
Comparative Analysis of Glycoprotein B (gB) of Equine Herpesvirus Type 1 and Type 4 (EHV-1 and EHV-4) in Cellular Tropism and Cell-to-Cell Transmission

Abstract
:1. Introduction

2. Materials and Methods
2.1. Viruses
2.2. Plasmids

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer | Product | Sequence |
|---|---|---|
| P1 | gB1 | aatctcgagatgtcctctggttgccgttc |
| P2 | gB1 | aactctagattaaaccattttttcatttt |
| P3 | gB4 | aatctcgagatgtccacttgttgccgtgc |
| P4 | gB4 | acatctagattaaaccattttttcgcttt |
| P5 | KanR 1 | accggatccaccgtcgtacgcatcgaaccaggatgacgacgataagtaggg |
| P6 | KanR 1 | ggtggatccggtaggcggtgggcaggtgtcaaccaattaaccaattctgattag |
| P7 | KanR 4 | actggatccacagttgtacgcattgaaccaggatgacgacgataagtaggg |
| P8 | KanR 4 | tgtggatccagtaggcggcgggcaggtgtcaaccaattaaccaattctgattag |
| P9 | gB1 deletion | agcgctgcgtgagcggcatttacataacctacgaggcgtcacatgtttaataaatattat aggatgacgacgataagtaggg |
| P10 | gB1 deletion | tcacactttgagtacgtgtcataatatttattaaacatgtgacgcctcgtaggttatgta caaccaattaaccaattctgattag |
| P11 | gB4 deletion | agcgctgcgctagcggcatttacataacatacgagacgtcaaatgttaaataaatatttt aggatgacgacgataagtaggg |
| P12 | gB4 deletion | tcaacccacaagtacgtgtcaaaatatttatttaacatttgacgtctcgtatgttatgta caaccaattaaccaattctgattag |
| P13 | gB4 KanR | agcggcgcacagcgctgcgtgagcggcatttacataacctacgaggcgtcatgtccacttgttgccgtgc |
| P14 | gB4 KanR | aaatatgaggtcacactttgagtacgtgtcataatatttattaaacatgtttaaaccattttttcgcttt |
| P15 | gB1 KanR | aacggcgcacagcgctgcgctagcggcatttacataacatacgagacgtcatgtcctctggttgccgttc |
| P16 | gB1 KanR | caaatatgagtcaacccacaagtacgtgtcaaaatatttatttaacatttttaaaccattttttcatttt |
| P17 | gBY336A | ctgtccaccggtgatattgtgtacgcgtctccgtttGCcggcctgagggctgccgctcgc aggatgacgacgataagtaggg |
| P18 | gBY336A | gtagctattgtgctctatgcgagcggcagccctcaggccgGCaaacggagacgcgtacac caaccaattaaccaattctgattag |
| P19 | Sequencing | ctcggttttccactgtggag |
| P20 | Sequencing | ggtgaatgaggatgaaacct |
| P21 | Sequencing | cgaccacgccaagccccccaac |
| P22 | Sequencing | cggcctcccccactttacccag |
| P23 | Sequencing | atcgaaccacctagaacttg |
| P24 | Sequencing | gtcagctggaactggac |
| P25 | Sequencing | gggcgggagtagcacgtgtt |
| P26 | Sequencing | agccccccaaatgggttgt |
| P27 | Sequencing | ccacggtcatgtcccaagtt |
| P28 | Sequencing | ttctcttcggttttccactg |
| P29 | Sequencing | ttggcaaaaatactaggctt |
2.3. Cells
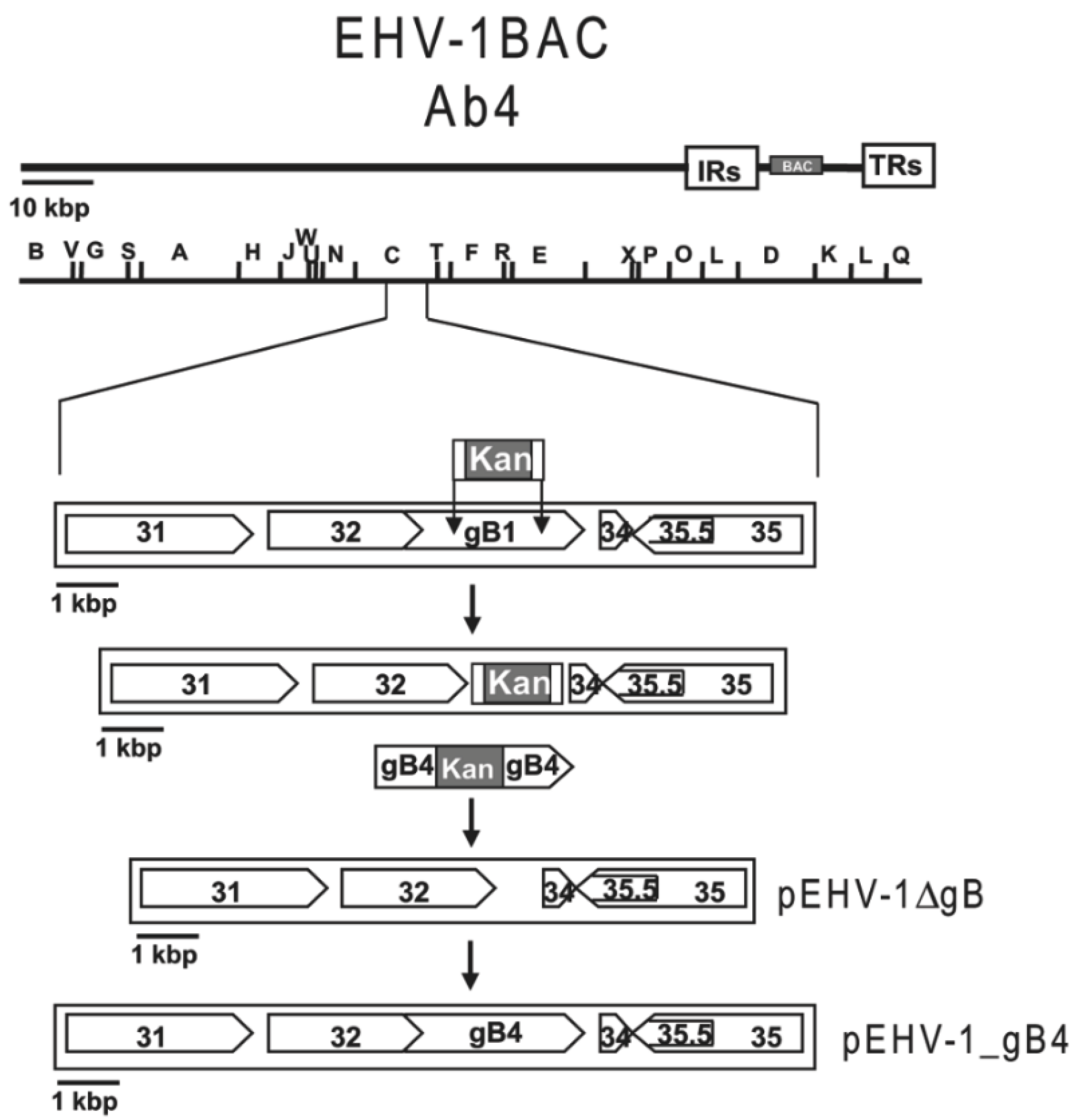
2.4. BAC Mutagenesis

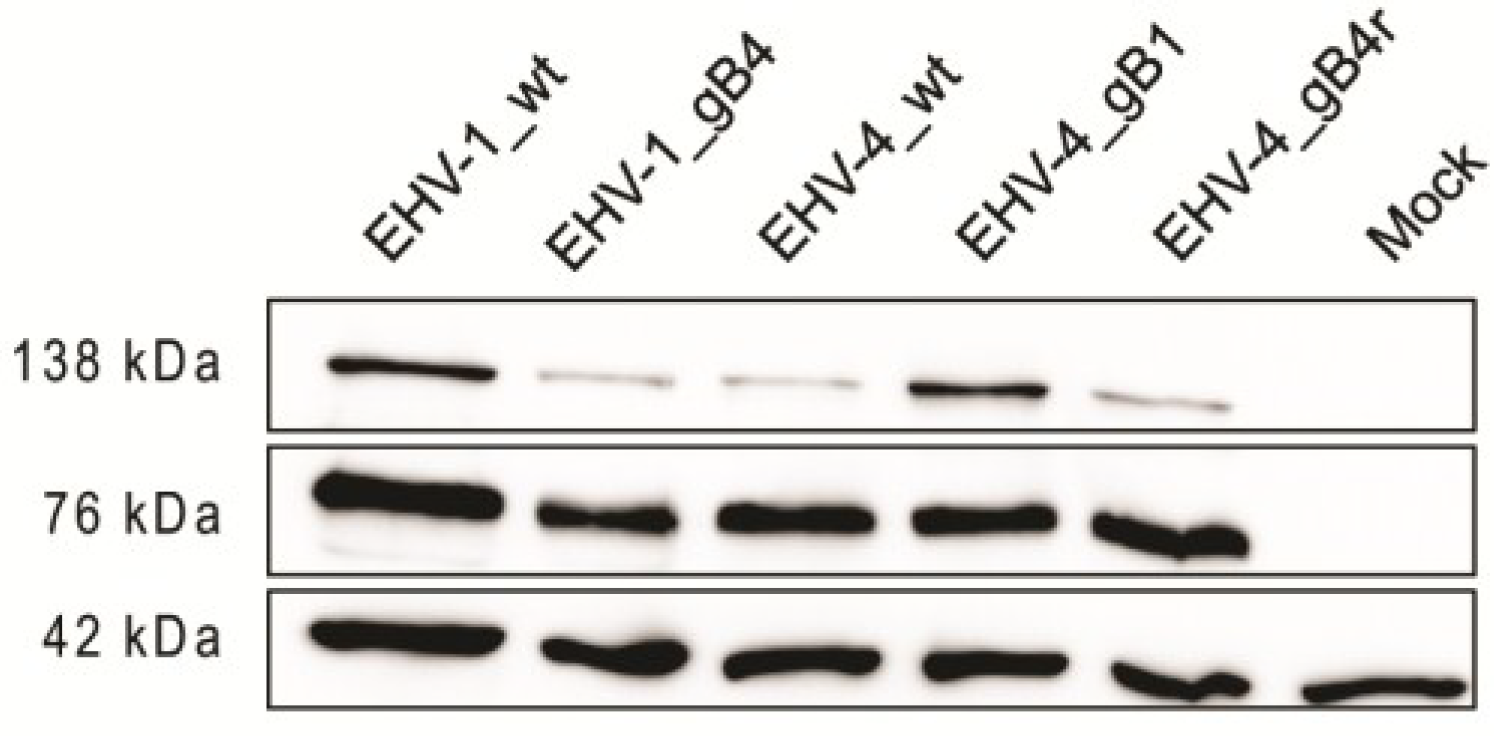
2.5. Western Blot Analysis
2.6. Virus Growth Assays
2.7. Virus Infection Assay
