
Genomic Mining Reveals Deep Evolutionary Relationships between Bornaviruses and Bats

Abstract
:1. Introduction
2. Materials and Methods
2.1. Genomic Mining
2.2. Phylogenetic Analysis

{kind=link}
{kind=link}
{kind=link}
| Bat Species | Suborder * | Abbreviation | Accession | Contig Location | E-Value Identity Coverage | Indels ** |
|---|---|---|---|---|---|---|
| Rhinolophus ferrumquinum | Yin | EBLN | ||||
| Rhf.N1 | AWHA01050524.1 | 4660–5799 | 2e-36, 77%, 50% | 2, 8 | ||
| Megaderma lyra | Yin | EBLN | ||||
| Mel.N1 | AWHB01421187.1 | 777–1623 | 4e-30, 78%, 43% | 3, 5 | ||
| Mel.N2 | AWHB01452047.1 | 298–714 | 2e-07, 40%, 29% | 0, 0 | ||
| Eidolon helvum | Yin | EBLN | ||||
| Eih.N1 | AWHC01264218.1 | 8841–9155 | 1e-04, 31%, 28% | 0, 2 | ||
| Pteronotus parnellii | Yang | EBLN | ||||
| Ptp.N1 | AWGZ01165285.1 | 928–1848 | 2e-21, 82%, 28% | 0, 10 | ||
| Ptp.N2 | AWGZ01398077.1 | 1–365 | 9e-19, 33%, 54% | 1, 3 | ||
| Ptp.N3 | AWGZ01350440.1 | 1486–1920 | 1e-13, 39%, 34% | 0, 1 | ||
| EBLM | ||||||
| Ptp.M1 | AWGZ01183839.1 | 1658–1996 | 2e-13, 76%, 35% | 0, 2 | ||
| EBLL | ||||||
| Ptp.L1 | AWGZ01393507.1 | 6559–10,179 | 8e-79, 56%, 36% | 6, 14 | ||
| Ptp.L2 | AWGZ01242856.1 | 1307–4498 | 8e-32, 43%, 41% | 11, 10 | ||
| Myotis brandtii | Yang | EBLN | ||||
| Myb.N1 | ANKR01245074.1 | 1445–1948 | 5e-14, 44%, 30% | 0, 0 | ||
| Myb.N2 | ANKR01266949.1 | 310–813 | 6e-14, 44%, 29% | 0, 1 | ||
| Myb.N3 | ANKR01225293.1 | 9897–10,340 | 3e-11, 38%, 30% | 0, 0 | ||
| Myb.N4 | ANKR01212309.1 | 7949–9532 | 2e-09, 28%, 33% | 1, 0 | ||
| Myb.N5 | ANKR01159012.1 | 25,939–26,232 | 9e-09, 26%, 39% | 0, 0 | ||
| EBLL | ||||||
| Myb.L1 | ANKR01212491.1 | 41,559–43,796 | 0.0, 43%, 44% | 0, 3 | ||
| Myb.L2 *** | ANKR01204699.1 | 20,384–40,610 | 2e-94, 71%, 38% | 21, 45 | ||
| Myb.L3 | ANKR01204701.1 | 25–1592 | 2e-56, 28%, 32% | 5, 10 | ||
| Myb.L4 | ANKR01225293.1 | 11,214–13,539 | 1e-42, 28%, 43% | 3, 1 | ||
