
DVGfinder: A Metasearch Tool for Identifying Defective Viral Genomes in RNA-Seq Data

 ,
,  ,
,  ,
,  ,
,  and
and 
Abstract
:1. Introduction
2. Materials and Methods
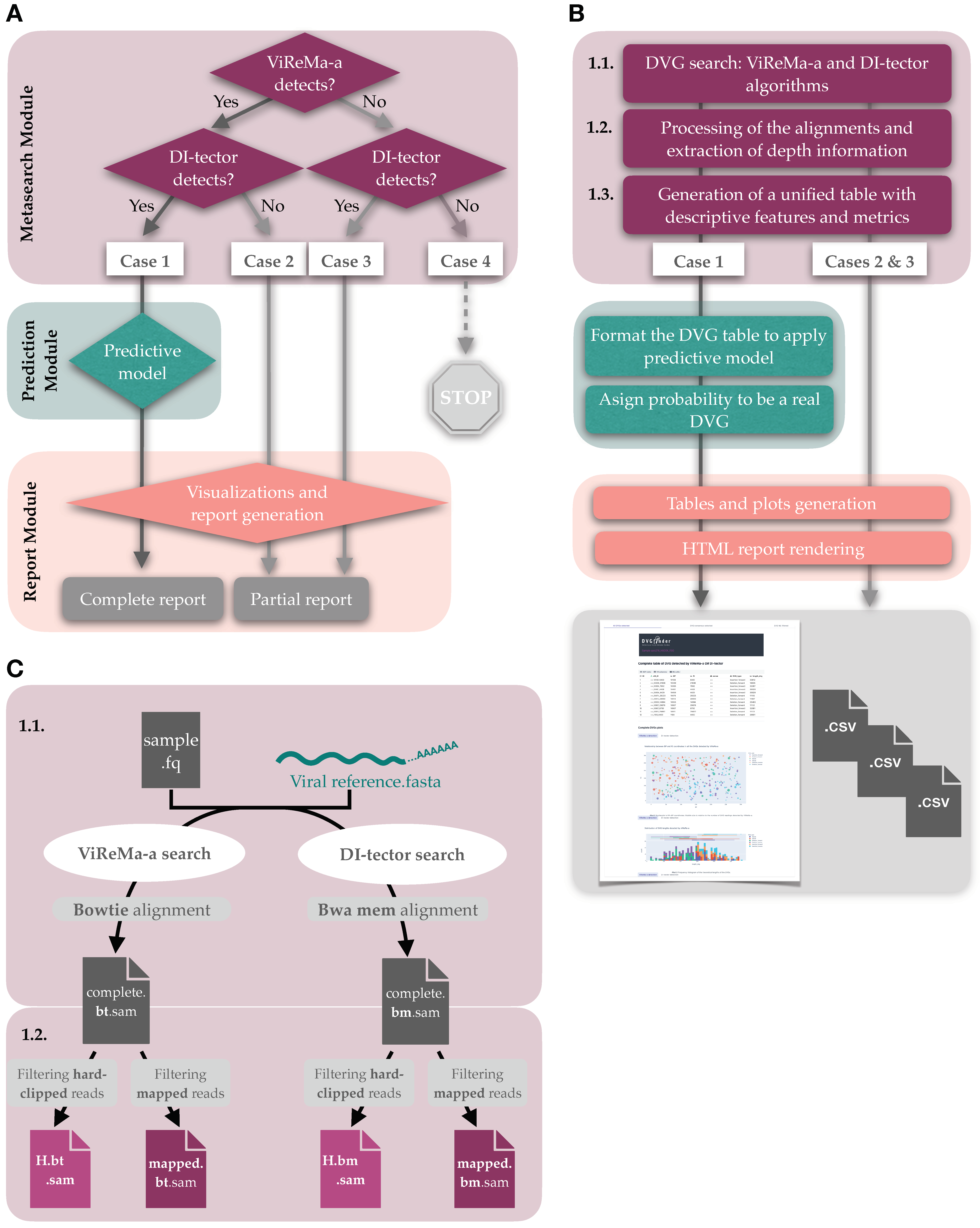
2.1. Implementation, Requirements, and Availability
2.2. Algorithm
2.3. Descriptive Features
- 1.
- RPHT, or reads per hundred thousand, relativizes the number of reads where the BP/RI junction has been found to the total number of reads that are correctly aligned with the reference sequence (mapped reads, first selection (Figure 2C, 1.2)). The use of RPHT was previously documented by Parker et al. [35] and, instead of hundred thousand, relative to million reads mapped (RPM) By Razelj et al. [2]:
- 2.
- pBP and pRI, or the proportion of reads in the BP and RI genome coordinates, normalizes the number of reads presenting the DVG with respect to the total number of mapped reads at these positions. The values can be greater than one because the search algorithms can find DVGs in reads that do not align with the reference in the first moment.where X ∈ [BP, RI].
- 3.