2.8. Flow Cytometry
2.9. Pharmacological Inhibitors
2.10. Statistical Analysis
3. Results
3.1. gB Expression by the Recombinant Viruses

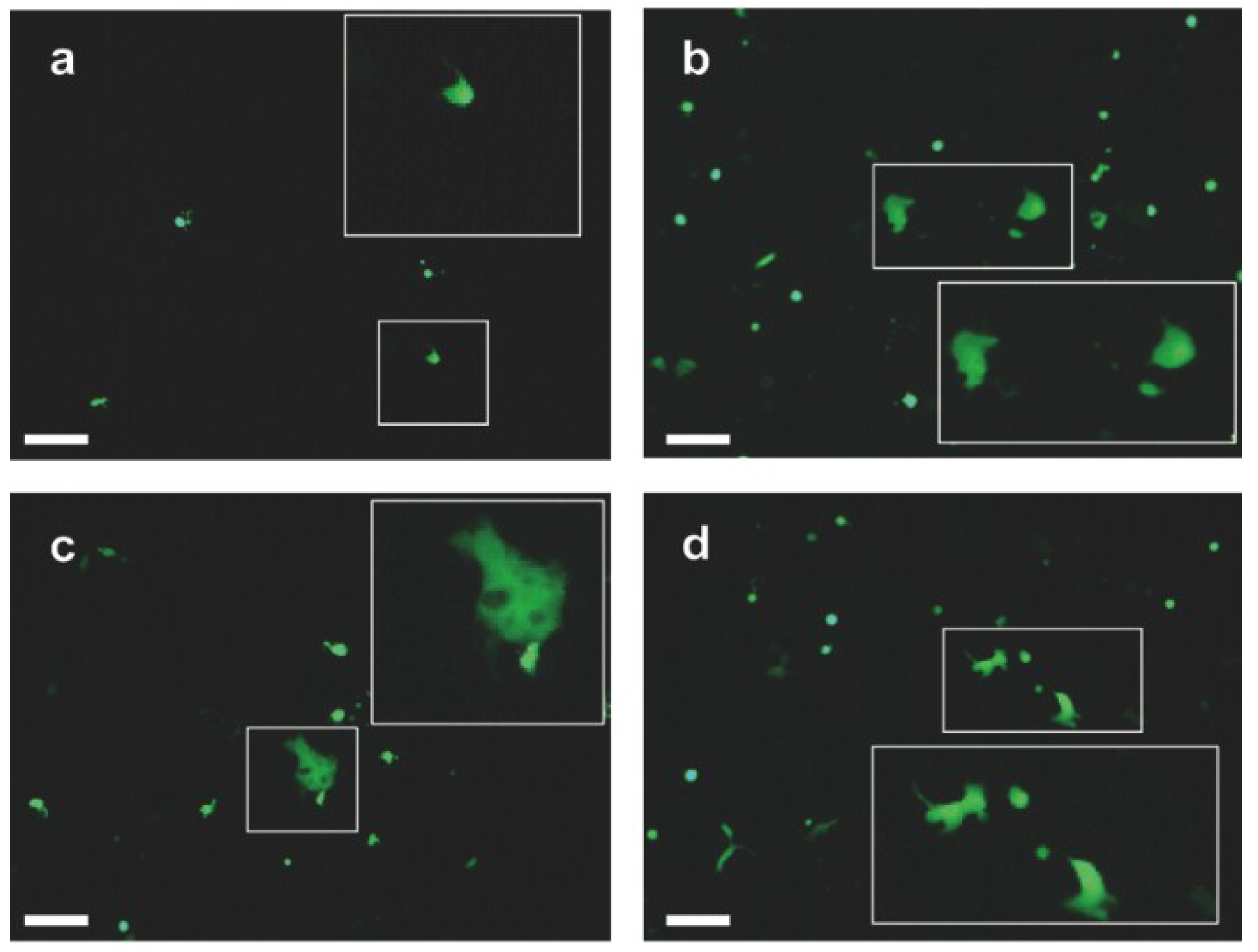
3.2. EHV-4 gB Is Essential for Viral Replication

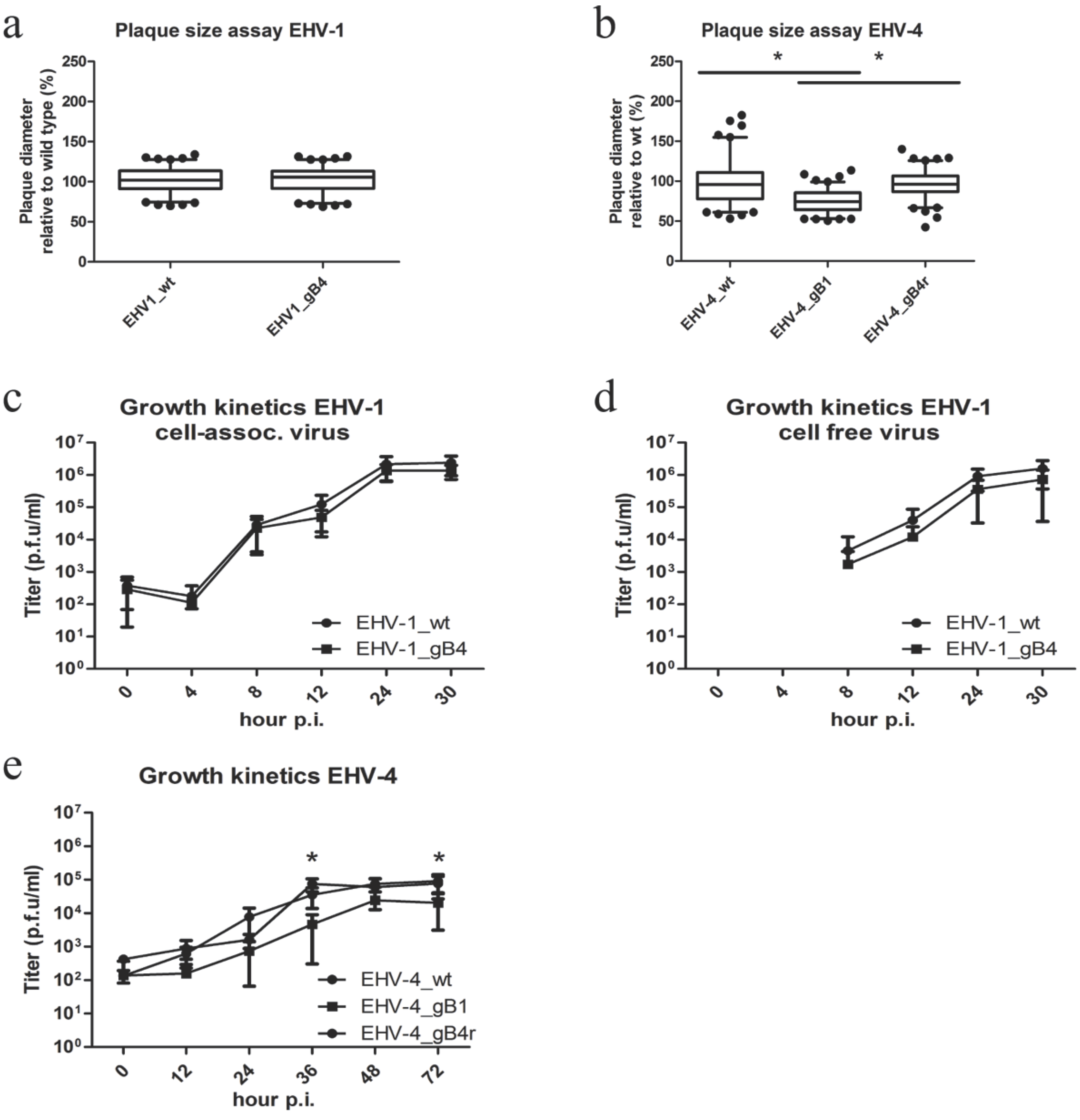
3.3. Virus Growth in Culture

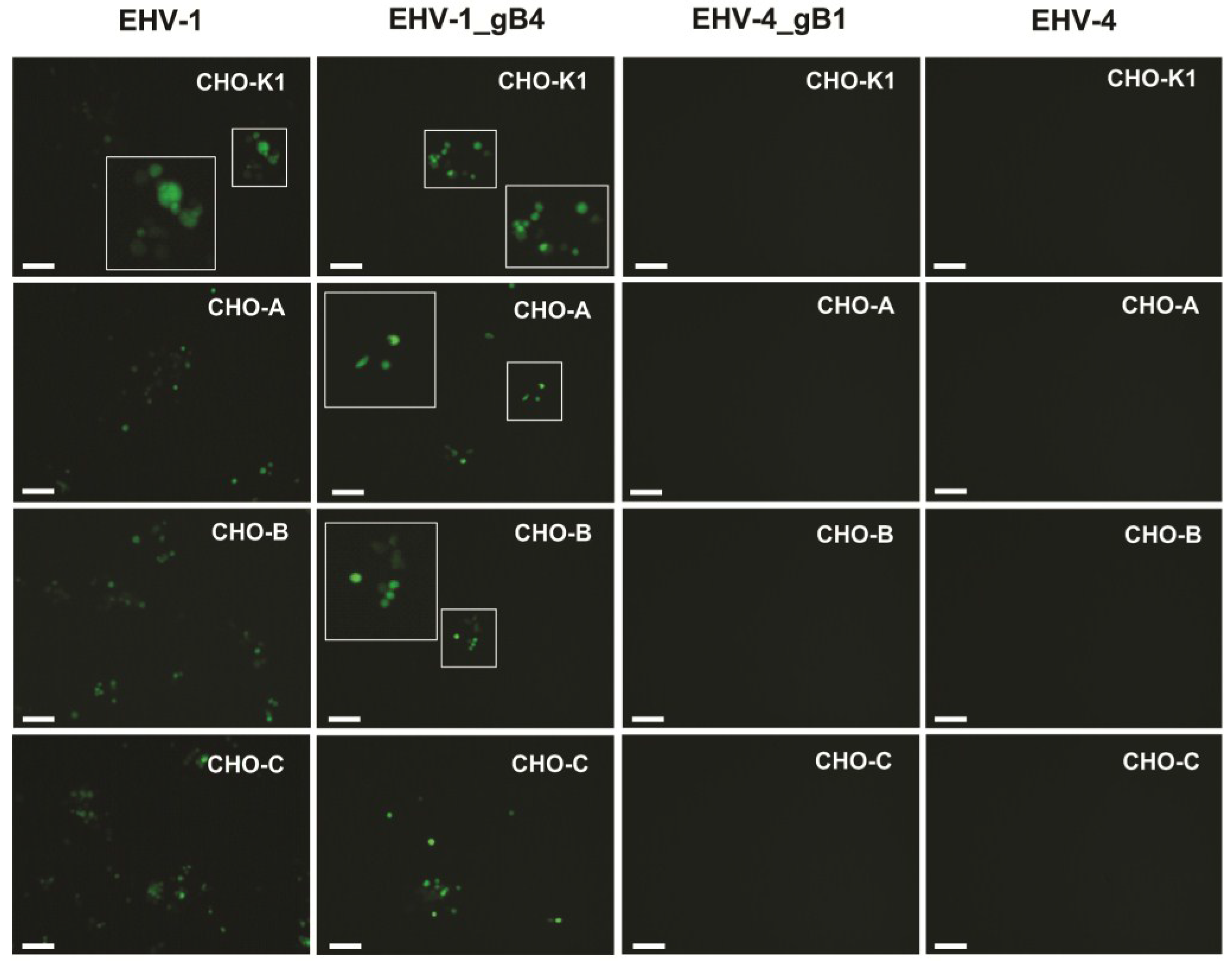
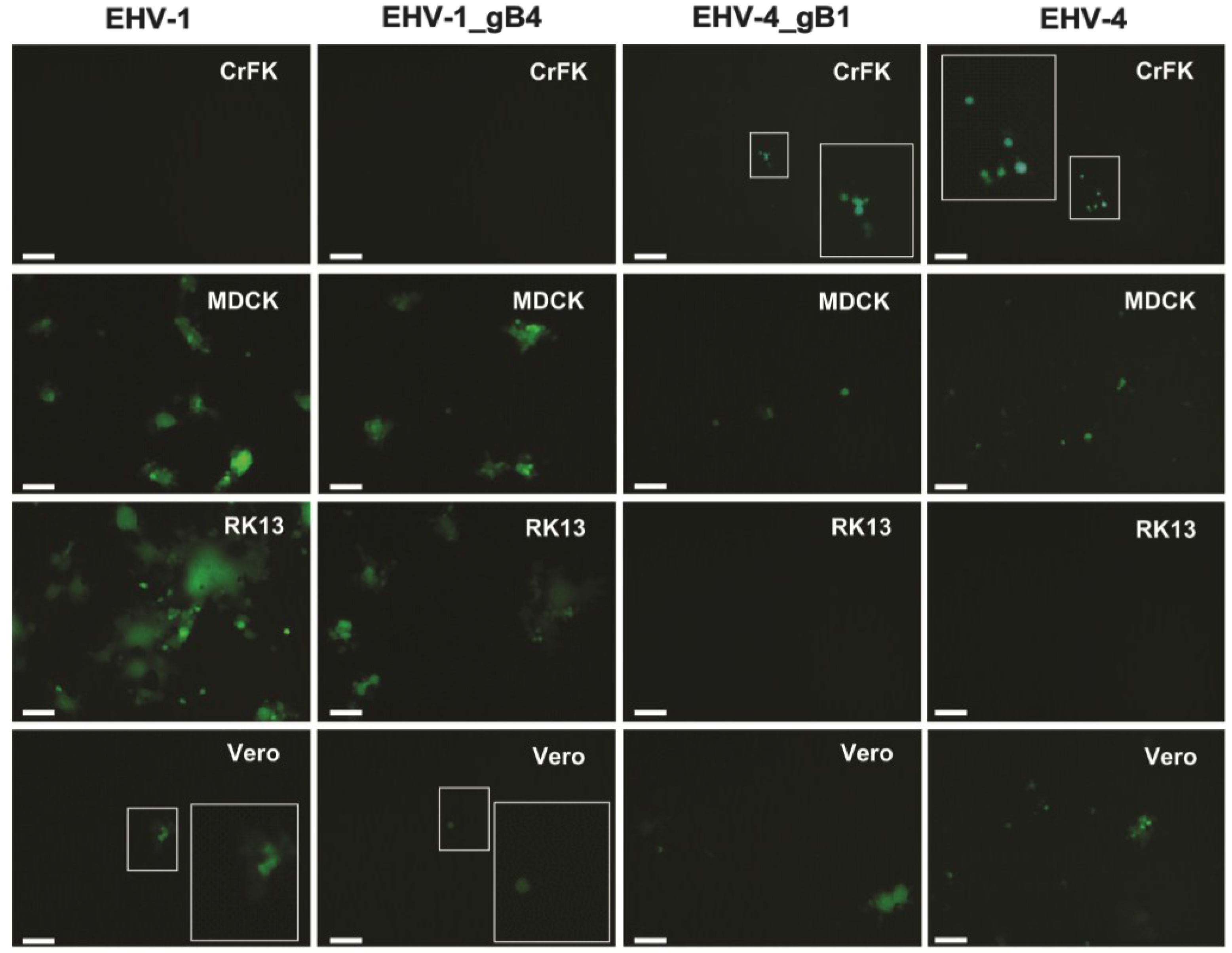
3.4. gB Has No Role in Determining the Host Range of EHV-1 and EHV-4 in Culture


| Cell line | EHV-1 | EHV-1 gB4 | EHV-4 | EHV-4 gB1 |
|---|---|---|---|---|
| CHO-K1 | + | + | − | − |
| CHO-A | + | + | − | − |
| CHO-B | + | + | − | − |
| CHO-C | + | + | − | − |
| CrFK | − | − | + | + |
| MDCK | + | + | + | + |
| RK13 | + | + | − | − |
| Vero | + | + | + | + |
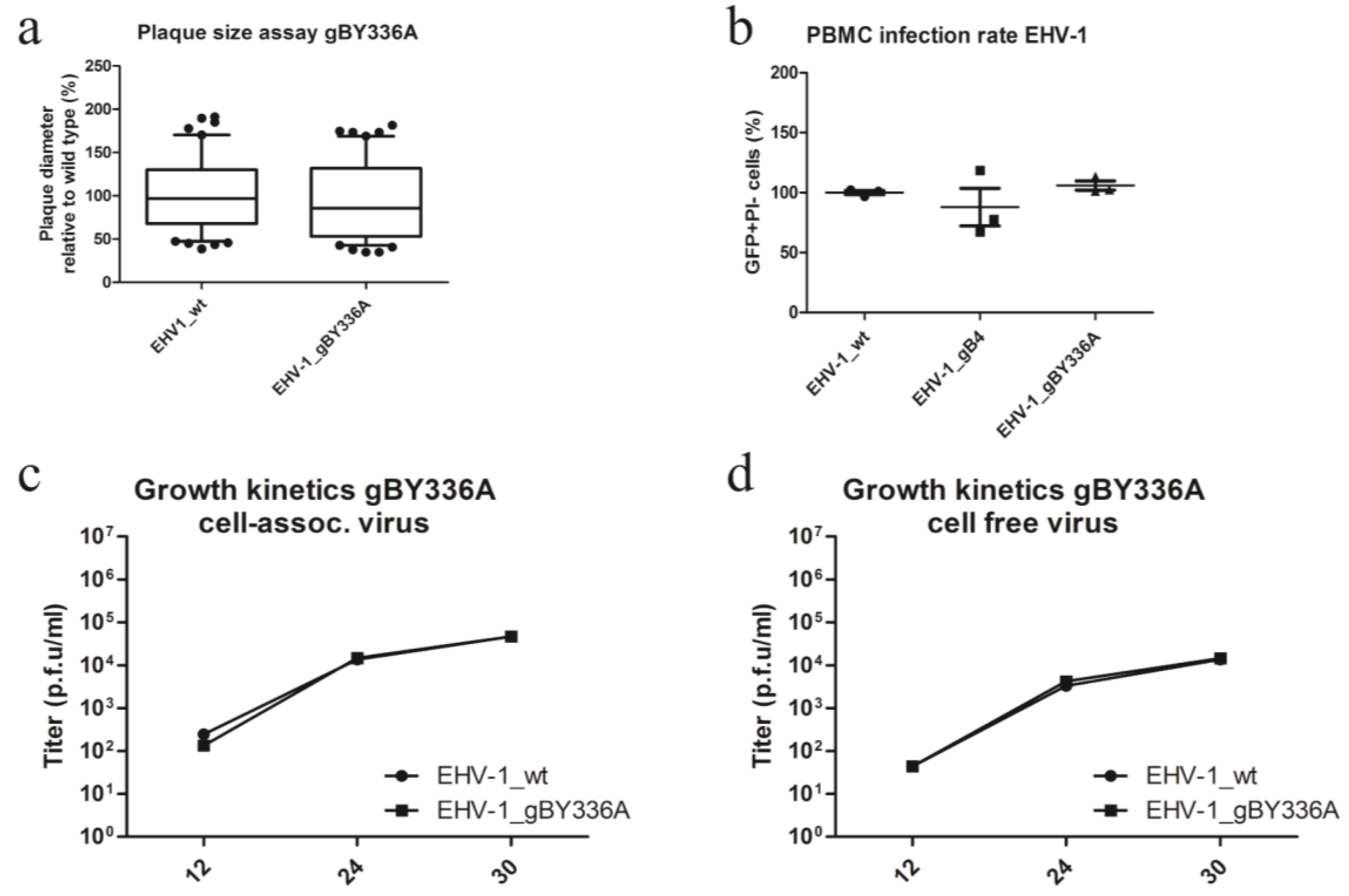
3.5. The Integrin-Binding Motif YGL Is Not Involved in EHV-1 Entry

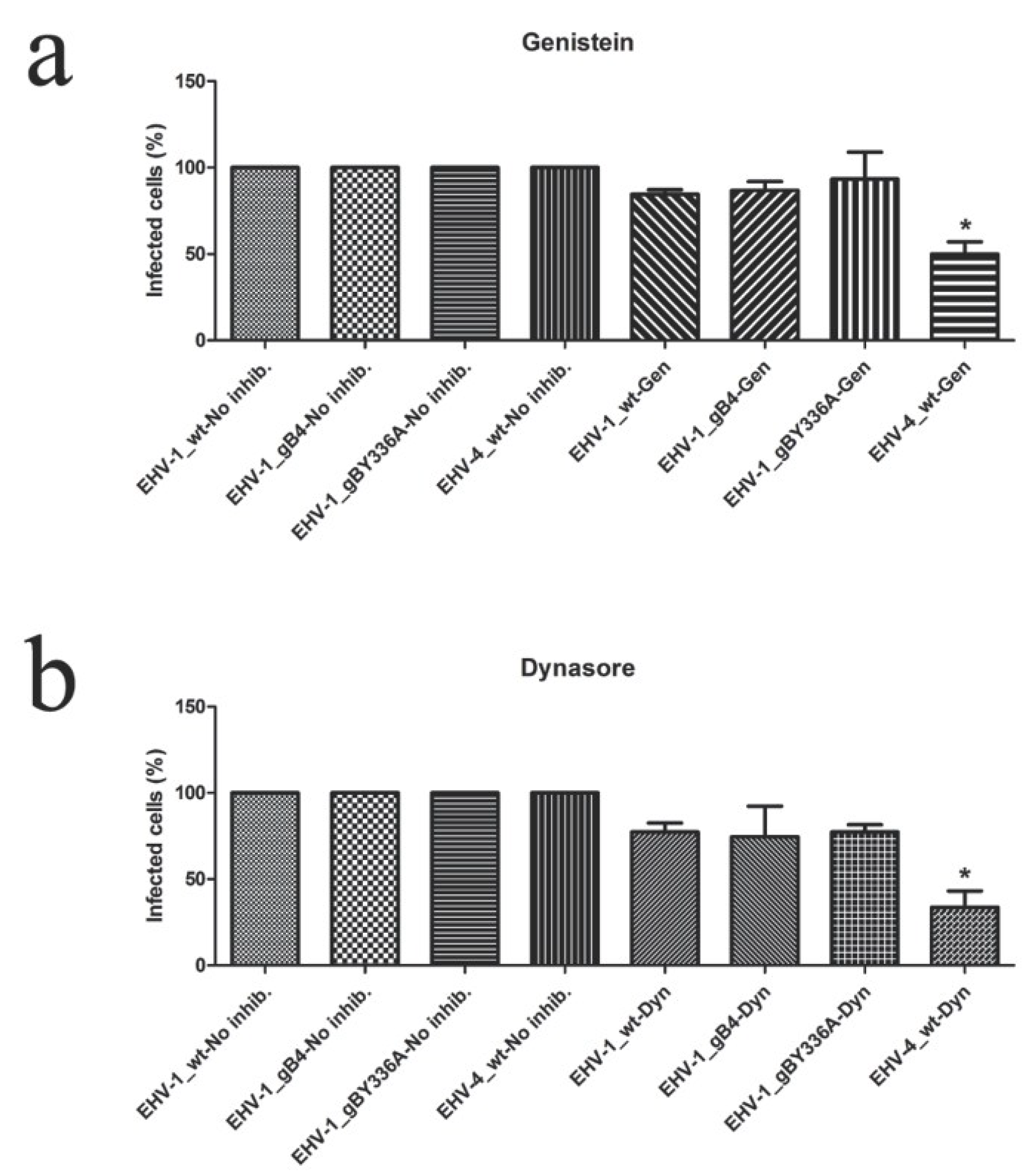
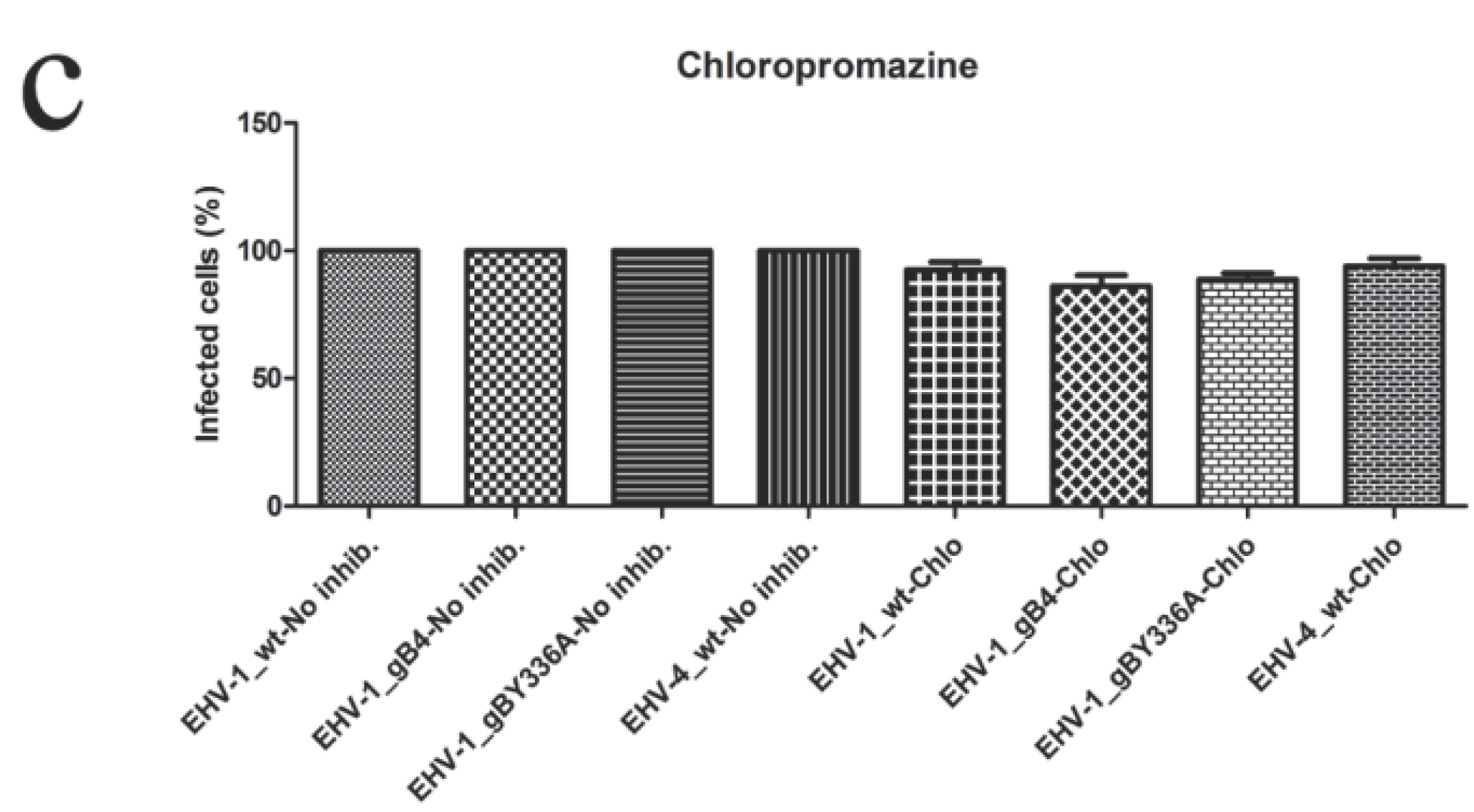
3.6. gB and YGL Do Not Play a Role in Determining the Cell Entry Pathway


4. Discussion
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Connolly, S.A.; Jackson, J.O.; Jardetzky, T.S.; Longnecker, R. Fusing structure and function: A structural view of the herpesvirus entry machinery. Nat. Rev. Microbiol. 2011, 9, 369–381. [Google Scholar] [CrossRef] [PubMed]
- Csellner, H.; Walker, C.; Wellington, J.E.; McLure, L.E.; Love, D.N.; Whalley, J.M. EHV-1 glycoprotein D (EHV-1 gD) is required for virus entry and cell-cell fusion, and an EHV-1 gD deletion mutant induces a protective immune response in mice. Arch. Virol. 2000, 145, 2371–2385. [Google Scholar] [CrossRef] [PubMed]
- Neubauer, A.; Braun, B.; Brandmuller, C.; Kaaden, O.R.; Osterrieder, N. Analysis of the contributions of the equine herpesvirus 1 glycoprotein gB homolog to virus entry and direct cell-to-cell spread. Virology 1997, 227, 281–294. [Google Scholar] [CrossRef] [PubMed]
- Osterrieder, N. Construction and characterization of an equine herpesvirus 1 glycoprotein C negative mutant. Virus Res. 1999, 59, 165–177. [Google Scholar] [CrossRef] [PubMed]
- Stokes, A.; Alber, D.G.; Greensill, J.; Amellal, B.; Carvalho, R.; Taylor, L.A.; Doel, T.R.; Killington, R.A.; Halliburton, I.W.; Meredith, D.M. The expression of the proteins of equine herpesvirus 1 which share homology with herpes simplex virus 1 glycoproteins H and L. Virus Res. 1996, 40, 91–107. [Google Scholar] [CrossRef] [PubMed]