| Myb.L5 | ANKR01212492.1 | 1625–3124 | 5e-26, 21%, 33% | 3, 0 | ||
| Myotis davidii | Yang | EBLN | ||||
| Myd.N1 | ALWT01306233.1 | 118–612 | 3e-15, 43%, 33% | 0, 0 | ||
| Myd.N2 | ALWT01173634.1 | 13,634–13,882 | 9e-10, 22%, 40% | 0, 0 | ||
| Myd.N3 | ALWT01316296.1 | 13,281–13,532 | 2e-08, 22%, 42% | 0, 0 | ||
| Myd.N4 | ALWT01050150.1 | 238–657 | 2e-07, 36%, 28% | 0, 0 | ||
| Myd.N5 | ALWT01072958.1 | 7199–7483 | 2e-07, 29%, 33% | 0, 1 | ||
| EBLL | ||||||
| Myd.L1 | ALWT01131278.1 | 3393–9913 | 4e-70, 64%, 36% | 10, 12 | ||
| Myd.L2 | ALWT01213390.1 | 1747–5042 | 2e-45, 26%, 39% | 4, 2 | ||
| Myd.L3 | ALWT01141698.1 | 1537–3741 | 2e-42, 21%, 40% | 3, 0 | ||
| Myd.L4 | ALWT01026930.1 | 16,010–18,092 | 5e-31, 27%, 32% | 4, 4 | ||
| Myd.L5 | ALWT01098736.1 | 1601–3530 | 1e-26, 21%, 36% | 4, 5 | ||
| Myd.L6 | ALWT01174464.1 | 1245–2467 | 6e-26, 21%, 38% | 3, 2 | ||
| Eptesicus fuscus | Yang | EBLN | ||||
| Epf.N1 | ALEH01023837.1 | 24,020–31,033 | 1e-12, 44%, 36% | 3, 2 | ||
| Epf.N2 | ALEH01041783.1 | 76,615–77,178 | 2e-12, 49%, 26% | 0, 0 | ||
| Epf.N3 | ALEH01151776.1 | 9973–10,473 | 5e-12, 40%, 31% | 0, 0 | ||
| Epf.N4 | ALEH01014408.1 | 3710–4336 | 1e-11, 55%, 30% | 0, 1 | ||
| Epf.N5 | ALEH01011989.1 | 69,678–69,995 | 2e-11, 28%, 35% | 0, 0 | ||
| Epf.N6 | ALEH01076397.1 | 50,180–50,776 | 1e-09, 53%, 27% | 0, 0 | ||
| Epf.N7 | ALEH01137033.1 | 9537–9971 | 1e-09, 38%, 32% | 0, 1 | ||
| Epf.N8 | ALEH01007189.1 | 306–707 | 6e-09, 34%, 29% | 0, 0 | ||
| Epf.N9 | ALEH01110526.1 | 1324–1632 | 1e-08, 27%, 34% | 0, 1 | ||
| Epf.N10 | ALEH01074910.1 | 824–1277 | 4e-08, 38%, 31% | 1, 1 | ||
| Epf.N11 | ALEH01010737.1 | 10,343–10,882 | 1e-06, 45%, 24% | 0, 1 | ||
| Epf.N12 | ALEH01037465.1 | 14,375–14,874 | 2e-06, 42%, 26% | 1, 0 | ||
| Epf.N13 | ALEH01154995.1 | 4103–4420 | 2e-05, 28%, 26% | 0, 0 | ||
