
Modulation of SIV and HIV DNA Vaccine Immunity by Fas-FasL Signaling


Abstract
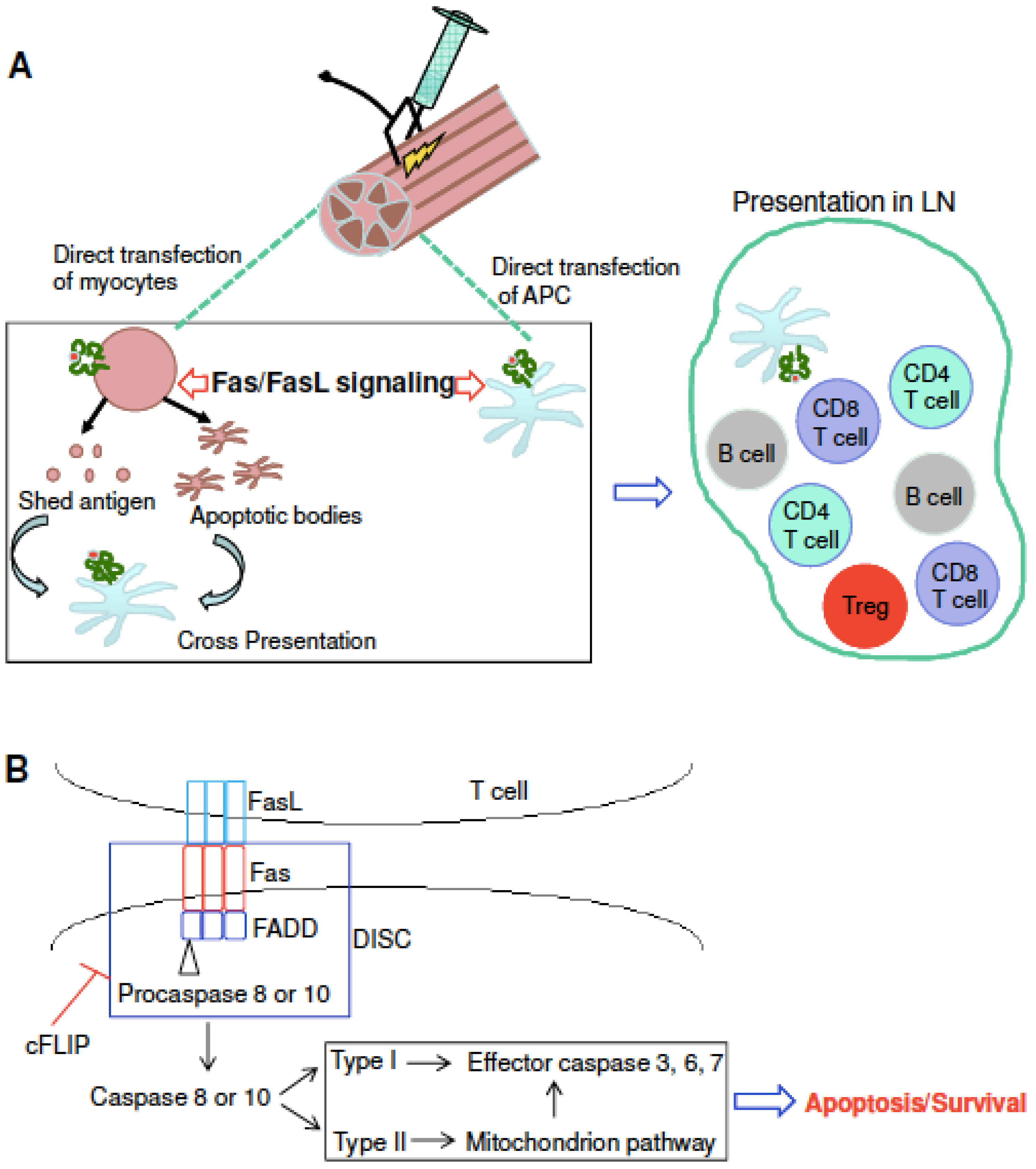
:1. Introduction
2. Materials and Methods
2.1. Plasmids and Preparation
2.2. Peptides, Antibodies, and Diagnostic Kits for Flow Cytometry and Western Blots
2.3. Assays for Gene Expression and Apoptosis in Cell Culture
2.4. In Vivo Immunizations
2.5. Cellular Immune Response Assays
2.6. Cell Proliferation Assays
2.7. Humoral Immune Response Assays
2.8. LCMV Infections, DNA Electroporations and CMI Assays
2.9. Statistical Analysis
3. Results
3.1. Plasmid Expression and Pro- or Anti-Apoptotic Function were Confirmed in Cell Culture
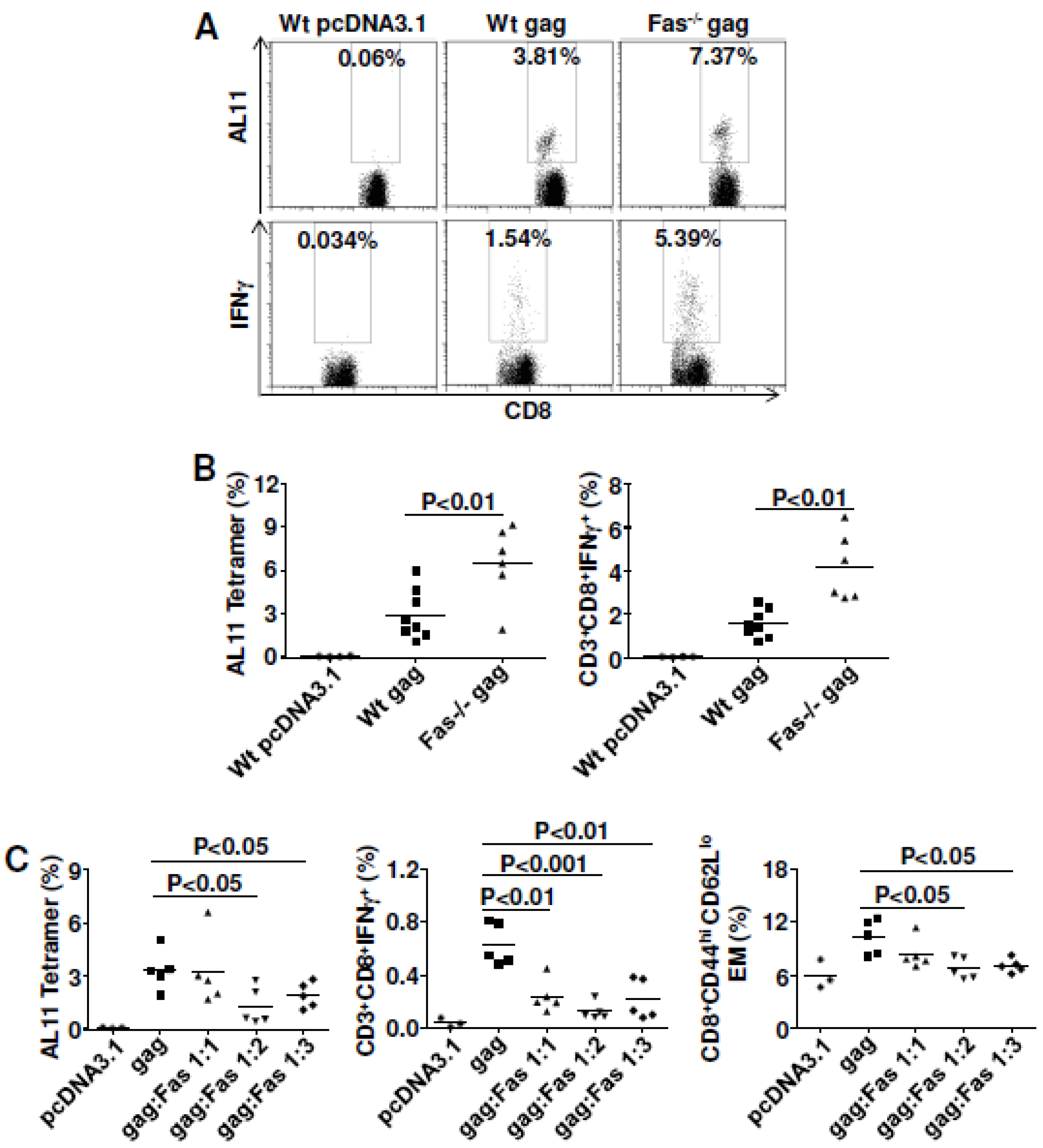
3.2. Fas Signaling Had a Negative Influence on DNA Vaccine Potency

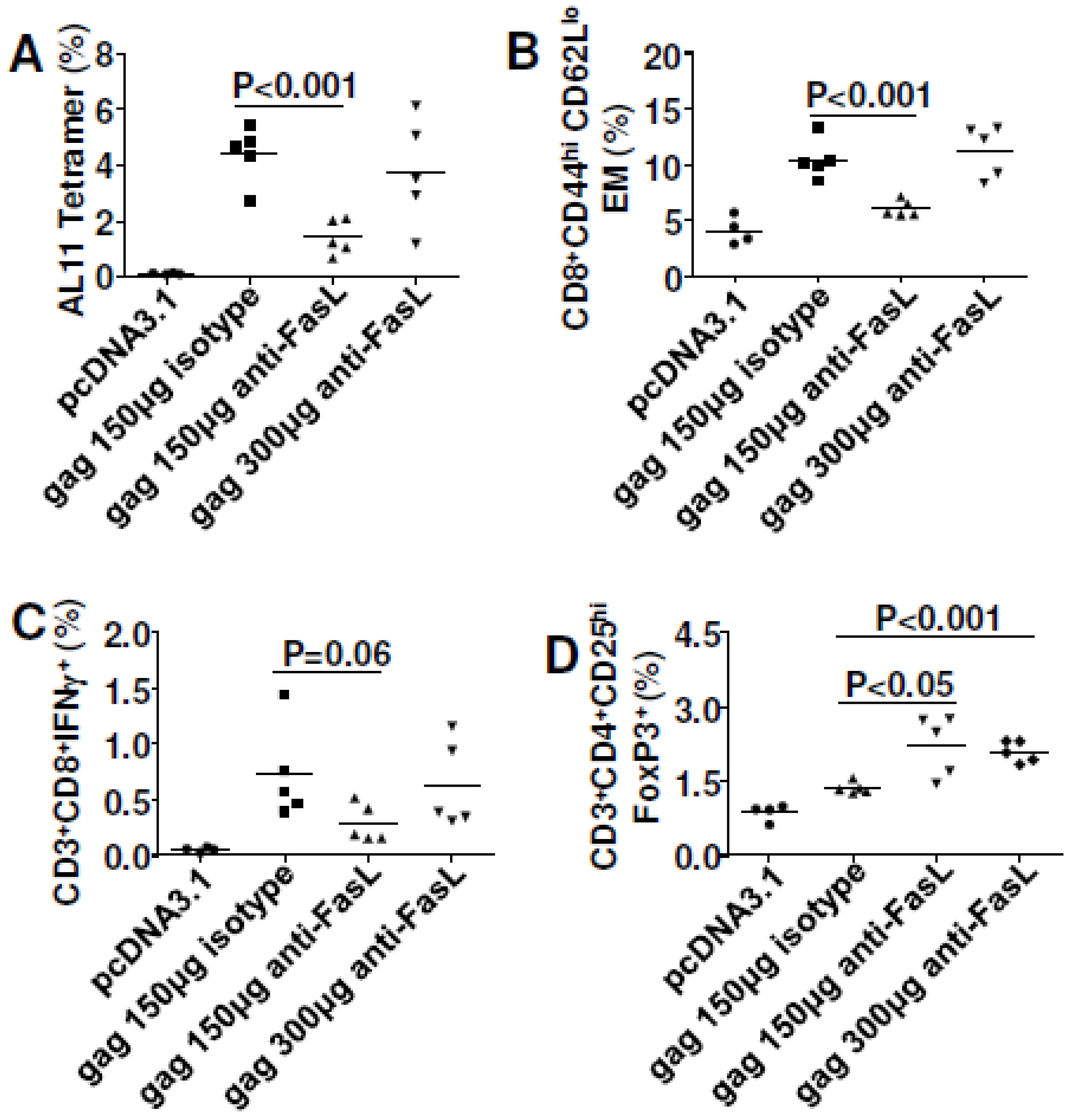
3.3. Inhibiting Fas or FasL (With shRNA or Antibody) Failed to Improve DNA Vaccine Immunity


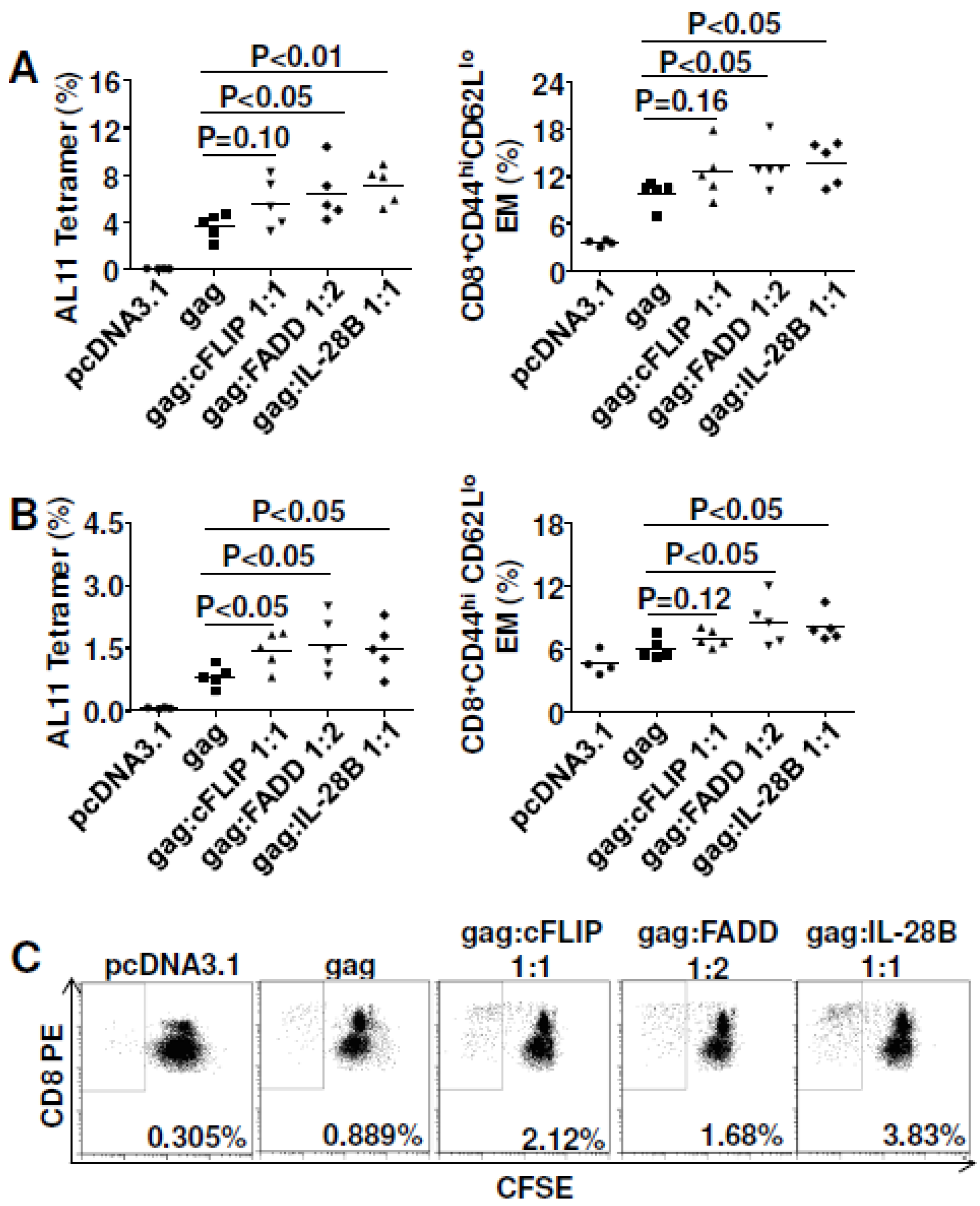
3.4. Pro-Apoptotic FADD Enhanced Cellular Immune Responses to Gag

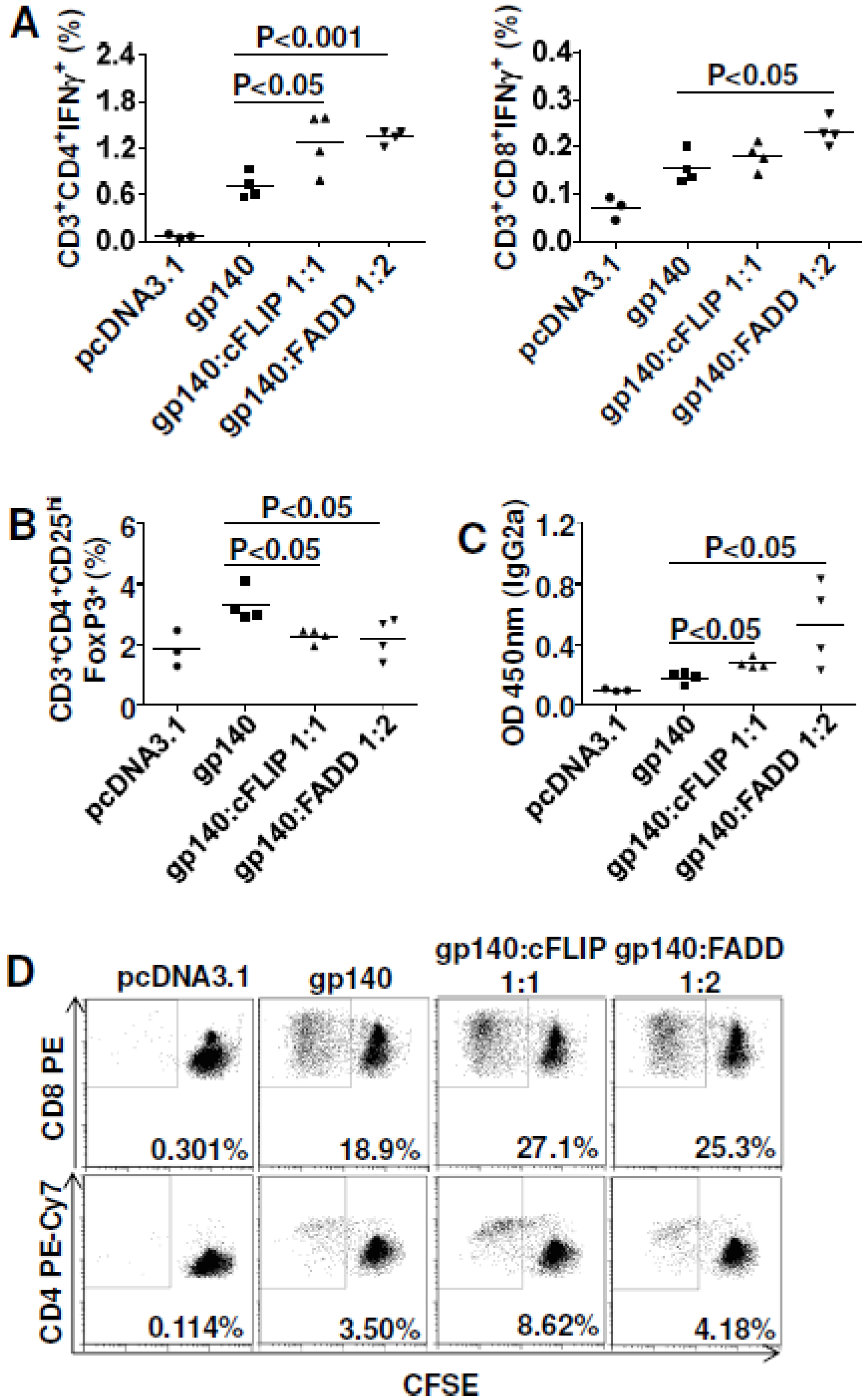
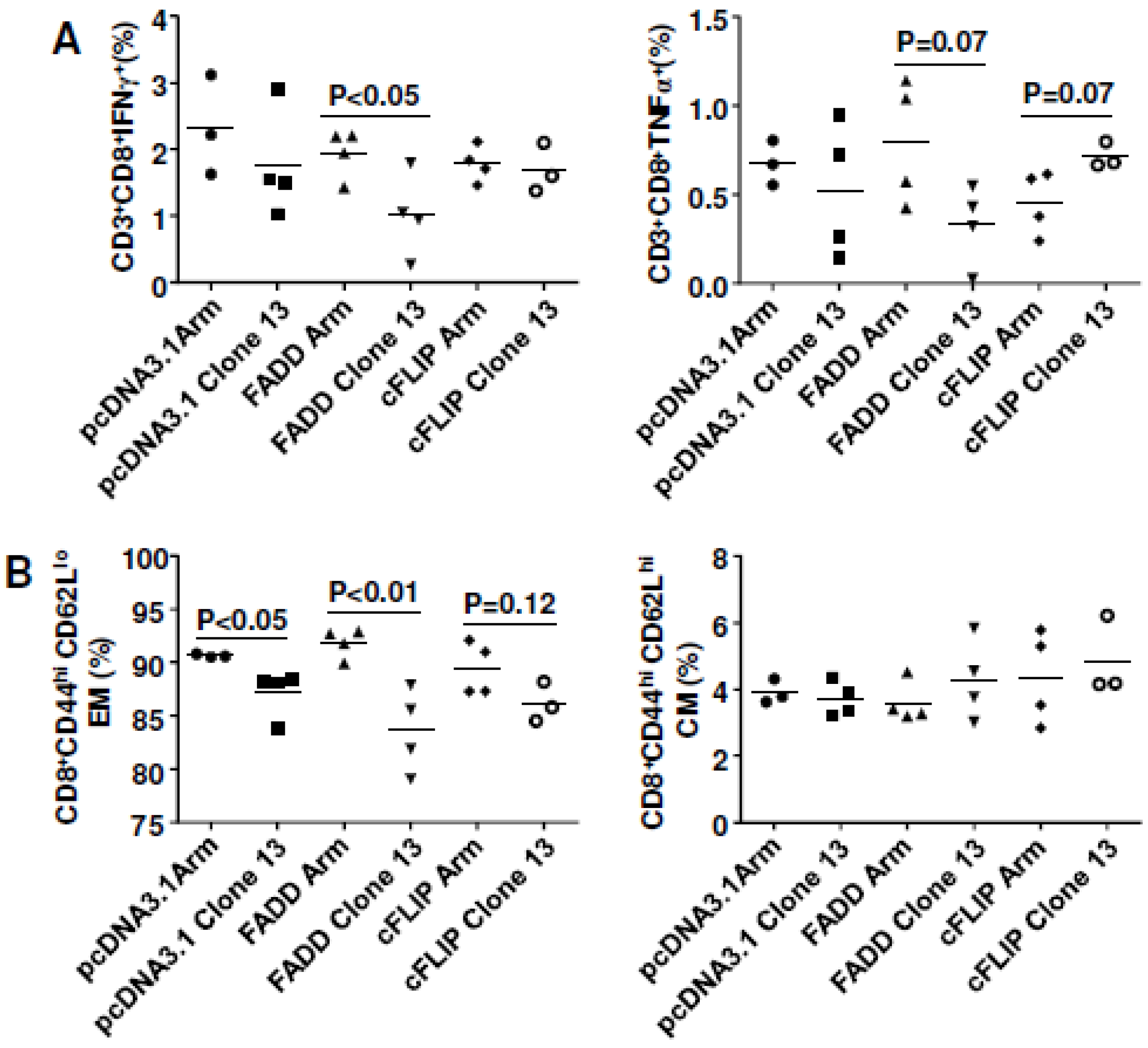