- Wellington, J.E.; Love, D.N.; Whalley, J.M. Evidence for involvement of equine herpesvirus 1 glycoprotein B in cell-cell fusion. Arch. Virol. 1996, 141, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Cai, W.H.; Gu, B.; Person, S. Role of glycoprotein B of herpes simplex virus type 1 in viral entry and cell fusion. J. Virol. 1988, 62, 2596–2604. [Google Scholar] [PubMed]
- Atanasiu, D.; Whitbeck, J.C.; Cairns, T.M.; Reilly, B.; Cohen, G.H.; Eisenberg, R.J. Bimolecular complementation reveals that glycoproteins gB and gH/gL of herpes simplex virus interact with each other during cell fusion. Proc. Natl. Acad. Sci. USA 2007, 104, 18718–18723. [Google Scholar] [CrossRef] [PubMed]
- Avitabile, E.; Forghieri, C.; Campadelli-Fiume, G. Complexes between herpes simplex virus glycoproteins gD, gB, and gH detected in cells by complementation of split enhanced green fluorescent protein. J. Virol. 2007, 81, 11532–11537. [Google Scholar] [CrossRef] [PubMed]
- Satoh, T.; Arii, J.; Suenaga, T.; Wang, J.; Kogure, A.; Uehori, J.; Arase, N.; Shiratori, I.; Tanaka, S.; Kawaguchi, Y.; et al. PILRalpha is a herpes simplex virus-1 entry coreceptor that associates with glycoprotein B. Cell 2008, 132, 935–944. [Google Scholar] [CrossRef] [PubMed]
- Arii, J.; Goto, H.; Suenaga, T.; Oyama, M.; Kozuka-Hata, H.; Imai, T.; Minowa, A.; Akashi, H.; Arase, H.; Kawaoka, Y.; et al. Non-muscle myosin IIA is a functional entry receptor for herpes simplex virus-1. Nature 2010, 467, 859–862. [Google Scholar] [CrossRef] [PubMed]
- Suenaga, T.; Satoh, T.; Somboonthum, P.; Kawaguchi, Y.; Mori, Y.; Arase, H. Myelin-associated glycoprotein mediates membrane fusion and entry of neurotropic herpesviruses. Proc. Natl. Acad. Sci. USA 2010, 107, 866–871. [Google Scholar] [CrossRef] [PubMed]
- Heldwein, E.E.; Lou, H.; Bender, F.C.; Cohen, G.H.; Eisenberg, R.J.; Harrison, S.C. Crystal structure of glycoprotein B from herpes simplex virus 1. Science 2006, 313, 217–220. [Google Scholar] [CrossRef] [PubMed]
- Backovic, M.; Longnecker, R.; Jardetzky, T.S. Structure of a trimeric variant of the Epstein-Barr virus glycoprotein B. Proc. Natl. Acad. Sci. USA 2009, 106, 2880–2885. [Google Scholar] [CrossRef] [PubMed]
- Roche, S.; Bressanelli, S.; Rey, F.A.; Gaudin, Y. Crystal structure of the low-pH form of the vesicular stomatitis virus glycoprotein G. Science 2006, 313, 187–191. [Google Scholar] [CrossRef] [PubMed]
- Kadlec, J.; Loureiro, S.; Abrescia, N.G.; Stuart, D.I.; Jones, I.M. The postfusion structure of baculovirus gp64 supports a unified view of viral fusion machines. Nat. Struct. Mol. Biol. 2008, 15, 1024–1030. [Google Scholar] [CrossRef] [PubMed]