| Epf.N14 | ALEH01155661.1 | 6639–6935 | 4e-05, 26%, 28% | 0, 0 | ||
| EBLG | ||||||
| Epf.G1 | ALEH01011989.1 | 67,661–68,359 | 2e-09, 47%, 23% | 0, 1 | ||
| EBLL | ||||||
| Epf.L1 | ALEH01013293.1 | 16,047–20,804 | 0.0, 91%, 37% | 0, 0 | ||
| Epf.L2 | ALEH01059268.1 | 10,200–12,479 | 2e-48,23%,51% | 4, 4 | ||
| Myotis lucifugus | Yang | EBLN | ||||
| Myl.N1 | AAPE02027471.1 | 113,136–113,495 | 1e-14, 31%, 38% | 2, 0 | ||
| Myl.N2 | AAPE02012651.1 | 118,026–118,529 | 6e-13, 44%, 29% | 0, 0 | ||
| Myl.N3 | AAPE02006259.1 | 24,888–25,331 | 5e-11, 38%, 29% | 0, 0 | ||
| Myl.N4 | AAPE02054433.1 | 11,820–13,638 | 2e-10, 39%, 32% | 2, 0 | ||
| Myl.N5 | AAPE02007546.1 | 82,644–82,937 | 1e-08, 26%, 38% | 0, 0 | ||
| EBLL | ||||||
| Myl.L1 | AAPE02025596.1 | 570–7767 | 0.0, 64%, 45% | 4, 5 | ||
| Myl.L2 *** | AAPE02049592.1 | 28,943–32,193 | 1e-95, 59%, 49% | 16, 30 | ||
| Myl.L3 | AAPE02020529.1 | 2038–3686 | 2e-27, 21%, 31% | 3, 0 |
2.3. Molecular Dating
3. Results and Discussion
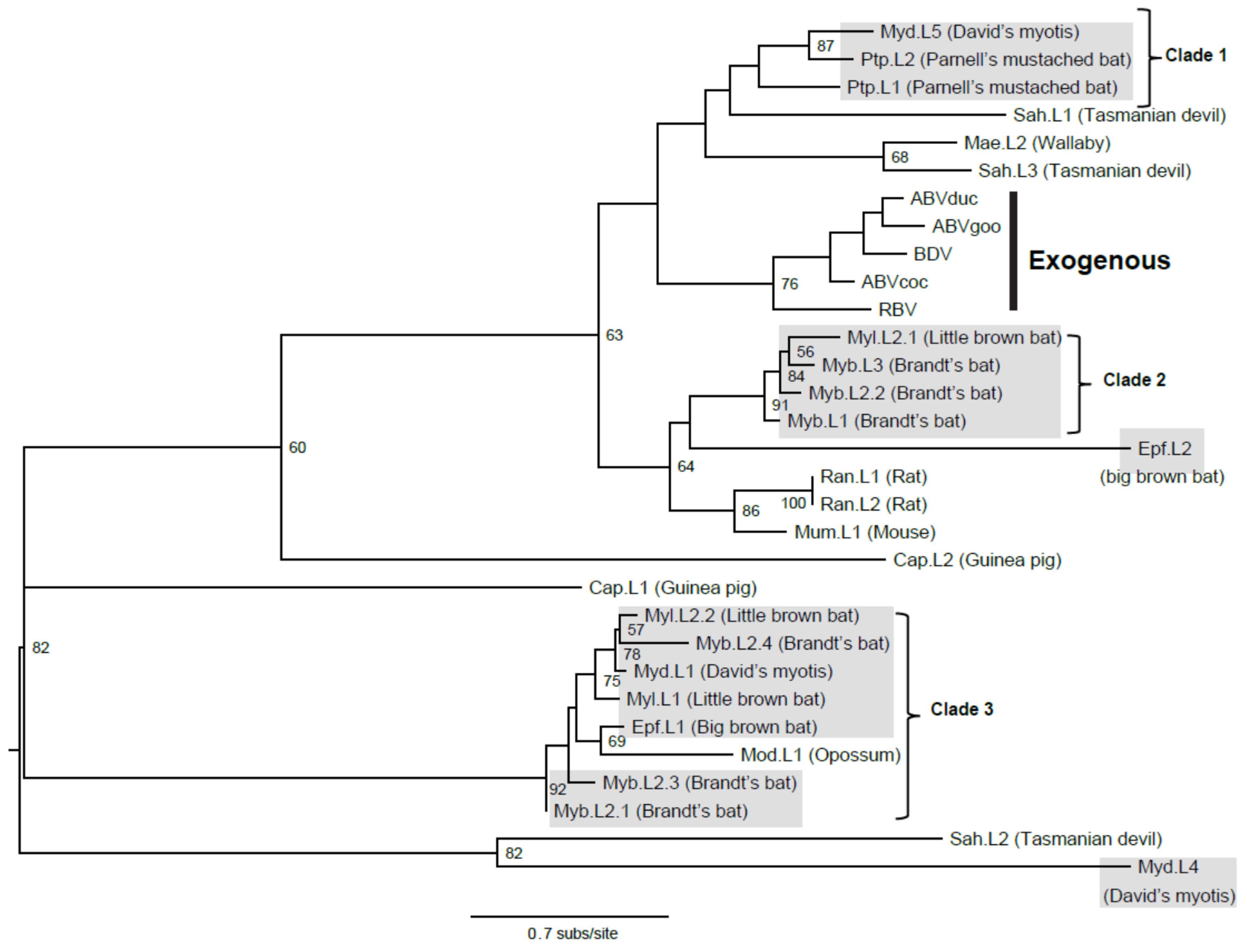
3.1. Bat Endogenous Bornaviruses
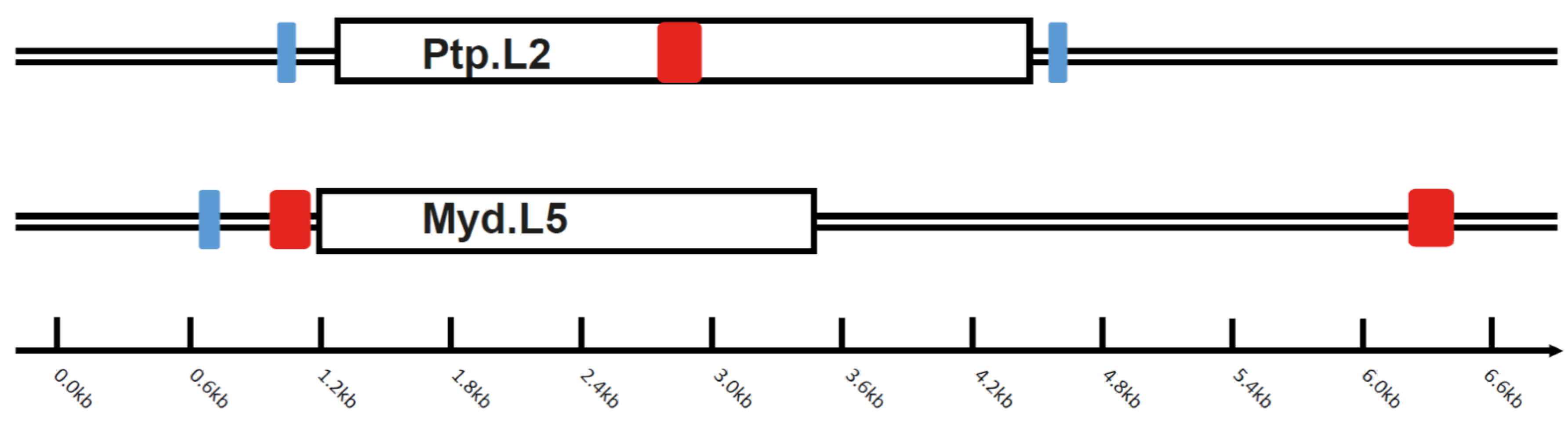
3.2. Intact Bat EBLL

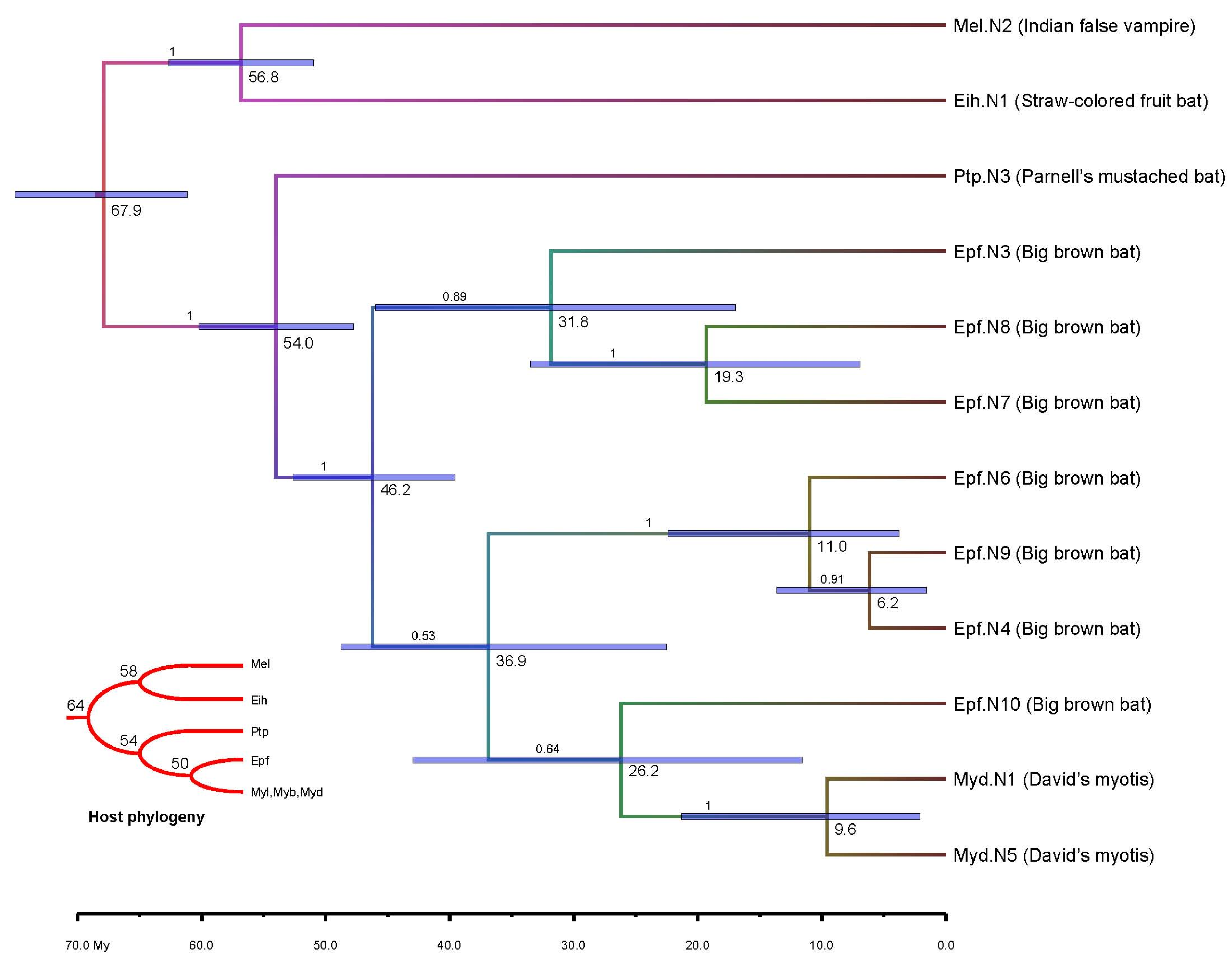
3.3. Viral Transmission
3.4. Molecular Dating


4. Conclusions
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Briese, T.; Schneemann, A.; Lewis, A.J.; Park, Y.S.; Kim, S.; Lipkin, W.I. Genomic organization of Borna disease virus. Proc. Natl. Acad. Sci. USA 1994, 91, 4362–4366. [Google Scholar] [CrossRef] [PubMed]
- Richt, J.A.; Pfeuffer, I.; Christ, M.; Frese, K.; Bechter, K.; Herzog, S. Borna disease virus infection in animals and humans. Emerg. Infect. Dis. 1997, 3, 343–352. [Google Scholar] [CrossRef] [PubMed]
- Staeheli, P.; Sauder, C.; Hausmann, J.; Ehrensperger, F.; Schwemmle, M. Epidemiology of Borna disease virus. J. Gen. Virol. 2000, 81, 2123–2135. [Google Scholar] [CrossRef] [PubMed]