3.5. Both cFLIP and FADD Improved Vaccine Efficacy to HIVBaL gp140

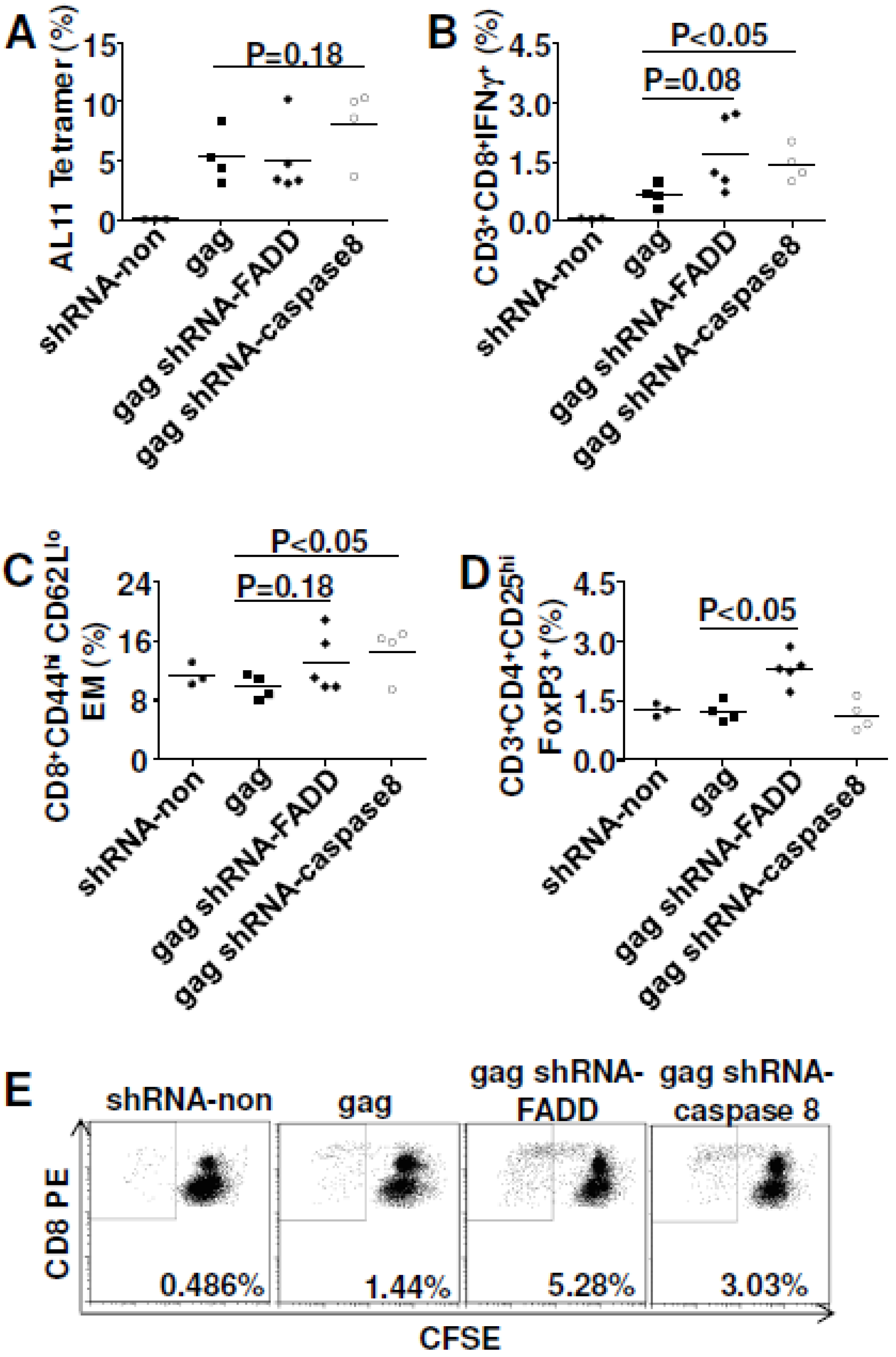
3.6. In Vivo Gene Silencing of Caspase 8 Enhanced DNA Vaccine Potency
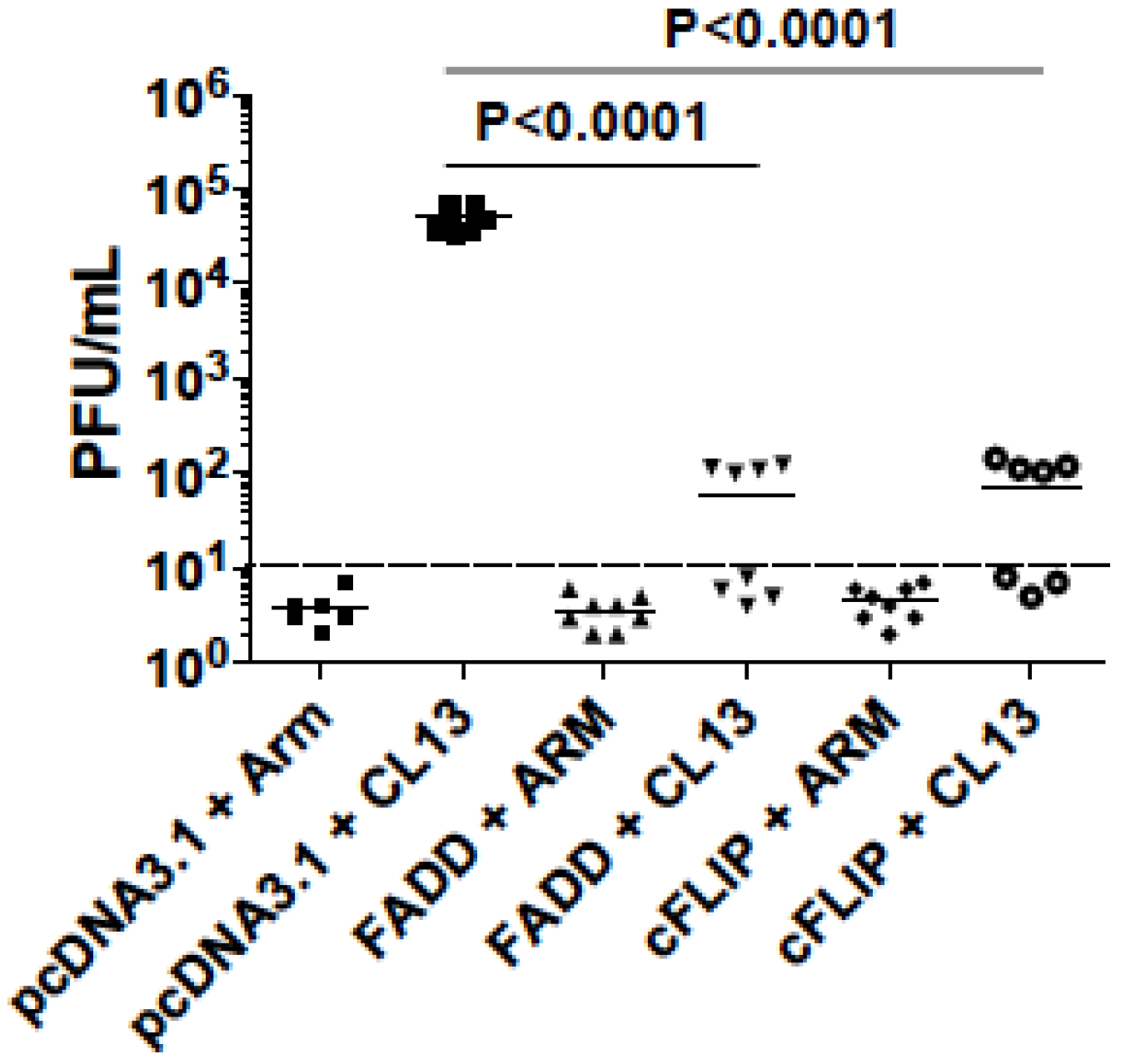
3.7. cFLIP and FADD Vaccination Helped Clear Chronic Virus LCMV-Clone13 in Mice



4. Discussion

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antigen | Plasmid-Expressed Molecules (Pro- or Anti-Apoptotic) | Ratio | Cellular Immune Responses (CMI) | |
|---|---|---|---|---|
| AL11 Tetramer | CD3CD8 IFNγ | |||
| SIVmac Gag | Fas (pro-) | 1:1 | no change | decrease |
| 1:2,1:3 | decrease | decrease | ||
| shRNA-Fas (anti-) | 1:1,1:4 | no change | decrease | |
| shRNA-FasL (anti-) | 1:1 | no change | decrease | |
| 1:4 | no change | no change | ||
| cFLIP ((anti-) | 1:1 | increase | increase | |
| FADD (pro-) | 1:2 | increase | increase | |
| shRNA-FADD (anti-) | 1:1 | no change | increase | |
| shRNA-caspase 8 (anti-) | 1:1 | increase | increase | |
| HIVBaL gp140 | cFLIP (anti-) | 1:1 | NA | increase |
| FADD (pro-) | 1:2 | NA | increase | |

Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ferraro, B.; Morrow, M.P.; Hutnick, N.A.; Shin, T.H.; Lucke, C.E.; Weiner, D.B. Clinical applications of DNA vaccines: Current progress. Clin. Infect. Dis. 2011, 53, 296–302. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.A. DNA vaccines: An historical perspective and view to the future. Immunol. Rev. 2011, 239, 62–84. [Google Scholar] [CrossRef] [PubMed]
- Rice, J.; Ottensmeier, C.H.; Stevenson, F.K. DNA vaccines: Precision tools for activating effective immunity against cancer. Nat. Rev. Cancer 2008, 8, 108–120. [Google Scholar] [CrossRef] [PubMed]
- Ishii, K.J.; Kawagoe, T.; Koyama, S.; Matsui, K.; Kumar, H.; Kawai, T.; Uematsu, S.; Takeuchi, O.; Takeshita, F.; Coban, C.; et al. TANK-binding kinase-1 delineates innate and adaptive immune responses to DNA vaccines. Nature 2008, 451, 725–729. [Google Scholar] [CrossRef] [PubMed]
- Sardesai, N.Y.; Weiner, D.B. Electroporation delivery of DNA vaccines: Prospects for success. Curr. Opin. Immunol. 2011, 23, 421–429. [Google Scholar] [CrossRef] [PubMed]