- SDRM stands for the semi-difference relative to the mean for positions a = depth in BP and b = depth in RI. SDRM can also be computed as the depth mean in the neighborhood of the coordinates of interests X and, in this case, a = mean pre-coordinate X depth and b = mean post-coordinate X depth:
2.4. Estimation of the Theoretical Length of DVG
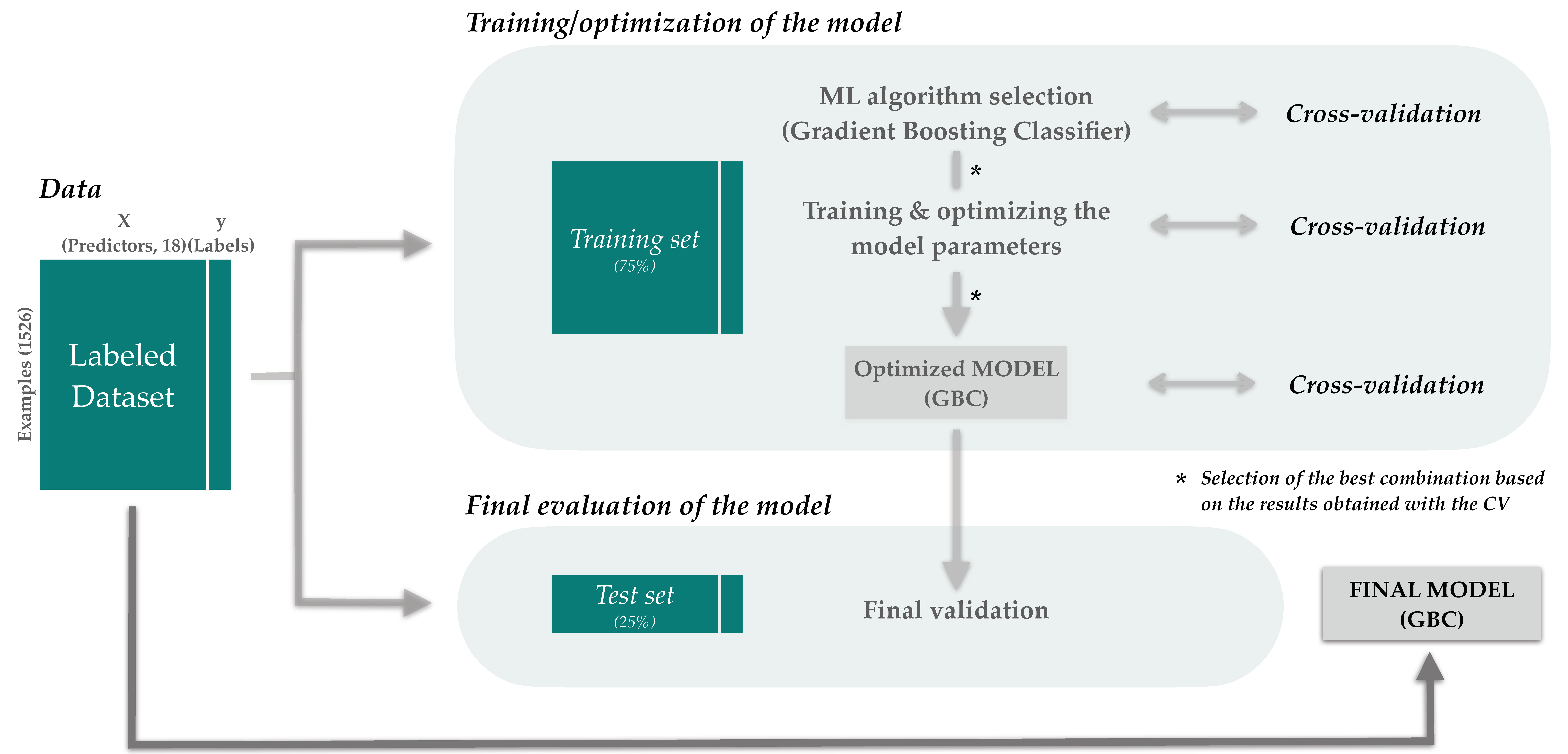
2.5. Model Generation
2.5.1. Data
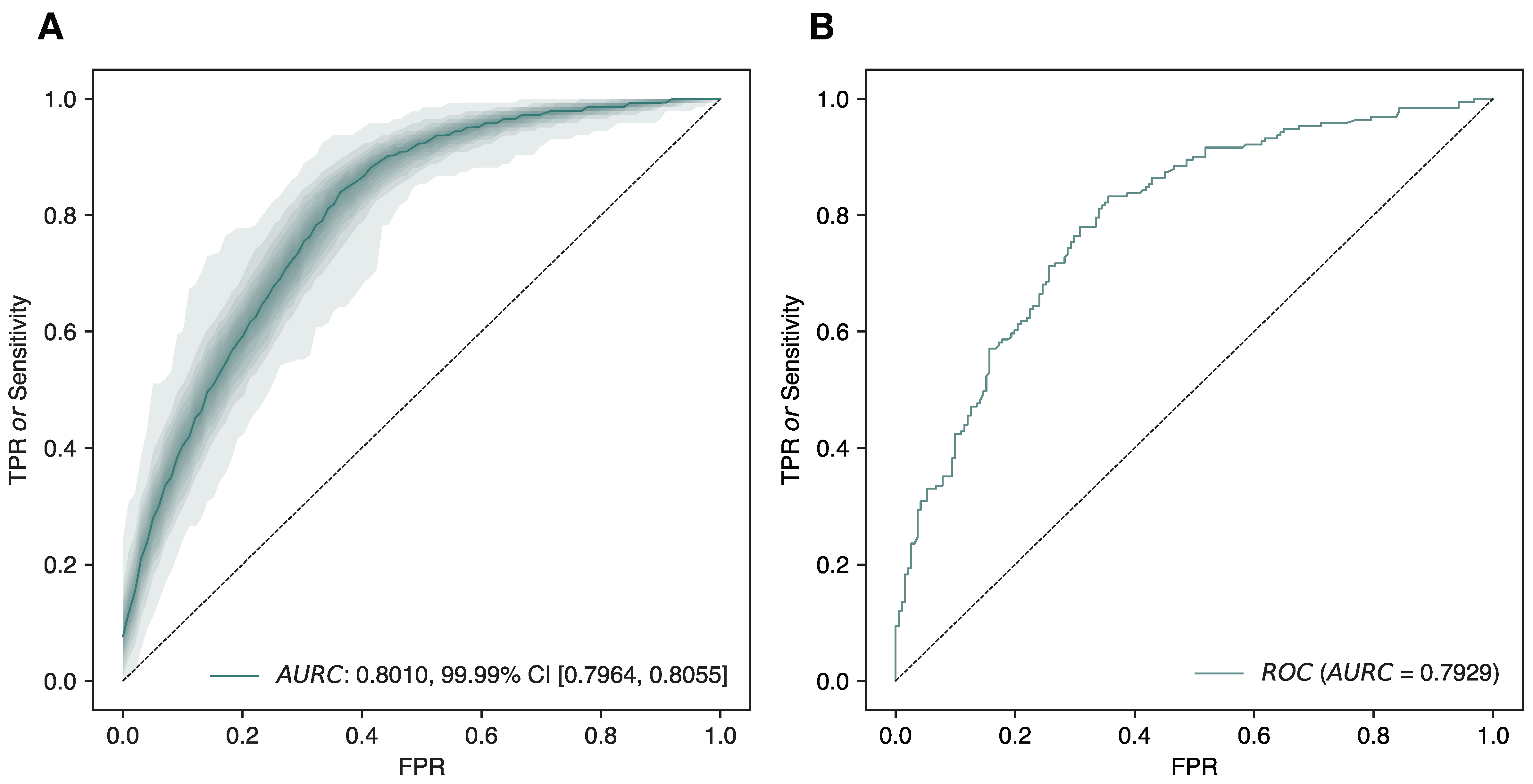
2.5.2. Training, Optimization, and Evaluation of the Model
2.6. Performance Metrics
2.7. Output of DVGfinder
2.7.1. HTML Interactive Report
2.7.2. Other Results
2.8. Recommended Pipeline
2.9. Evaluation Synthetic Datasets
2.10. Assessment of the Program Performance with Synthetic Datasets
2.11. Statistical Evaluation
2.12. Assesment of the Program Performance with Real HTS Samples
3. Results
3.1. Evaluating DVGfinder Performance
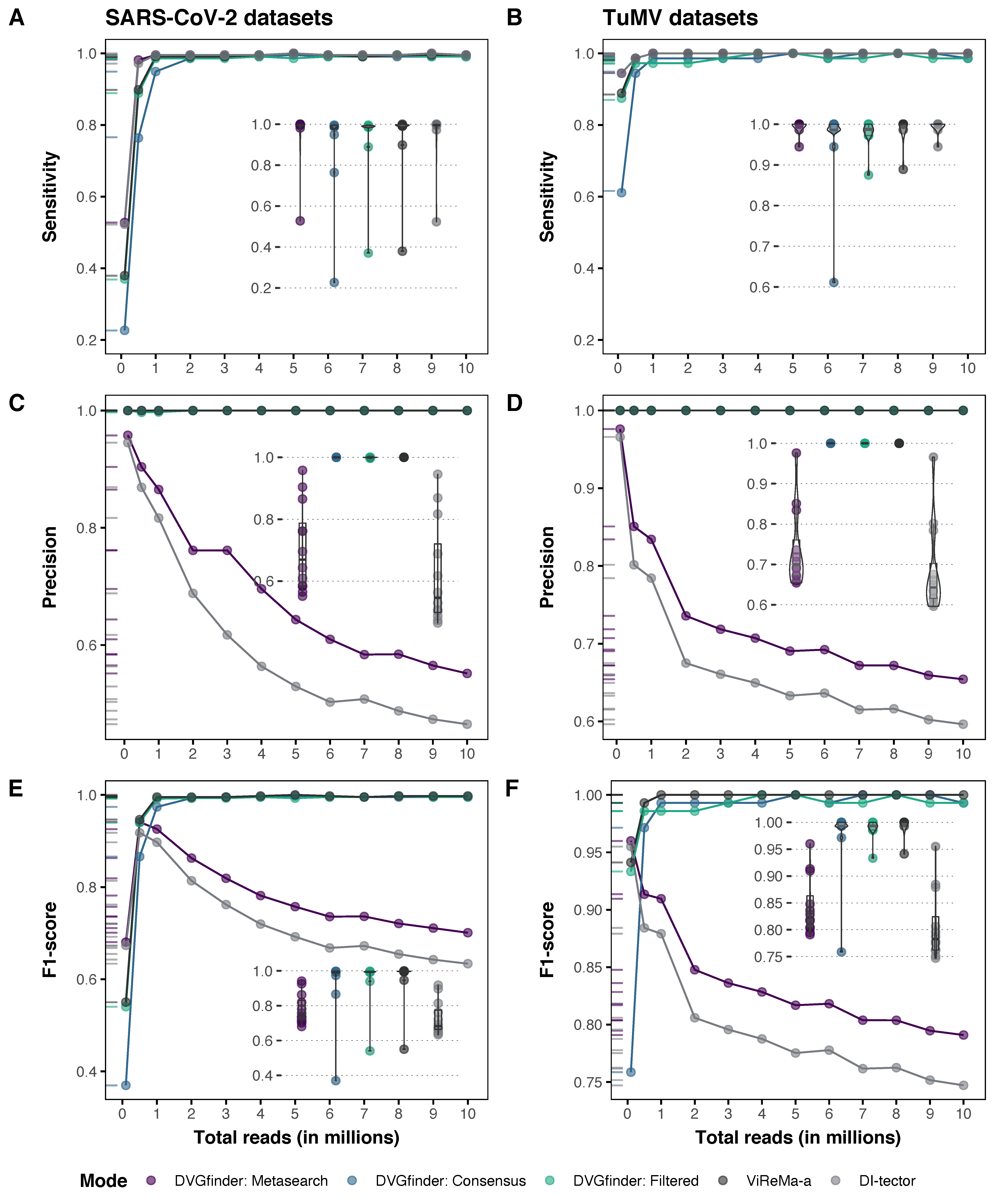
3.1.1. Sensitivity
3.1.2. Precision
3.1.3. F1 Score
3.2. DVGfinder Performance in Real Samples Containing Experimentally Validated DVGs
3.3. Final Report
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sola, I.; Almazán, F.; Zúñiga, S.; Enjuanes, L. Continuous and discontinuous RNA synthesis in coronaviruses. Annu. Rev. Virol. 2015, 2, 265–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rezelj, V.V.; Carrau, L.; Merwaiss, F.; Levi, L.I.; Erazo, D.; Tran, Q.D.; Henrion-Lacritick, A.; Gausson, V.; Suzuki, Y.; Shengjuler, D.; et al. Defective viral genomes as therapeutic interfering particles against flavivirus infection in mammalian and mosquito hosts. Nat. Commun. 2021, 12, 2290. [Google Scholar] [CrossRef] [PubMed]
- Von Magnus, P.; Gard, S. Studies on interference in experimental influenza. II. Purification and centrifugation experiments. Ark. Kem. Mineral. Geol. 1947, 8, 4. [Google Scholar]
- Ziegler, C.M.; Botten, J.W. Defective interfering particles of negative-strand RNA viruses. Trends Microbiol. 2020, 28, 554–565. [Google Scholar] [CrossRef]
- Vignuzzi, M.; López, C.B. Defective viral genomes are key drivers of the virus–host interaction. Nat. Microbiol. 2019, 4, 1075–1087. [Google Scholar] [CrossRef]
- Genoyer, E.; López, C.B. The impact of defective viruses on infection and immunity. Annu. Rev. Virol. 2019, 6, 547–566. [Google Scholar] [CrossRef]
- Shrestha, N.; Bujarski, J.J. Long noncoding RNAs in plant viroids and viruses: A review. Pathogens 2020, 9, 765. [Google Scholar] [CrossRef]