- Kistler, A.L.; Gancz, A.; Clubb, S.; Skewes-Cox, P.; Fischer, K.; Sorber, K.; Chiu, C.Y.; Lublin, A.; Mechani, S.; Farnoushi, Y. Recovery of divergent avian bornaviruses from cases of proventricular dilatation disease, identification of a candidate etiologic agent. Virol. J. 2008, 5, e88. [Google Scholar] [CrossRef] [PubMed]
- Stenglein, M.D.; Leavitt, E.B.; Abramovitch, M.A.; McGuire, J.A.; DeRisi, J.L. Genome sequence of a bornavirus recovered from an African garter snake (Elapsoidea loveridgei). Genome Announc. 2014, 2, e00779-14. [Google Scholar] [CrossRef] [PubMed]
- Feschotte, C.; Gilbert, C. Endogenous viruses: Insights into viral evolution and impact on host biology. Nat. Rev. Genet. 2012, 13, 283–296. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Zhao, W.; Huang, Z.; Jarvis, E.D.; Gilbert, M.; Walker, P.J.; Holmes, E.C.; Zhang, G. Low frequency of paleoviral infiltration across the avian phylogeny. Genome Biol. 2014, 15, e539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horie, M.; Honda, T.; Suzuki, Y.; Kobayashi, Y.; Daito, T.; Oshida, T.; Ikuta, K.; Jern, P.; Gojobori, T.; Coffin, J.M.; et al. Endogenous non-retroviral RNA virus elements in mammalian genomes. Nature 2010, 463, 84–87. [Google Scholar] [CrossRef] [PubMed]
- Belyi, V.A.; Levine, A.J.; Skalka, A.M. Unexpected inheritance: Multiple integrations of ancient bornavirus and ebolavirus/marburgvirus sequences in vertebrate genomes. PLoS Pathog. 2010, 6, e1001030. [Google Scholar] [CrossRef] [PubMed]
- Katzourakis, A.; Gifford, R.J. Endogenous viral elements in animal genomes. PLoS Genet. 2010, 6, e1001191. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, Y.; Horie, M.; Tomonaga, K.; Suzuki, Y. No evidence for natural selection on endogenous borna-like nucleoprotein elements after the divergence of Old World and New World monkeys. PLoS ONE 2011, 6, e24403. [Google Scholar] [CrossRef] [PubMed]
