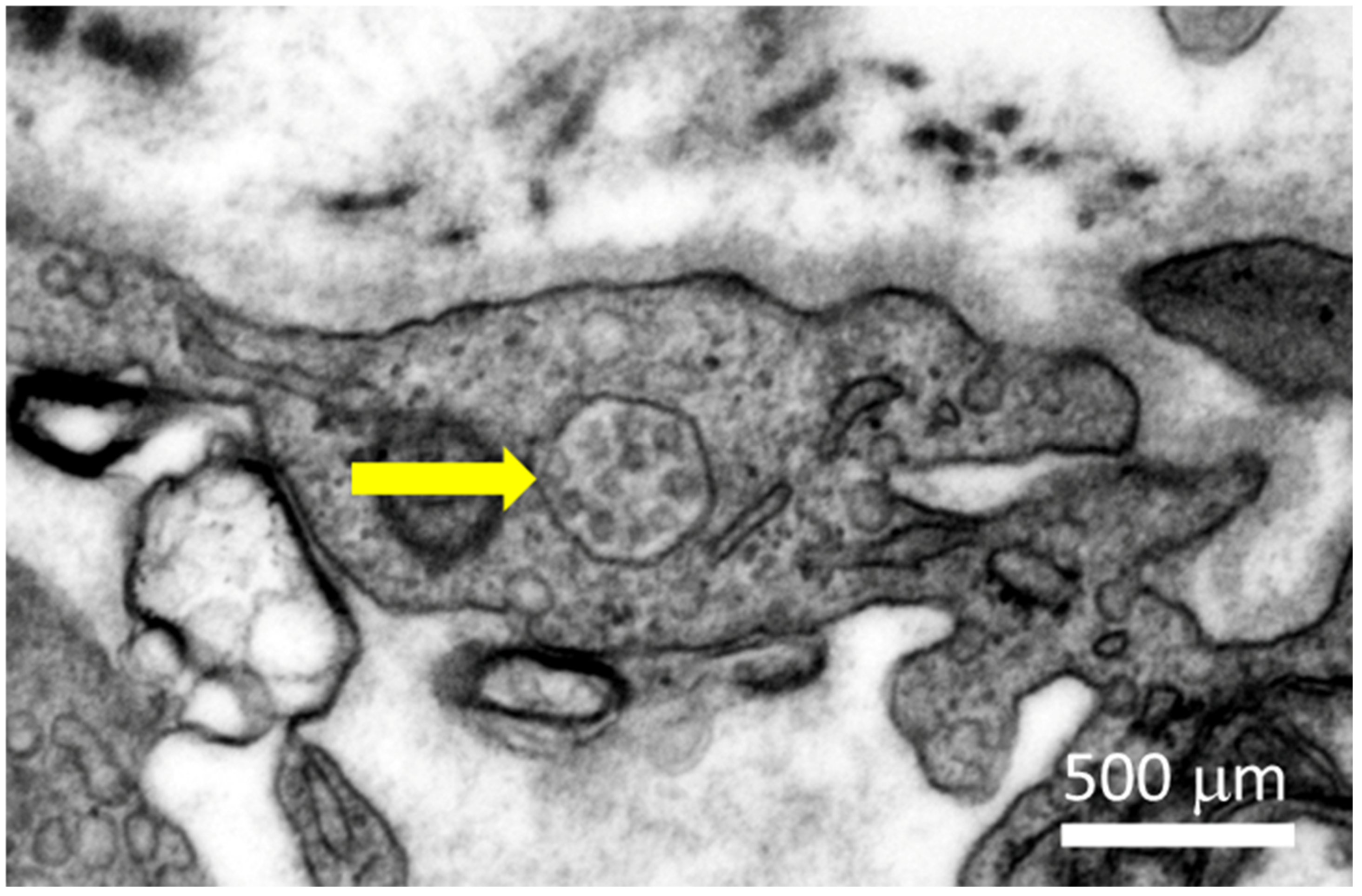{kind=link}