- Atanasiu, D.; Whitbeck, J.C.; de Leon, M.P.; Lou, H.; Hannah, B.P.; Cohen, G.H.; Eisenberg, R.J. Bimolecular complementation defines functional regions of Herpes simplex virus gB that are involved with gH/gL as a necessary step leading to cell fusion. J. Virol. 2010, 84, 3825–3834. [Google Scholar] [CrossRef] [PubMed]
- Backovic, M.; DuBois, R.M.; Cockburn, J.J.; Sharff, A.J.; Vaney, M.C.; Granzow, H.; Klupp, B.G.; Bricogne, G.; Mettenleiter, T.C.; Rey, F.A. Structure of a core fragment of glycoprotein H from pseudorabies virus in complex with antibody. Proc. Natl. Acad. Sci. USA 2010, 107, 22635–22640. [Google Scholar] [CrossRef] [PubMed]
- Chowdary, T.K.; Cairns, T.M.; Atanasiu, D.; Cohen, G.H.; Eisenberg, R.J.; Heldwein, E.E. Crystal structure of the conserved herpesvirus fusion regulator complex gH-gL. Nat. Struct. Mol. Biol. 2010, 17, 882–888. [Google Scholar] [CrossRef] [PubMed]
- Matsuura, H.; Kirschner, A.N.; Longnecker, R.; Jardetzky, T.S. Crystal structure of the Epstein-Barr virus (EBV) glycoprotein H/glycoprotein L (gH/gL) complex. Proc. Natl. Acad. Sci. USA 2010, 107, 22641–22646. [Google Scholar] [CrossRef] [PubMed]
- Snowden, B.W.; Kinchington, P.R.; Powell, K.L.; Halliburton, I.W. Antigenic and biochemical analysis of gB of herpes simplex virus type 1 and type 2 and of cross-reacting glycoproteins induced by bovine mammillitis virus and equine herpesvirus type 1. J. Gen. Virol. 1985, 66 (Pt 2), 231–247. [Google Scholar] [CrossRef] [PubMed]
- Telford, E.A.; Watson, M.S.; McBride, K.; Davison, A.J. The DNA sequence of equine herpesvirus-1. Virology 1992, 189, 304–316. [Google Scholar] [CrossRef] [PubMed]
- Telford, E.A.; Watson, M.S.; Perry, J.; Cullinane, A.A.; Davison, A.J. The DNA sequence of equine herpesvirus-4. J. Gen. Virol. 1998, 79, 1197–1203. [Google Scholar] [PubMed]
- Patel, J.R.; Heldens, J. Equine herpesviruses 1 (EHV-1) and 4 (EHV-4)—Epidemiology, disease and immunoprophylaxis: A brief review. Vet. J. 2005, 170, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Osterrieder, N.; van de Walle, G.R. Pathogenic potential of equine alphaherpesviruses: The importance of the mononuclear cell compartment in disease outcome. Vet. Microbiol. 2010, 143, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Azab, W.; Kato, K.; Abdel-Gawad, A.; Tohya, Y.; Akashi, H. Equine herpesvirus 4: Recent advances using BAC technology. Vet. Microbial. 2011, 150, 1–14. [Google Scholar] [CrossRef]