- Calisher, C.H.; Childs, J.E.; Field, H.E.; Holmes, K.V.; Schountz, T. Bats: Important reservoir hosts of emerging viruses. Clin. Microbiol. Rev. 2006, 19, 531–545. [Google Scholar] [CrossRef] [PubMed]
- Taylor, D.J.; Dittmar, K.; Ballinger, M.J.; Bruenn, J.A. Evolutionary maintenance of filovirus-like genes in bat genomes. BMC Evol. Biol. 2011, 11, e336. [Google Scholar] [CrossRef] [PubMed]
- Dacheux, L.; Cervantes-Gonzalez, M.; Guigon, G.; Thiberge, J.M.; Vandenbogaert, M.; Maufrais, C.; Caro, V.; Bourhy, H. A preliminary study of viral metagenomics of French bat species in contact with humans: Identification of new mammalian viruses. PLoS ONE 2014, 9, e87194. [Google Scholar] [CrossRef] [PubMed]
- Horie, M.; Kobayashi, Y.; Suzuki, Y.; Tomonaga, K. Comprehensive analysis of endogenous bornavirus-like elements in eukaryote genomes. Phil. Trans. R. Soc. B 2013, 368, e20120499. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Cowled, C.; Shi, Z.; Huang, Z.; Bishop-Lilly, K.A.; Fang, X.; Wynne, J.W.; Xiong, Z.; Baker, M.L.; Zhao, W.; et al. Comparative analysis of bat genomes provides insight into the evolution of flight and immunity. Science 2013, 339, 456–460. [Google Scholar] [CrossRef] [PubMed]
- Seim, I.; Fang, X.; Xiong, Z.; Lobanov, A.V.; Huang, Z.; Ma, S.; Feng, Y.; Turanov, A.A.; Zhu, Y.; Lenz, T.L.; et al. Genome analysis reveals insights into physiology and longevity of the Brandt’s bat Myotis brandtii. Nat. Commun. 2013, 4, 2212. [Google Scholar] [CrossRef] [PubMed]
- Parker, J.; Tsagkogeorga, G.; Cotton, J.A.; Liu, Y.; Provero, P.; Stupka, E.; Rossiter, S.J. Genome-wide signatures of convergent evolution in echolocating mammals. Nature 2013, 502, 228–231. [Google Scholar] [CrossRef] [PubMed]