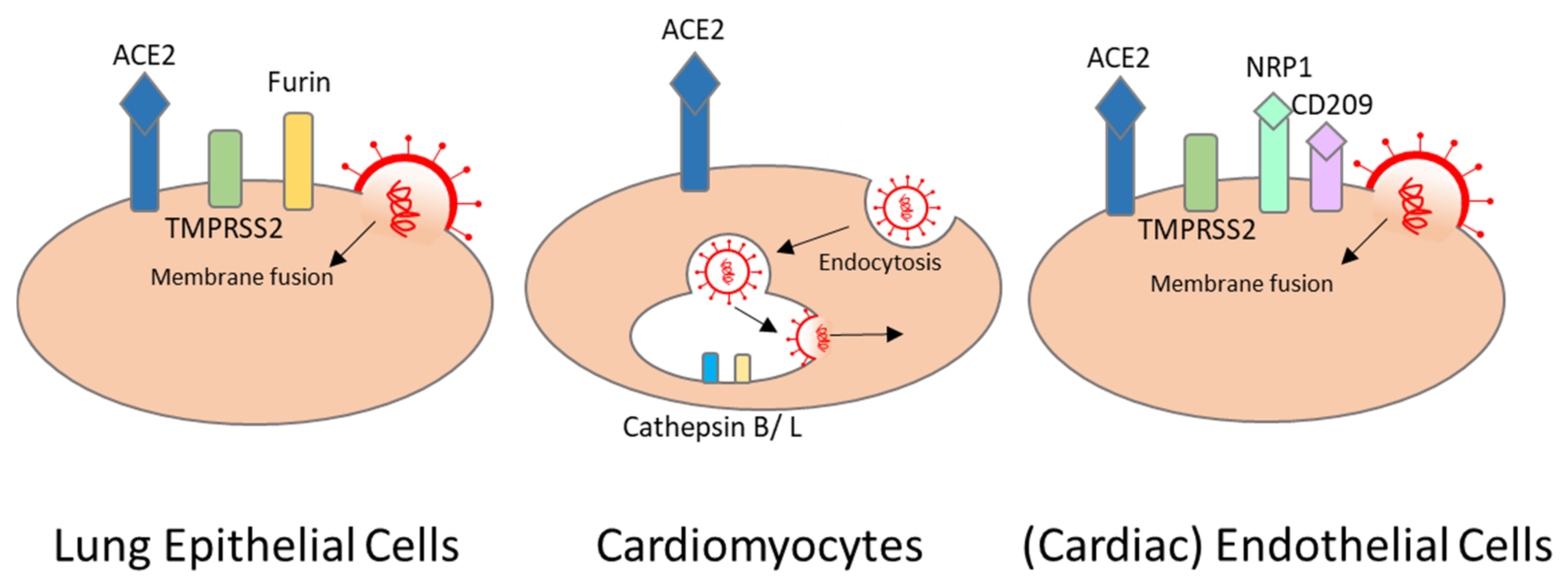{kind=link}
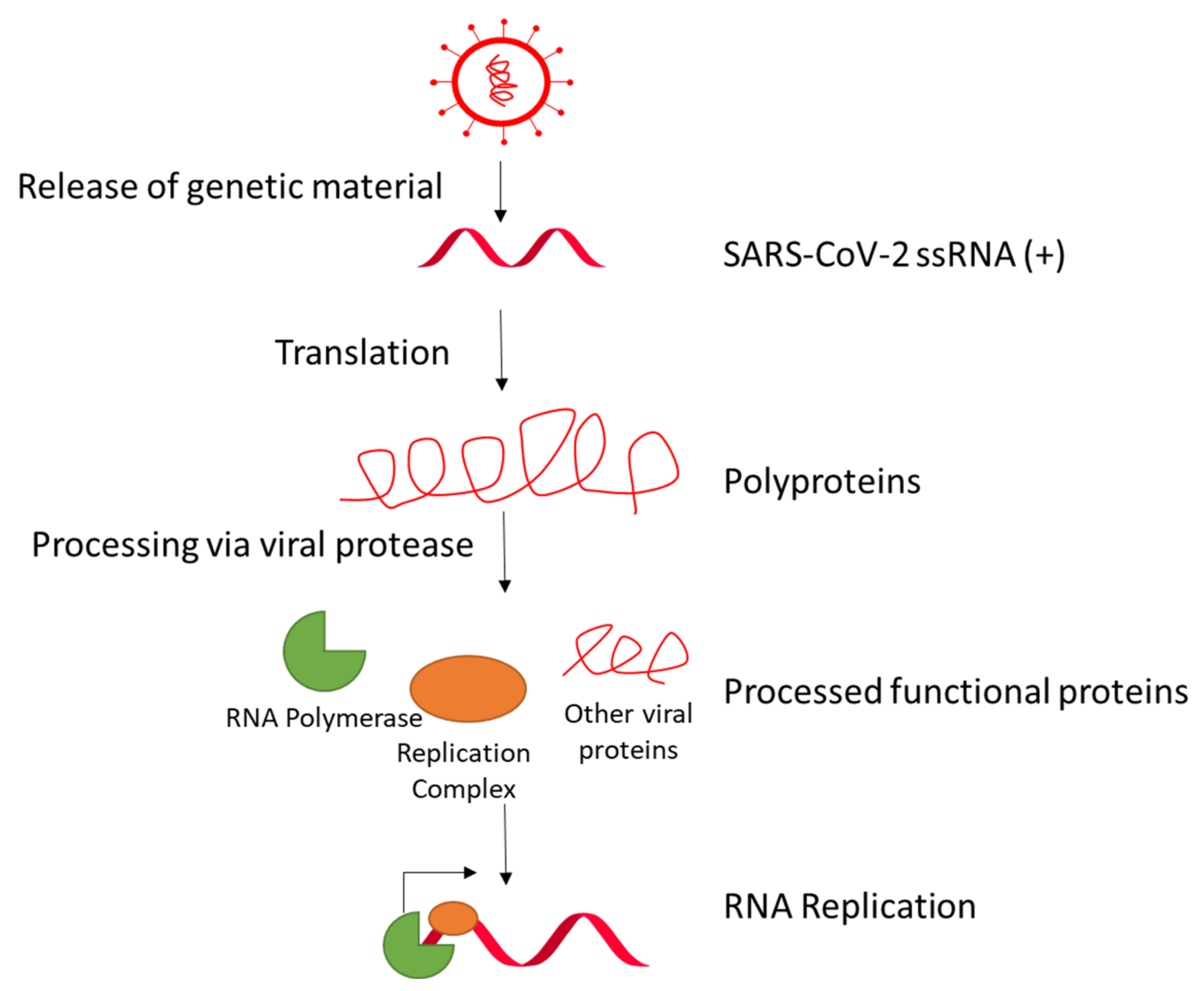