- Chaturvedi, S.; Vasen, G.; Pablo, M.; Chen, X.; Beutler, N.; Kumar, A.; Tanner, E.; Illouz, S.; Rahgoshay, D.; Burnett, J.; et al. Identification of a therapeutic interfering particle—A single-dose SARS-CoV-2 antiviral intervention with a high barrier to resistance. Cell 2021, 184, 6022–6036.e18. [Google Scholar] [CrossRef]
- Huang, A.S.; Baltimore, D. Defective viral particles and viral disease processes. Nature 1970, 226, 325–327. [Google Scholar] [CrossRef]
- Yang, Y.; Lyu, T.; Zhou, R.; He, X.; Ye, K.; Xie, Q.; Zhu, L.; Chen, T.; Shen, C.; Wu, Q.; et al. The antiviral and antitumor effects of defective interfering particles/genomes and their mechanisms. Front. Microbiol. 2019, 10, 1852. [Google Scholar] [CrossRef] [Green Version]
- Wignall-Fleming, E.B.; Vasou, A.; Young, D.; Short, J.A.L.; Hughes, D.J.; Goodbourn, S.; Randall, R.E. Innate intracellular antiviral responses restrict the amplification of defective virus genomes of parainfluenza virus 5. J. Virol. 2020, 94, e00246-20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Sun, Y.; Li, Y.; Ruthel, G.; Weiss, S.R.; Raj, A.; Beiting, D.; López, C.B. Replication defective viral genomes exploit a cellular pro-survival mechanism to establish paramyxovirus persistence. Nat. Commun. 2017, 8, 799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vodovar, N.; Goic, B.; Blanc, H.; Saleh, M.C. In silico reconstruction of viral genomes from small RNAs improves virus-derived small interfering RNA profiling. J. Virol. 2011, 85, 11016–11021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Routh, A.; Johnson, J.E. Discovery of functional genomic motifs in viruses with ViReMa-a virus recombination mapper-for analysis of next-generation sequencing data. Nucleic Acids Res. 2014, 42, e11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beauclair, G.; Mura, M.; Combredet, C.; Tangy, F.; Jouvenet, N.; Komarova, A.V. DI-tector: Defective interfering viral genomes’ detector for next-generation sequencing data. RNA 2018, 24, 1285–1296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gribble, J.; Stevens, L.J.; Agostini, M.L.; Anderson-Daniels, J.; Chappell, J.D.; Lu, X.; Pruijssers, A.J.; Routh, A.L.; Denison, M.R. The coronavirus proofreading exoribonuclease mediates extensive viral recombination. PLoS Pathog. 2021, 17, e1009226. [Google Scholar] [CrossRef] [PubMed]
- Muruato, A.; Vu, M.N.; Johnson, B.A.; Davis-Gardner, M.E.; Vanderheiden, A.; Lokugamage, K.; Schindewolf, C.; Crocquet-Valdes, P.A.; Langsjoen, R.M.; Plante, J.A.; et al. Mouse adapted SARS-CoV-2 protects animals from lethal SARS-CoV challenge. PLoS Biol. 2021, 19, e3001284. [Google Scholar] [CrossRef]