- Best, S.R.; Peng, S.; Juang, C.M.; Hung, C.F.; Hannaman, D.; Saunders, J.R.; Wu, T.C.; Pai, S.I. Administration of HPV DNA vaccine via electroporation elicits the strongest CD8+ T cell immune responses compared to intramuscular injection and intradermal gene gun delivery. Vaccine 2009, 27, 5450–5459. [Google Scholar] [CrossRef] [PubMed]
- Hirao, L.A.; Wu, L.; Satishchandran, A.; Khan, A.S.; Draghia-Akli, R.; Finnefrock, A.C.; Bett, A.J.; Betts, M.R.; Casimiro, D.R.; Sardesai, N.Y.; et al. Comparative analysis of immune responses induced by vaccination with SIV antigens by recombinant Ad5 vector or plasmid DNA in rhesus macaques. Mol. Ther. 2010, 18, 1568–1576. [Google Scholar] [CrossRef] [PubMed]
- Morrow, M.P.; Pankhong, P.; Laddy, D.J.; Schoenly, K.A.; Yan, J.; Cisper, N.; Weiner, D.B. Comparative ability of IL-12 and IL-28B to regulate Treg populations and enhance adaptive cellular immunity. Blood 2009, 113, 5868–5877. [Google Scholar] [CrossRef] [PubMed]