{kind=link}
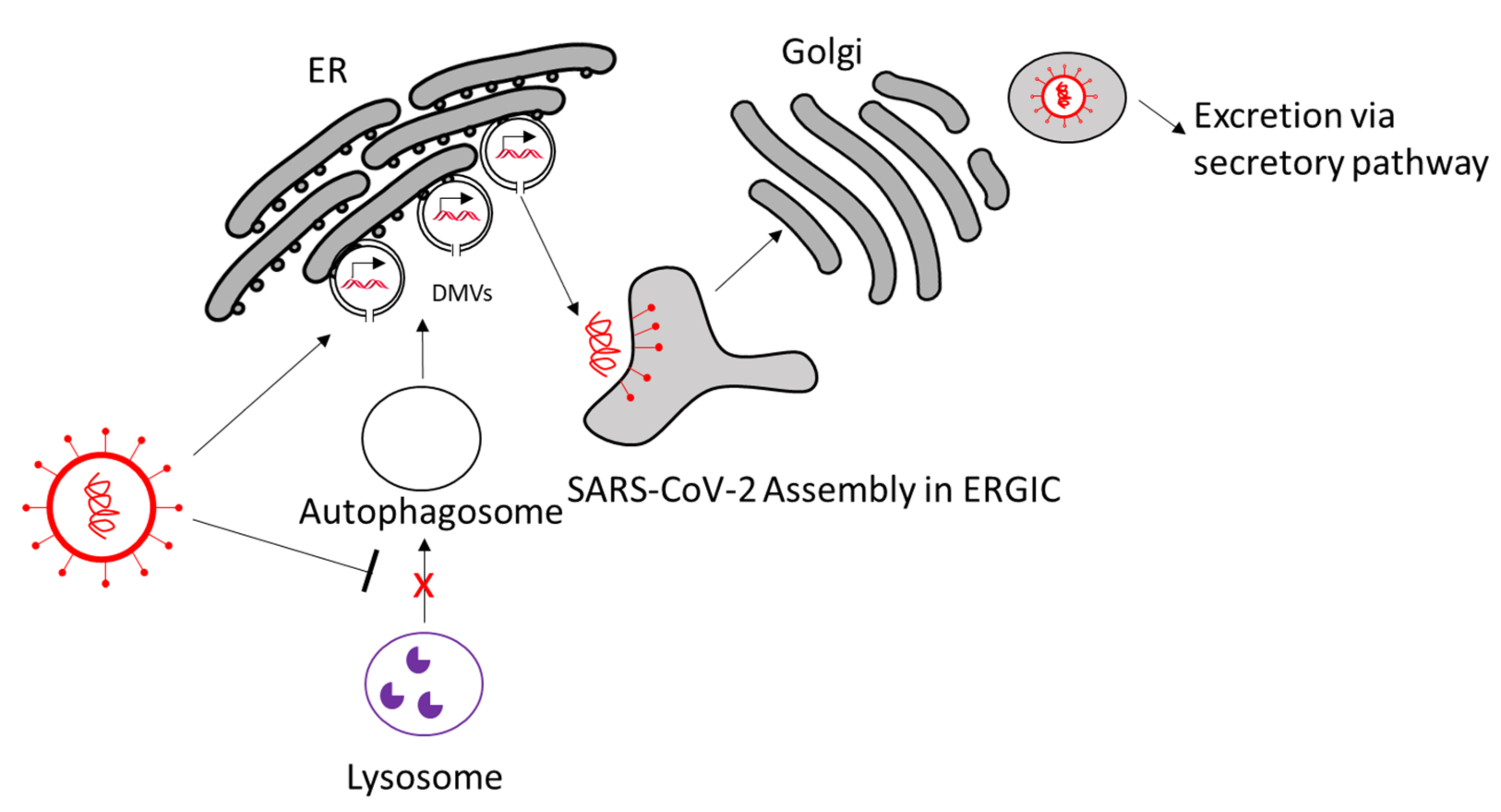