- Jaworski, E.; Langsjoen, R.M.; Mitchell, B.; Judy, B.; Newman, P.; Plante, J.A.; Plante, K.S.; Miller, A.L.; Zhou, Y.; Swetnam, D.; et al. Tiled-ClickSeq for targeted sequencing of complete coronavirus genomes with simultaneous capture of RNA recombination and minority variants. eLife 2021, 10, e68479. [Google Scholar] [CrossRef]
- Nilsson-Payant, B.E.; Blanco-Melo, D.; Uhl, S.; Escudero-Pérez, B.; Olschewski, S.; Thibault, P.; Panis, M.; Rosenthal, M.; Muñoz-Fontela, C.; Lee, B.; et al. Reduced nucleoprotein availability impairs negative-sense RNA virus replication and promotes host recognition. J. Virol. 2021, 95, e02274-20. [Google Scholar] [CrossRef]
- Smith, S.C.; Gribble, J.; Diller, J.R.; Wiebe, M.A.; Thoner, T.W., Jr.; Denison, M.R.; Ogden, K.M. Reovirus RNA recombination is sequence directed and generates internally deleted defective genome segments during passage. J. Virol. 2021, 95, e02181-20. [Google Scholar] [CrossRef]
- Langsjoen, R.M.; Muruato, A.E.; Kunkel, S.R.; Jaworski, E.; Routh, A. Differential alphavirus defective RNA diversity between intracellular and extracellular compartments is driven by subgenomic recombination events. mBio 2020, 11, e00731-20. [Google Scholar] [CrossRef] [PubMed]
- Kautz, T.F.; Jaworski, E.; Routh, A.; Forrester, N.L. A low fidelity virus shows increased recombination during the removal of an alphavirus reporter gene. Viruses 2020, 12, 660. [Google Scholar] [CrossRef] [PubMed]
- Alnaji, F.G.; Holmes, J.R.; Rendon, G.; Vera, J.C.; Fields, C.J.; Martin, B.E.; Brooke, C.B. Sequencing framework for the sensitive detection and precise mapping of defective interfering particle-associated deletions across influenza A and B viruses. J. Virol. 2019, 93, e00354-19. [Google Scholar] [CrossRef] [Green Version]
- Jaworski, E.; Routh, A. Parallel ClickSeq and Nanopore sequencing elucidates the rapid evolution of defective-interfering RNAs in Flock House virus. PLoS Pathog. 2017, 13, e1006365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, C.; Sun, X.; Taylor, A.; Jiao, C.; Xu, Y.; Cai, X.; Wang, X.; Ge, C.; Pan, G.; Wang, Q.; et al. Diversity, distribution, and evolution of tomato viruses in China uncovered by small RNA sequencing. J. Virol. 2017, 91, e00173-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bifani, A.M.; Choy, M.M.; Tan, H.C.; Ooi, E.E. Attenuated dengue viruses are genetically more diverse than their respective wild-type parents. NPJ Vaccines 2021, 6, 76. [Google Scholar] [CrossRef]