- Loudon, P.T.; Yager, E.J.; Lynch, D.T.; Narendran, A.; Stagnar, C.; Franchini, A.M.; Fuller, J.T.; White, P.A.; Nyuandi, J.; Wiley, C.A.; et al. GM-CSF increases mucosal and systemic immunogenicity of an H1N1 influenza DNA vaccine administered into the epidermis of non-human primates. PLoS One 2010, 5, e11021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sui, Y.; Zhu, Q.; Gagnon, S.; Dzutsev, A.; Terabe, M.; Vaccari, M.; Venzon, D.; Klinman, D.; Strober, W.; Kelsall, B.; et al. Innate and adaptive immune correlates of vaccine and adjuvant-induced control of mucosal transmission of SIV in macaques. Proc. Natl. Acad. Sci. USA 2010, 107, 9843–9848. [Google Scholar] [CrossRef] [PubMed]
- Poonia, B.; Pauza, C.D.; Salvato, M.S. Role of the Fas/FasL pathway in HIV or SIV disease. Retrovirology 2009, 6, 91. [Google Scholar] [CrossRef] [PubMed]
- Cummins, N.W.; Badley, A.D. Mechanisms of HIV-associated lymphocyte apoptosis: 2010. Cell Death Dis. 2010, 1, e99. [Google Scholar] [CrossRef] [PubMed]
- Greenland, J.R.; Geiben, R.; Ghosh, S.; Pastor, W.A.; Letvin, N.L. Plasmid DNA vaccine-elicited cellular immune responses limit in vivo vaccine antigen expression through Fas-mediated apoptosis. J. Immunol. 2007, 178, 5652–5658. [Google Scholar] [CrossRef] [PubMed]