- Smith, K.C.; Whitwell, K.E.; Blunden, A.S.; Bestbier, M.E.; Scase, T.J.; Geraghty, R.J.; Nugent, J.; Davis-Poynter, N.J.; Cardwell, J.M. Equine herpesvirus-1 abortion: Atypical cases with lesions largely or wholly restricted to the placenta. Equine Vet. J. 2004, 36, 79–82. [Google Scholar] [CrossRef] [PubMed]
- Smith, K.C.; Borchers, K. A study of the pathogenesis of equid herpesvirus-1 (EHV-1) abortion by DNA in-situ hybridization. J. Comp. Pathol. 2001, 125, 304–310. [Google Scholar] [CrossRef] [PubMed]
- Gilkerson, J.R.; Love, D.N.; Whalley, J.M. Epidemiology of equine herpesvirus abortion: Searching for clues to the future. Aust. Vet. J. 1998, 76, 675–676. [Google Scholar] [CrossRef] [PubMed]
- Pusterla, N.; David Wilson, W.; Madigan, J.E.; Ferraro, G.L. Equine herpesvirus-1 myeloencephalopathy: A review of recent developments. Vet. J. 2009, 180, 279–289. [Google Scholar] [CrossRef] [PubMed]
- Borchers, K.; Thein, R.; Sterner-Kock, A. Pathogenesis of equine herpesvirus-associated neurological disease: A revised explanation. Equine Vet. J. 2006, 38, 283–287. [Google Scholar] [CrossRef] [PubMed]
- Azab, W.; Lehmann, M.J.; Osterrieder, N. Glycoprotein H and α4β1 Integrins Determine the Entry Pathway of Alphaherpesviruses. J. Virol. 2013, 87, 5937–5948. [Google Scholar] [CrossRef] [PubMed]
- Azab, W.; Osterrieder, N. Glycoproteins D of equine herpesvirus type 1 (EHV-1) and EHV-4 determine cellular tropism independently of integrins. J. Virol. 2012, 86, 2031–2044. [Google Scholar] [CrossRef] [PubMed]
- Azab, W.; Tsujimura, K.; Maeda, K.; Kobayashi, K.; Mohamed, Y.M.; Kato, K.; Matsumura, T.; Akashi, H. Glycoprotein C of equine herpesvirus 4 plays a role in viral binding to cell surface heparan sulfate. Virus Res. 2010, 151, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Azab, W.; Zajic, L.; Osterrieder, N. The role of glycoprotein H of equine herpesviruses 1 and 4 (EHV-1 and EHV-4) in cellular host range and integrin binding. Vet. Res. 2012, 43, 61. [Google Scholar] [CrossRef] [PubMed]
- Graham, K.L.; Fleming, F.E.; Halasz, P.; Hewish, M.J.; Nagesha, H.S.; Holmes, I.H.; Takada, Y.; Coulson, B.S. Rotaviruses interact with α4β7 and α4β1 integrins by binding the same integrin domains as natural ligands. J. Gen. Virol. 2005, 86 (Pt 12), 3397–3408. [Google Scholar] [CrossRef] [PubMed]
- Crowhurst, F.A.; Dickinson, G.; Burrows, R. An outbreak of paresis in mares and geldings associated with equid herpesvirus 1. Vet. Rec. 1981, 109, 527–528. [Google Scholar] [PubMed]