- Bosworth, A.; Rickett, N.Y.; Dong, X.; Ng, L.F.P.; García-Dorival, I.; Matthews, D.A.; Fletcher, T.; Jacobs, M.; Thomson, E.C.; Carroll, M.W.; et al. Analysis of an Ebola virus disease survivor whose host and viral markers were predictive of death indicates the effectiveness of medical countermeasures and supportive care. Genome Med. 2021, 13, 5. [Google Scholar] [CrossRef]
- Bosma, T.J.; Karagiannis, K.; Santana-Quintero, L.; Ilyushina, N.; Zagarodnyaya, T.; Petrovskaya, S.; Laassri, M.; Donnelly, R.P.; Rubin, S.; Simonyan, V.; et al. Identification and quantification of defective virus genomes in high throughput sequencing data using DVG-profiler, a novel post-sequence alignment processing algorithm. PLoS ONE 2019, 14, e0216944. [Google Scholar] [CrossRef] [Green Version]
- Johnson, R.I.; Boczkowska, B.; Alfson, K.; Weary, T.; Menzie, H.; Delgado, J.; Rodriguez, G.; Carrion Jr, R.; Griffiths, A. Identification and characterization of defective viral genomes in Ebola virus-infected rhesus macaques. J. Virol. 2021, 95, e00714-21. [Google Scholar] [CrossRef]
- Addetia, A.; Phung, Q.; Bradley, B.T.; Lin, M.J.; Zhu, H.; Xie, H.; Huang, M.L.; Greninger, A.L. In vivo generation of BK and JC polyomavirus defective viral genomes in human urine samples associated with higher viral loads. J. Virol. 2021, 95, e00250-21. [Google Scholar] [CrossRef]
- Sun, Y.; Kim, E.J.; Felt, S.A.; Taylor, L.J.; Agarwal, D.; Grant, G.R.; López, C.B. A specific sequence in the genome of respiratory syncytial virus regulates the generation of copy-back defective viral genomes. PLoS Pathog. 2019, 15, e1008099. [Google Scholar]
- Felt, S.A.; Sun, Y.; Jozwik, A.; Paras, A.; Habibi, M.X.; Nickle, D.; Anderson, L.; Achouri, E.; Feemster, K.A.; Cárdenas, A.M.; et al. Detection of respiratory syncytial virus defective genomes in nasal secretions is associated with distinct clinical outcomes. Nat. Microbiol. 2021, 6, 672–681. [Google Scholar] [CrossRef] [PubMed]
- Boussier, J.; Munier, S.; Achouri, E.; Meyer, B.; Crescenzo-Chaigne, B.; Behillil, S.; Enouf, V.; Vignuzzi, M.; van der Werf, S.; Naffakh, N. RNA-seq accuracy and reproducibility for the mapping and quantification of influenza defective viral genomes. RNA 2020, 26, 1905–1918. [Google Scholar] [CrossRef] [PubMed]
- DVGfinder Repository. Available online: https://github.com/MJmaolu/DVGfinder (accessed on 20 May 2022).