{kind=link}
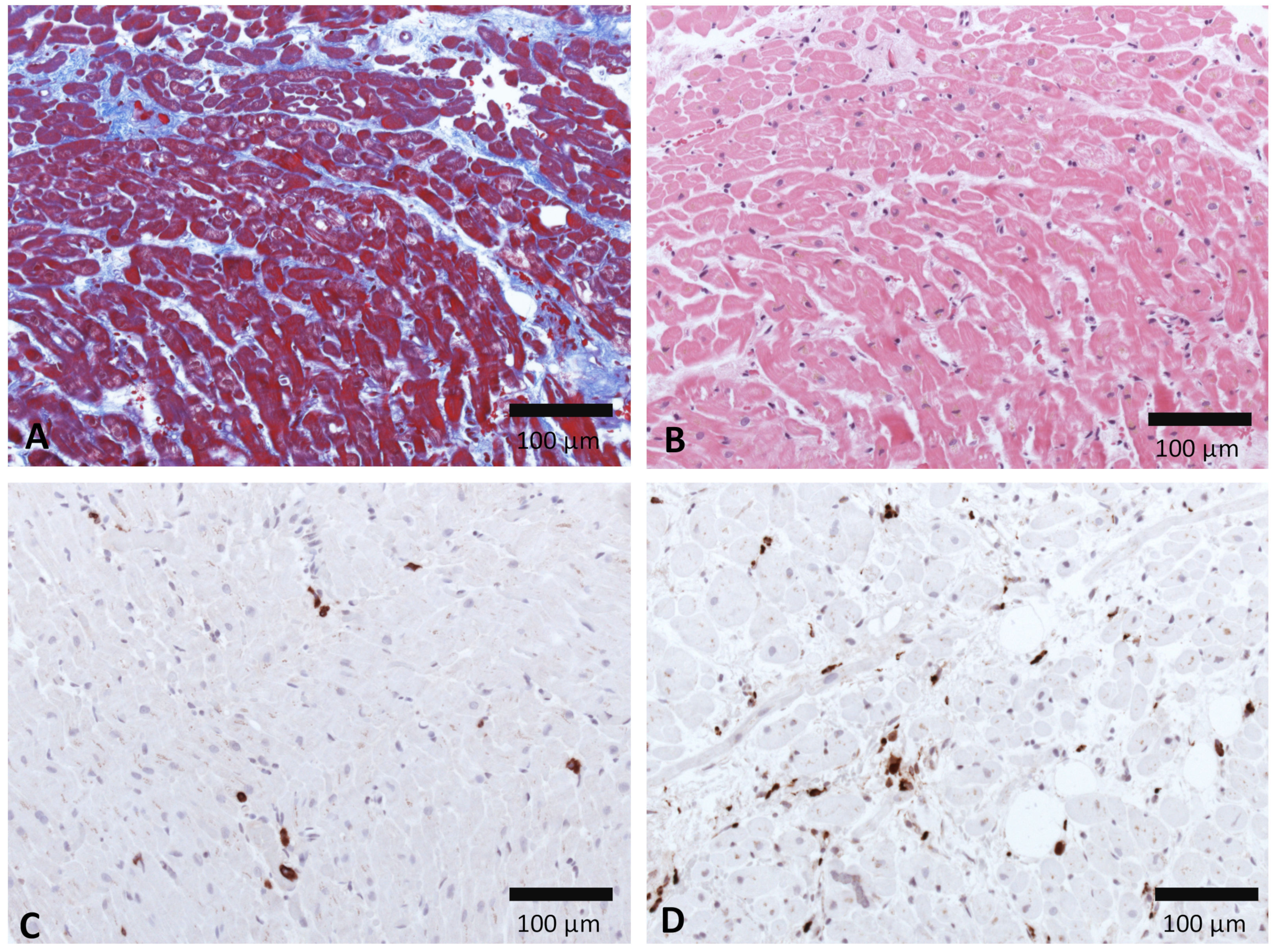{kind=link}