- Geiben-Lynn, R.; Greenland, J.R.; Frimpong-Boateng, K.; van Rooijen, N.; Hovav, A.H.; Letvin, N.L. CD4+ T lymphocytes mediate in vivo clearance of plasmid DNA vaccine antigen expression and potentiate CD8+ T-cell immune responses. Blood 2008, 112, 4585–4590. [Google Scholar] [CrossRef] [PubMed]
- Leitner, W.W.; Restifo, N.P. DNA vaccines and apoptosis: To kill or not to kill? J. Clin. Invest. 2003, 112, 22–24. [Google Scholar] [CrossRef] [PubMed]
- Chattergoon, M.A.; Kim, J.J.; Yang, J.S.; Robinson, T.M.; Lee, D.J.; Dentchev, T.; Wilson, D.M.; Ayyavoo, V.; Weiner, D.B. Targeted antigen delivery to antigen-presenting cells including dendritic cells by engineered Fas-mediated apoptosis. Nat. Biotechnol. 2000, 18, 974–979. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, S.; Amara, R.R.; Oran, A.E.; Smith, J.M.; Robinson, H.L. Apoptosis-mediated enhancement of DNA-raised immune responses by mutant caspases. Nat. Biotechnol. 2001, 19, 543–547. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.W.; Hung, C.F.; Ling, M.; Juang, J.; He, L.; Hardwick, J.M.; Kumar, S.; Wu, T.C. Enhancing DNA vaccine potency by coadministration of DNA encoding antiapoptotic proteins. J. Clin. Invest. 2003, 112, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Hughes, P.D.; Belz, G.T.; Fortner, K.A.; Budd, R.C.; Strasser, A.; Bouillet, P. Apoptosis regulators Fas and Bim cooperate in shutdown of chronic immune responses and prevention of autoimmunity. Immunity 2008, 28, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Hutcheson, J.; Scatizzi, J.C.; Siddiqui, A.M.; Haines, G.K., 3rd; Wu, T.; Li, Q.Z.; Davis, L.S.; Mohan, C.; Perlman, H. Combined deficiency of proapoptotic regulators Bim and Fas results in the early onset of systemic autoimmunity. Immunity 2008, 28, 206–217. [Google Scholar] [CrossRef] [PubMed]
- Weant, A.E.; Michalek, R.D.; Khan, I.U.; Holbrook, B.C.; Willingham, M.C.; Grayson, J.M. Apoptosis regulators Bim and Fas function concurrently to control autoimmunity and CD8+ T cell contraction. Immunity 2008, 28, 218–230. [Google Scholar] [CrossRef] [PubMed]