- Parker, M.D.; Lindsey, B.B.; Leary, S.; Gaudieri, S.; Chopra, A.; Wyles, M.; Angyal, A.; Green, L.R.; Parsons, P.; Tucker, R.M.; et al. Subgenomic RNA identification in SARS-CoV-2 genomic sequencing data. Genome Res. 2021, 31, 645–658. [Google Scholar] [CrossRef]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Walsh, I.; Fishman, D.; Garcia-Gasulla, D.; Titma, T.; Pollastri, G.; ELIXIR Machine Learning Focus Group; Harrow, J.; Psomopoulos, F.E.; Tosatto, S.C.E. DOME: Recommendations for supervised machine learning validation in biology. Nat. Methods 2021, 18, 1122–1127. [Google Scholar] [CrossRef]
- Labeled Dataset, DVGfinder Repository. Available online: https://github.com/MJmaolu/DVGfinder/blob/v3.1/630N5Ml100_v2_metrics_labeledDataset.csv (accessed on 20 May 2022).
- Datapane. Available online: https://github.com/datapane/datapane (accessed on 29 December 2021).
- Plotly Technologies Inc. Collaborative Data Science; Plotly Technologies Inc.: Montreal, QC, Canada, 2015. [Google Scholar]
- Wu, H.Y.; Brian, D.A. Subgenomic messenger RNA amplification in coronaviruses. Proc. Natl. Acad. Sci. USA 2010, 107, 12257–12262. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Lee, J.Y.; Yang, J.S.; Kim, J.W.; Kim, V.N.; Chang, H. The architecture of SARS-CoV-2 transcriptome. Cell 2020, 181, 914–921.e10. [Google Scholar] [CrossRef]
- Di Gioacchino, A.; Legendre, R.; Rahou, Y.; Najburg, V.; Charneau, P.; Greenbaum, B.D.; Tangy, F.; van der Werf, S.; Cocco, S.; Komarova, A.V. sgDI-tector: Defective interfering viral genome bioinformatics for detection of coronavirus subgenomic RNAs. RNA 2022, 28, 277–289. [Google Scholar] [CrossRef]
- Mura, M.; Combredet, C.; Najburg, V.; David, R.Y.S.; Tangy, F.; Komarova, A.V. Nonencapsidated 5′ copy-back defective interfering genomes produced by recombinant measles viruses are recognized by RIG-I and LGP2 but not MDA5. J. Virol. 2017, 91, 643–660. [Google Scholar] [CrossRef] [Green Version]
- Synthetic Samples Repository. Available online: https://github.com/MJmaolu/SyntheticSamplesWithDVGs (accessed on 20 May 2022).
- Synthetic Viral Samples with DVGs. Available online: https://0-doi-org.brum.beds.ac.uk/10.5281/zenodo.6411689 (accessed on 20 May 2022). [CrossRef]
- Olmo-Uceda, M.J.; Muñoz-Sánchez, J.C.; Lasso-Giraldo, W.; Arnau, V.; Díaz-Villanueva, W.; Elena, S.F. DVGfinder: A metasearch engine for identifying defective viral genomes in RNA-seq data. Preprints 2022. [Google Scholar] [CrossRef]
- Sokal, R.R.; Rohlf, F.J. Biometry, 3rd ed.; W.H. Freeman & Co: New York, NY, USA, 1995. [Google Scholar]
- Results-DVGfinder. Available online: https://github.com/MJmaolu/results-DVGfinder (accessed on 20 May 2022).
- Example Report. Available online: http://147.156.206.144/appweb/tumvas72_N100K_l100_report.html (accessed on 20 May 2022).
- Results DVGfinder on Real Samples. Available online: https://github.com/MJmaolu/results-DVGfinder (accessed on 20 May 2022).