- Osterrieder, N.; Schumacher, D.; Trapp, S.; Beer, M.; von Einem, J.; Tischer, K. Establishment and use of infectious bacterial artificial chromosome (BAC) DNA clones of animal herpesviruses. Berl. und Munch. tierarztl. Wochenschr. 2003, 116, 373–380. [Google Scholar]
- Von Einem, J.; Wellington, J.; Whalley, J.M.; Osterrieder, K.; O’Callaghan, D.J.; Osterrieder, N. The truncated form of glycoprotein gp2 of equine herpesvirus 1 (EHV-1) vaccine strain KyA is not functionally equivalent to full-length gp2 encoded by EHV-1 wild-type strain RacL11. J. Virol. 2004, 78, 3003–3013. [Google Scholar]
- Goodman, L.B.; Loregian, A.; Perkins, G.A.; Nugent, J.; Buckles, E.L.; Mercorelli, B.; Kydd, J.H.; Palu, G.; Smith, K.C.; Osterrieder, N.; et al. A point mutation in a herpesvirus polymerase determines neuropathogenicity. PLoS Pathog. 2007, 3, e160. [Google Scholar] [CrossRef] [PubMed]
- Azab, W.; Kato, K.; Arii, J.; Tsujimura, K.; Yamane, D.; Tohya, Y.; Matsumura, T.; Akashi, H. Cloning of the genome of equine herpesvirus 4 strain TH20p as an infectious bacterial artificial chromosome. Arch. Virol. 2009, 154, 833–842. [Google Scholar] [CrossRef] [PubMed]
- Azab, W.; Tsujimura, K.; Kato, K.; Arii, J.; Morimoto, T.; Kawaguchi, Y.; Tohya, Y.; Matsumura, T.; Akashi, H. Characterization of a thymidine kinase-deficient mutant of equine herpesvirus 4 and in vitro susceptibility of the virus to antiviral agents. Antivir. Res. 2010, 85, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Rudolph, J.; O’Callaghan, D.J.; Osterrieder, N. Cloning of the genomes of equine herpesvirus type 1 (EHV-1) strains KyA and racL11 as bacterial artificial chromosomes (BAC). J. Vet. Med. B Infect. Dis. Vet. Public Health 2002, 49, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.C.; Yu, D.; Martinez de Velasco, J.; Tessarollo, L.; Swing, D.A.; Court, D.L.; Jenkins, N.A.; Copeland, N.G. A highly efficient Escherichia coli-based chromosome engineering system adapted for recombinogenic targeting and subcloning of BAC DNA. Genomics 2001, 73, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Tischer, B.K.; von Einem, J.; Kaufer, B.; Osterrieder, N. Two-step red-mediated recombination for versatile high-efficiency markerless DNA manipulation in Escherichia coli. Biotechniques 2006, 40, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Von Einem, J.; Smith, P.M.; Van de Walle, G.R.; O’Callaghan, D.J.; Osterrieder, N. In vitro and in vivo characterization of equine herpesvirus type 1 (EHV-1) mutants devoid of the viral chemokine-binding glycoprotein G (gG). Virology 2007, 362, 151–162. [Google Scholar]