- Lindblad-Toh, K.; Garber, M.; Zuk, O.; Lin, M.F.; Parker, B.J.; Washietl, S.; Kheradpour, P.; Ernst, J.; Jordan, G.; Mauceli, E.; et al. A high-resolution map of human evolutionary constraint using 29 mammals. Nature 2011, 478, 476–482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Abascal, F.; Zardoya, R.; Posada, D. ProtTest: Selection of best-fit models of protein evolution. Bioinformatics 2005, 21, 2104–2105. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Subramanian, S. Mutation rates in mammalian genomes. Proc. Natl. Acad. Sci. USA 2002, 99, 803–808. [Google Scholar] [CrossRef] [PubMed]
- Drummond, A.J.; Suchard, M.A.; Xie, D.; Rambaut, A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 2012, 29, 1969–1973. [Google Scholar] [CrossRef] [PubMed]
- Teeling, E.C.; Springer, M.S.; Madsen, O.; Bates, P.; O’Brien, S.J.; Murphy, W.J. A molecular phylogeny of bats illuminates biogeography and fossil record. Science 2005, 307, 580–584. [Google Scholar] [CrossRef] [PubMed]
- Cantrell, M.A.; Scott, L.; Brown, C.J.; Martinez, A.R.; Wichman, H.A. Loss of LINE-1 activity in megabats. Genetics 2008, 178, 393–404. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed]
- Fujino, K.; Horie, M.; Honda, T.; Merriman, D.K.; Tomonaga, K. Inhibition of Borna disease virus replication by an endogenous bornavirus-like element in the ground squirrel genome. Proc. Natl. Acad. Sci. USA 2014, 111, 13175–13180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cui, J.; Wang, L.-F. Genomic Mining Reveals Deep Evolutionary Relationships between Bornaviruses and Bats. Viruses 2015, 7, 5792-5800. https://0-doi-org.brum.beds.ac.uk/10.3390/v7112906
Cui J, Wang L-F. Genomic Mining Reveals Deep Evolutionary Relationships between Bornaviruses and Bats. Viruses. 2015; 7(11):5792-5800. https://0-doi-org.brum.beds.ac.uk/10.3390/v7112906
Chicago/Turabian StyleCui, Jie, and Lin-Fa Wang. 2015. "Genomic Mining Reveals Deep Evolutionary Relationships between Bornaviruses and Bats" Viruses 7, no. 11: 5792-5800. https://0-doi-org.brum.beds.ac.uk/10.3390/v7112906