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Program | Language | DVG Type Identified | Reference | Number of Citations | Used by |
|---|---|---|---|---|---|
| ViReMa-a | Python | D, I, cb/sb | [14] | 31 | [16,17,18,19,20,21,22,23,24,25] |
| DI-tector | Python | D, I, cb/sb | [15] | 9 | [26,27,28,29,30] |
| VODKA | Perl | cb/sb | [31] | 13 | [32] |
| DVG-profiler | C++ | D, I, cb/sb | [28] | 4 | - |
| DG-seq | R | D, I, cb/sb | [33] | 5 | - |
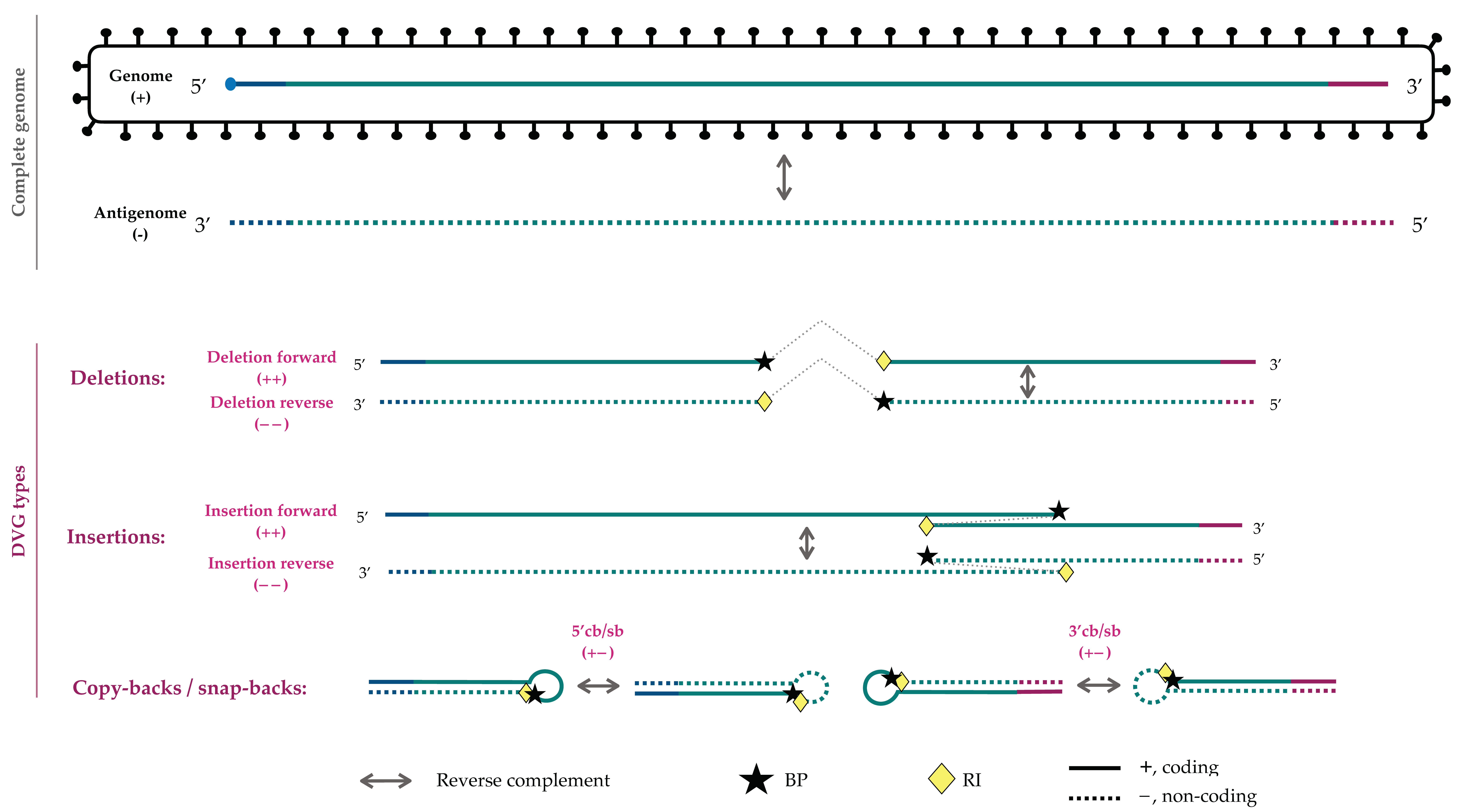
| DVG Type | Fragment | Sense | Coordinates Extracted from Reference |
|---|---|---|---|
| Deletion forward and insertion forward | pre-BP post-RI | + + | [start, BP] [RI, end] |
| Deletion reverse and insertion reverse | pre-BP post-RI | − − | [BP, end] [start, RI] |
| 5′ cb/sb | pre-BP post-RI | + − | [start, BP] [start, RI] |
| 3′ cb/sb | pre-BP post-RI | − + | [BP, end] [RI, end] |
| SARS-CoV-2 Dataset | ||
| Performance Index | Friedman Statistic | p |
| Sensitivity (TPR) | 40.6030 | 3.248 × 10−8 |
| Precision (PPV) | 46.7879 | 1.688 × 10−9 |
| F1 score | 30.6725 | 3.570 × 10−6 |
| TuMV Dataset | ||
| Performance Index | Friedman Statistic | p |
| Sensitivity (TPR) | 30.7887 | 3.381 × 10−6 |
| Precision (PPV) | 39.6123 | 5.206 × 10−8 |
| F1 score | 32.9735 | 1.210 × 10−6 |
| SARS-CoV-2 Dataset | |||
| Performance Index | Contrast | p | FDR |
| Sensitivity (TPR) | Metasearch-ViReMa-a | 0.0089 | 0.0153 |
| Sensitivity (TPR) | Metasearch-DI-tector | 0.1855 | 0.2783 |
| TuMV Dataset | |||
| Performance Index | Contrast | p | FDR |
| Sensitivity (TPR) | Metasearch-ViReMa-a | 0.5000 | 0.8571 |
| Sensitivity (TPR) | Metasearch-DI-tector | 1.0000 | 1.0000 |
| SARS-CoV-2 Dataset | |||
| Performance Index | Contrast | p | FDR |
| Precision (PPV) | Consensus-ViReMa-a | 1.0000 | 1.0000 |
| Precision (PPV) | Consensus-DI-tector | 0.0013 | 0.0038 |
| Precision (PPV) | Filtered-ViReMa-a | 0.9632 | 1.0000 |
| Precision (PPV) | Filtered-DI-tector | 0.0013 | 0.0038 |
| Precision (PPV) | Metasearch–DI-tector | 0.0013 | 0.0038 |
| TuMV Dataset | |||
| Performance Index | Contrast | p | FDR |
| Precision (PPV) | Consensus-ViReMa-a | 1.0000 | 1.0000 |
| Precision (PPV) | Consensus-DI-tector | 0.0013 | 0.0038 |
| Precision (PPV) | Filtered-ViReMa-a | 1.0000 | 1.0000 |
| Precision (PPV) | Filtered-DI-tector | 0.0013 | 0.0038 |
| Precision (PPV) | Metasearch–DI-tector | 0.0013 | 0.0038 |
| SARS-CoV-2 Dataset | |||
| Performance Index | Contrast | p | FDR |
| F1 score | Consensus-ViReMa-a | 0.9981 | 1.0000 |
| F1 score | Consensus-DI-tector | 0.0067 | 0.0135 |
| F1 score | Filtered-ViReMa-a | 0.9980 | 1.0000 |
| F1 score | Filtered-DI-tector | 0.0027 | 0.0064 |
| F1 score | Metasearch-DI-tector | 0.0013 | 0.0038 |
| TuMV dataset | |||
| Performance Index | Contrast | p | FDR |
| F1 score | Consensus-ViReMa-a | 0.9966 | 1.0000 |
| F1 score | Consensus-DI-tector | 0.0034 | 0.0068 |