{kind=link}
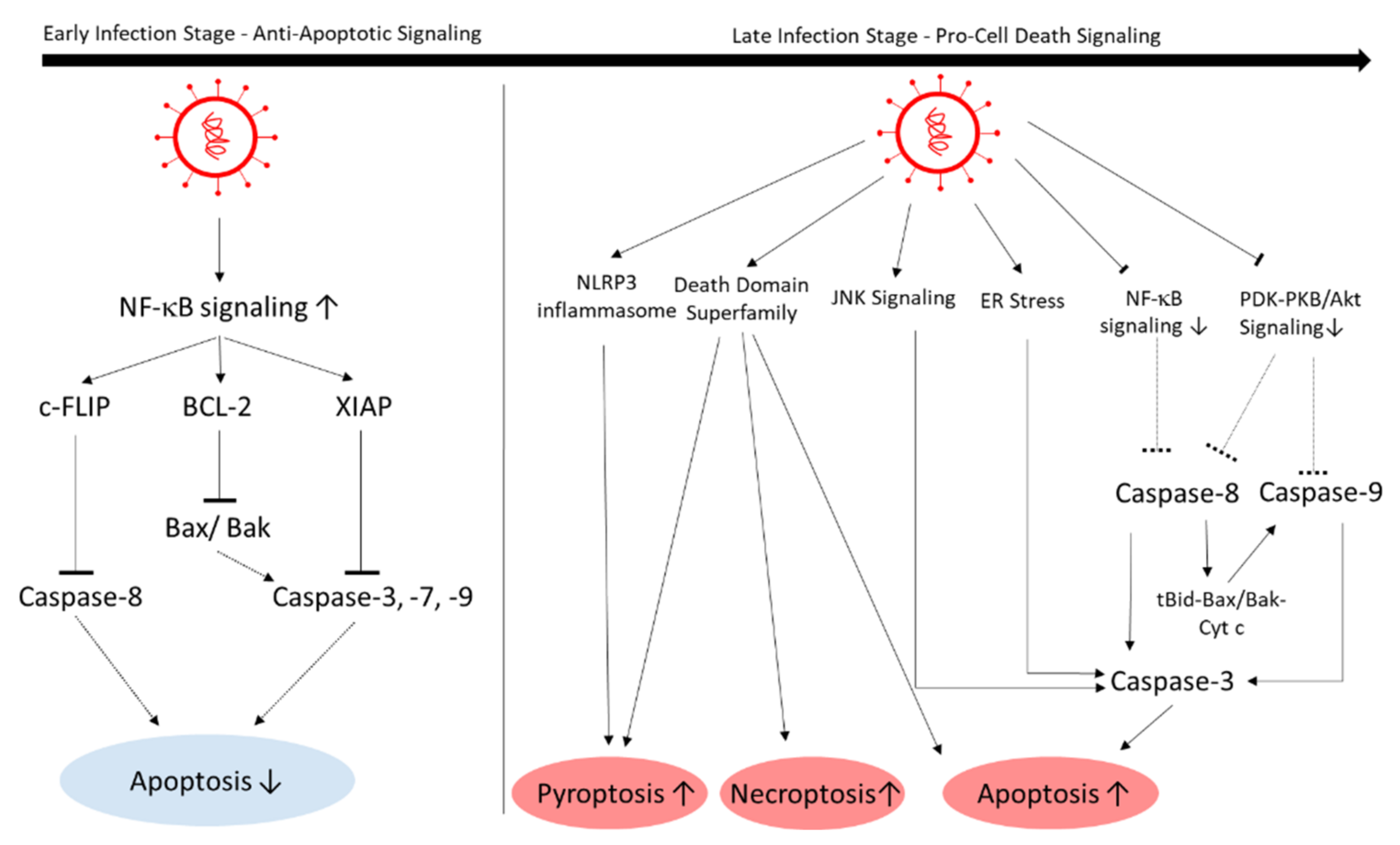{kind=link}