- Allen, G.P.; Yeargan, M.R. Use of lambda gt11 and monoclonal antibodies to map the genes for the six major glycoproteins of equine herpesvirus 1. J. Virol. 1987, 61, 2454–2461. [Google Scholar] [PubMed]
- Sullivan, D.C.; Allen, G.P.; O’Callaghan, D.J. Synthesis and processing of equine herpesvirus type 1 glycoprotein 14. Virology 1989, 173, 638–646. [Google Scholar] [CrossRef] [PubMed]
- Whalley, J.M.; Ruitenberg, K.M.; Sullivan, K.; Seshadri, L.; Hansen, K.; Birch, D.; Gilkerson, J.R.; Wellington, J.E. Host cell tropism of equine herpesviruses: Glycoprotein D of EHV-1 enables EHV-4 to infect a non-permissive cell line. Arch. Virol. 2007, 152, 717–725. [Google Scholar] [CrossRef] [PubMed]
- Nabi, I.R.; Le, P.U. Caveolae/raft-dependent endocytosis. J. Cell Biol. 2003, 161, 673–677. [Google Scholar] [CrossRef] [PubMed]
- Macia, E.; Ehrlich, M.; Massol, R.; Boucrot, E.; Brunner, C.; Kirchhausen, T. Dynasore, a cell-permeable inhibitor of dynamin. Dev. Cell 2006, 10, 839–850. [Google Scholar] [CrossRef] [PubMed]
- Pelkmans, L.; Puntener, D.; Helenius, A. Local actin polymerization and dynamin recruitment in SV40-induced internalization of caveolae. Science 2002, 296, 535–539. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.H.; Rothberg, K.G.; Anderson, R.G. Mis-assembly of clathrin lattices on endosomes reveals a regulatory switch for coated pit formation. J. Cell Biol. 1993, 123, 1107–1117. [Google Scholar] [CrossRef] [PubMed]
- Rauh, I.; Mettenleiter, T.C. Pseudorabies virus glycoproteins gII and gp50 are essential for virus penetration. J. Virol. 1991, 65, 5348–5356. [Google Scholar] [PubMed]
- Rauh, I.; Weiland, F.; Fehler, F.; Keil, G.M.; Mettenleiter, T.C. Pseudorabies virus mutants lacking the essential glycoprotein gII can be complemented by glycoprotein gI of bovine herpesvirus 1. J. Virol. 1991, 65, 621–631. [Google Scholar] [PubMed]
- Kopp, A.; Blewett, E.; Misra, V.; Mettenleiter, T.C. Proteolytic cleavage of bovine herpesvirus 1 (BHV-1) glycoprotein gB is not necessary for its function in BHV-1 or pseudorabies virus. J. Virol. 1994, 68, 1667–1674. [Google Scholar] [PubMed]
- Packiarajah, P.; Walker, C.; Gilkerson, J.; Whalley, J.M.; Love, D.N. Immune responses and protective efficacy of recombinant baculovirus-expressed glycoproteins of equine herpesvirus 1 (EHV-1) gB, gC and gD alone or in combinations in BALB/c mice. Vet. Microbial. 1998, 61, 261–278. [Google Scholar] [CrossRef]