| F1 score | Filtered-ViReMa-a | 0.9973 | 1.0000 |
| F1 score | Filtered-DI-tector | 0.0016 | 0.0039 |
| F1 score | Metasearch-DI-tector | 0.0013 | 0.0038 |
| Sample | BP | RI | Predicted Size | ViReMa-a | DI-Tector | DVGfinder Metasearch |
|---|---|---|---|---|---|---|
| 1 | 13,811–13,812 | 14,697–14,696 | 2262 | Found | Found | Found |
| 13,580 | 14,964 | 2262 | Found | Found * | Found | |
| 13,865 | 14,964 | 1941 | Found * | Found * | Found * | |
| 14,575 | 14,934 | 1261 | Not found | Not found | Not found | |
| 14,770 | 15,004 | 997 | Found | Found | Found | |
| 14,874 | 15,104 | 792 | Found | Found | Found | |
| 2 | 13,308 | 14,864 | 2598 | Found | Found | Found |
| 13,566–13,568 | 14,678–14,676 | 2526 | Found * | Found | Found | |
| 13,617 | 14,633 | 2490 | Not found | Not found | Not found | |
| 13,316 | 15,144 | 2310 | Not found | Found | Found | |
| 13,339 | 15,131 | 2300 | Found * | Found * | Found * | |
| 13,347 | 15,239 | 2184 | Not found | Found * | Found * | |
| 13,480 | 15,064 | 1905 | Found | Found | Found | |
| 13,907–13,909 | 15,279–15,277 | 1584 | Found | Found | Found | |
| 14,456 | 14,885 | 1429 | Not found | Found | Found | |
| 14,223–14,224 | 15,166–15,165 | 1381 | Found | Found | Found | |
| 14,342 | 15,108 | 1320 | Found | Found | Found | |
| 14,596 | 14,896 | 1278 | Not found | Not found | Not found | |
| 14,761 | 14,917 | 1092 | Found | Found | Found | |
| 14,730–14,733 | 15,026–15,023 | 1014 | Found | Found | Found | |
| 14,868 | 15,032 | 870 | Found | Found | Found | |
| 14,856–14,863 | 15,043–15,036 | 870 | Found * | Found | Found | |
| 14,861–14,862 | 15,178–15,177 | 731 | Found * | Found * | Found * | |
| 14,947–14,950 | 15,145–15,142 | 678 | Found | Found | Found | |
| 14,864 | 15,198 | 708 | Found | Found | Found | |
| 14,934 | 15,272 | 564 | Found | Found | Found | |
| 3 | 14,706 | 14,930 | 1134 | Found | Found | Found |
| 12,661 | 15,231 | 2878 | Not found | Not found | Not found | |
| 21/28 | 24/28 | 24/28 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Olmo-Uceda, M.J.; Muñoz-Sánchez, J.C.; Lasso-Giraldo, W.; Arnau, V.; Díaz-Villanueva, W.; Elena, S.F. DVGfinder: A Metasearch Tool for Identifying Defective Viral Genomes in RNA-Seq Data. Viruses 2022, 14, 1114. https://0-doi-org.brum.beds.ac.uk/10.3390/v14051114
Olmo-Uceda MJ, Muñoz-Sánchez JC, Lasso-Giraldo W, Arnau V, Díaz-Villanueva W, Elena SF. DVGfinder: A Metasearch Tool for Identifying Defective Viral Genomes in RNA-Seq Data. Viruses. 2022; 14(5):1114. https://0-doi-org.brum.beds.ac.uk/10.3390/v14051114
Chicago/Turabian StyleOlmo-Uceda, Maria J., Juan C. Muñoz-Sánchez, Wilberth Lasso-Giraldo, Vicente Arnau, Wladimiro Díaz-Villanueva, and Santiago F. Elena. 2022. "DVGfinder: A Metasearch Tool for Identifying Defective Viral Genomes in RNA-Seq Data" Viruses 14, no. 5: 1114. https://0-doi-org.brum.beds.ac.uk/10.3390/v14051114