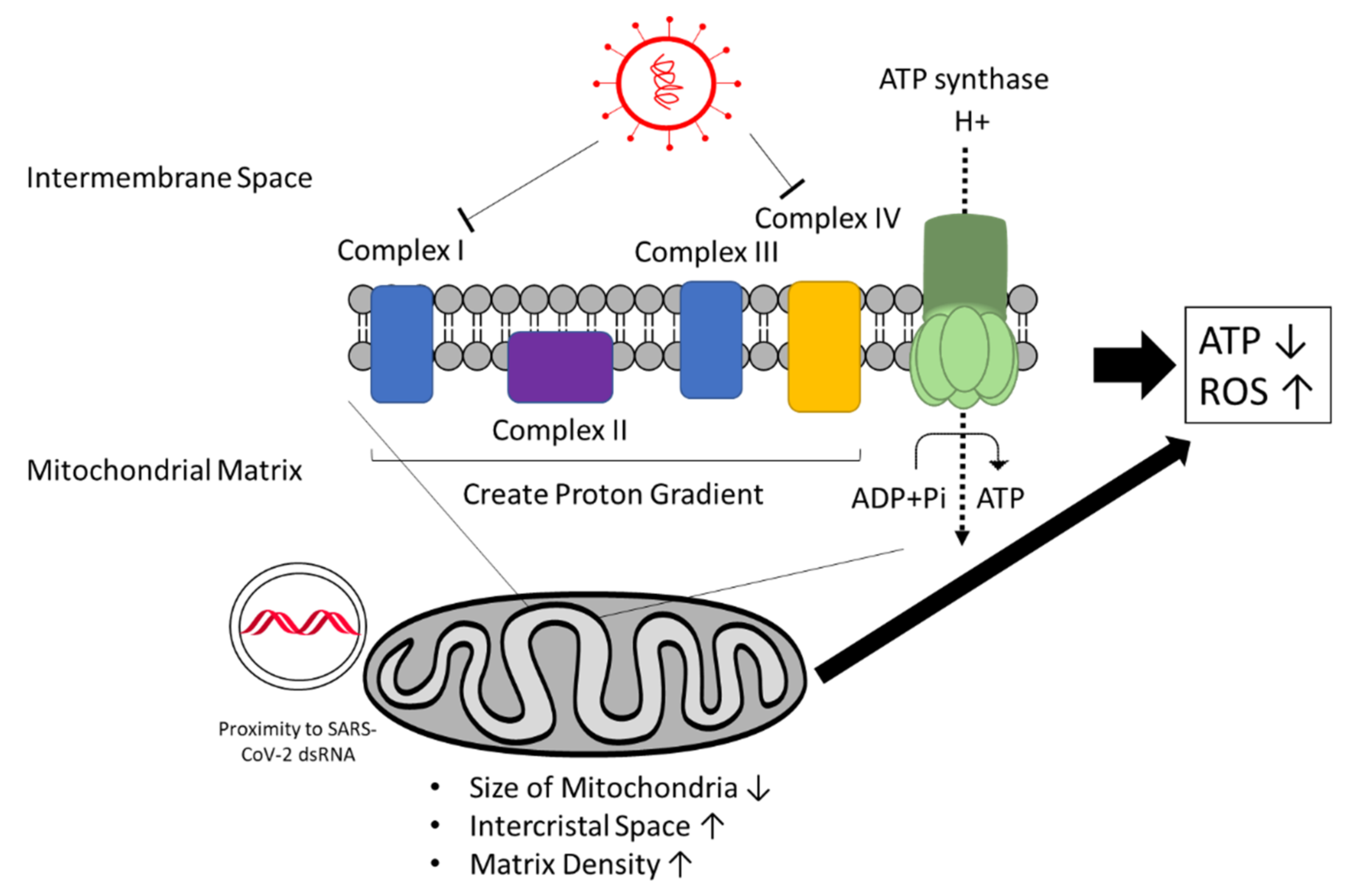{kind=link}
{kind=link}