- Maresova, L.; Pasieka, T.J.; Homan, E.; Gerday, E.; Grose, C. Incorporation of three endocytosed varicella-zoster virus glycoproteins, gE, gH, and gB, into the virion envelope. J. Virol. 2005, 79, 997–1007. [Google Scholar] [CrossRef] [PubMed]
- Farnsworth, A.; Wisner, T.W.; Webb, M.; Roller, R.; Cohen, G.; Eisenberg, R.; Johnson, D.C. Herpes simplex virus glycoproteins gB and gH function in fusion between the virion envelope and the outer nuclear membrane. Proc. Natl. Acad. Sci. USA 2007, 104, 10187–10192. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, H.H.; Sharma-Walia, N.; Zeng, L.; Gao, S.J.; Chandran, B. Envelope glycoprotein gB of Kaposi’s sarcoma-associated herpesvirus is essential for egress from infected cells. J. Virol. 2005, 79, 10952–10967. [Google Scholar] [CrossRef] [PubMed]
- Kopp, A.; Mettenleiter, T.C. Stable rescue of a glycoprotein gII deletion mutant of pseudorabies virus by glycoprotein gI of bovine herpesvirus 1. J. Virol. 1992, 66, 2754–2762. [Google Scholar] [PubMed]
- Fan, Q.; Longnecker, R.; Connolly, S.A. Substitution of herpes simplex virus 1 entry glycoproteins with those of saimiriine herpesvirus 1 reveals a gD-gH/gL functional interaction and a region within the gD profusion domain that is critical for fusion. J. Virol. 2014, 88, 6470–6482. [Google Scholar] [CrossRef] [PubMed]
- Reed, S.M.; Toribio, R.E. Equine herpesvirus 1 and 4. Vet. Clin. N. Am. Equine Pract. 2004, 20, 631–642. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Spiesschaert, B.; Osterrieder, N.; Azab, W. Comparative Analysis of Glycoprotein B (gB) of Equine Herpesvirus Type 1 and Type 4 (EHV-1 and EHV-4) in Cellular Tropism and Cell-to-Cell Transmission. Viruses 2015, 7, 522-542. https://0-doi-org.brum.beds.ac.uk/10.3390/v7020522
Spiesschaert B, Osterrieder N, Azab W. Comparative Analysis of Glycoprotein B (gB) of Equine Herpesvirus Type 1 and Type 4 (EHV-1 and EHV-4) in Cellular Tropism and Cell-to-Cell Transmission. Viruses. 2015; 7(2):522-542. https://0-doi-org.brum.beds.ac.uk/10.3390/v7020522
Chicago/Turabian StyleSpiesschaert, Bart, Nikolaus Osterrieder, and Walid Azab. 2015. "Comparative Analysis of Glycoprotein B (gB) of Equine Herpesvirus Type 1 and Type 4 (EHV-1 and EHV-4) in Cellular Tropism and Cell-to-Cell Transmission" Viruses 7, no. 2: 522-542. https://0-doi-org.brum.beds.ac.uk/10.3390/v7020522