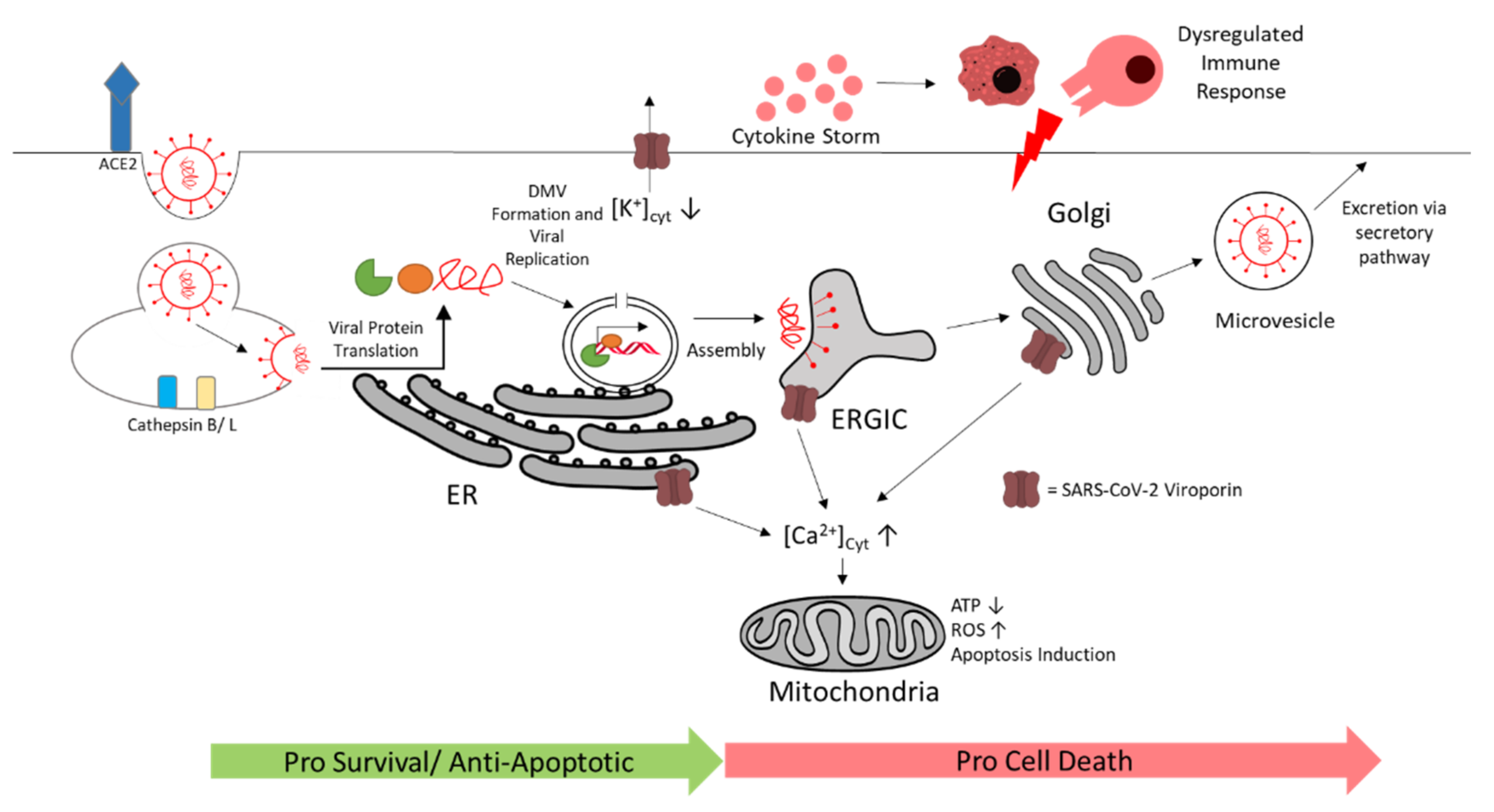{kind=link}
Table 1.
Glossary of key terms used in this review.
| Key Term | Definition |
|---|---|
| Cardiomyocytes | Heart muscle cells. Responsible for mechanical contractions. |
| Endothelial cells | Cells that form the interior wall of blood vessels. |
| Inflammation | Local response of the body against (alleged) foreign bodies such as pathogens. In this process, the immune system tries to remove the pathogens, which can be harmful when the reaction is excessive. Characteristic reactions are heat, pain, swelling due to capillary dilation, and leukocyte infiltration. |
| Myocarditis | Inflammation of the myocardium. Characteristics include flulike symptoms, left ventricle systolic dysfunction, enhanced numbers of interstitial immune cells, and an increase in creatine kinase biomarker and cardiac troponin levels in the blood. |
| Endotheliitis | Inflammation of the endothelium in the blood vessels. |
| Cytokine storm | Reaction by the innate immune system in which an excessive amount of proinflammatory signals; i.e., cytokines, are released. |
| Spike S protein | A specific protein structure of SARS-CoV-2 located on the surface of the viral envelope. It is used by the virus to dock onto the host cell. |
| Angiotensin-converting-enzyme 2 (ACE2) | An enzyme located at the surface of specific cells. It catalyzes angiotensin II to angiotensin (1–7), which leads to vasodilation. |
| Viral protease | Proteins expressed by the virus that catalyze the cleavage of specific proteins. Viral proteases are usually used to process their own precursor polyproteins and manipulate host proteins. |
| Autophagy | Cell mechanism to remove misfolded proteins, organelles, and pathogens within the cell. This process usually includes the formation of vesicles called autophagosomes as the site for digestion. |
| Double-membrane vesicle | Vesicles that appear to have two membranes made of components of intracellular organelles such as the ER, ERGIC, or Golgi. They are created and used by specific RNA viruses as a site of replication. |
| Inflammasome | Oligomers secreted by the innate immune system that activate specific inflammatory reactions. |
| Apoptosis, necrosis, and pyroptosis | Apoptosis is the process of programmed cell death. The apoptotic cell creates cell fragments that are removed by phagocytes. Necrosis describes an uncontrolled cell death in which the membrane ruptures and intracellular contents are released. Pyroptosis is also a form of programmed cell death that is triggered by inflammatory reactions. |
| Respiratory chain | Known additionally as the electron-transport chain. It is composed of 5 protein complexes in mitochondria, transports electrons and creates a proton gradient to generate ATP. |
| Viroporin | A viral protein that can selectively conduct ions similar to ion channels. They promote viral replication and manipulate biological processes in the host cell. |
Table 2.
Comparison of several virus–host interactions and viral mechanisms between SARS-CoV-2, CVB3, and PVB19 in myocarditis.
Table 2.
Comparison of several virus–host interactions and viral mechanisms between SARS-CoV-2, CVB3, and PVB19 in myocarditis.
| SARS-CoV-2 | CVB3 | PVB19 | |
|---|---|---|---|
| Viral Genome | ssRNA(+) [34] | ssRNA(+) [164] | ssDNA [165] |
| Infected cardiac cell types |
| Cardiomyocytes [166] | Cardiac endothelial cells [167] |
| Receptors/primers for viral entry | |||
| Possesses RNA-dependent RNA polymerase | Yes [68] | Yes [173] | No |
| Creates DMVs for viral replication | Yes, closed conformation with molecular pores [86] | Yes, open vaselike and closed conformation [174] | No |
| Cytokine activation in myocardium |
|
| Interferon-γ, TNF-α, IL-6, IL-8 [176] |
| Dysregulated immune-cell activation | Infiltration of macrophages and cytotoxic T cells [178] | ||
| Modulation of cell death | Activates anti- and proapoptotic signaling [115] | Activates anti- and proapoptotic signaling [113] | So far, only proapoptotic signal activation in later stages has been reported [179] |
| Energy metabolism | |||
| Viroporin | E and Nsp3a viroporins: Ca2+, K+, and Na+ conducting [101,151,154,156] | 2B viroporin: Ca2+ conducting [181] | NA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Ho, H.T.; Peischard, S.; Strutz-Seebohm, N.; Klingel, K.; Seebohm, G. Myocardial Damage by SARS-CoV-2: Emerging Mechanisms and Therapies. Viruses 2021, 13, 1880. https://0-doi-org.brum.beds.ac.uk/10.3390/v13091880
AMA Style
Ho HT, Peischard S, Strutz-Seebohm N, Klingel K, Seebohm G. Myocardial Damage by SARS-CoV-2: Emerging Mechanisms and Therapies. Viruses. 2021; 13(9):1880. https://0-doi-org.brum.beds.ac.uk/10.3390/v13091880
Chicago/Turabian StyleHo, Huyen Tran, Stefan Peischard, Nathalie Strutz-Seebohm, Karin Klingel, and Guiscard Seebohm. 2021. "Myocardial Damage by SARS-CoV-2: Emerging Mechanisms and Therapies" Viruses 13, no. 9: 1880. https://0-doi-org.brum.beds.ac.uk/10.3390/v13091880
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.