- Strasser, A.; Jost, P.J.; Nagata, S. The many roles of FAS receptor signaling in the immune system. Immunity 2009, 30, 180–192. [Google Scholar] [CrossRef] [PubMed]
- Barouch, D.H.; Pau, M.G.; Custers, J.H.; Koudstaal, W.; Kostense, S.; Havenga, M.J.; Truitt, D.M.; Sumida, S.M.; Kishko, M.G.; Arthur, J.C.; et al. Immunogenicity of recombinant adenovirus serotype 35 vaccine in the presence of pre-existing anti-Ad5 immunity. J. Immunol. 2004, 172, 6290–6297. [Google Scholar] [CrossRef] [PubMed]
- Widera, G.; Austin, M.; Rabussay, D.; Goldbeck, C.; Barnett, S.W.; Chen, M.; Leung, L.; Otten, G.R.; Thudium, K.; Selby, M.J.; et al. Increased DNA vaccine delivery and immunogenicity by electroporation in vivo. J. Immunol. 2000, 164, 4635–4640. [Google Scholar] [CrossRef] [PubMed]
- Martin, R.M.; Brady, J.L.; Lew, A.M. The need for IgG2c specific antiserum when isotyping antibodies from C57BL/6 and NOD mice. J. Immunol. Methods 1998, 212, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.T.; Anderson, A.C.; Tan, W.G.; West, E.E.; Ha, S.J.; Araki, K.; Freeman, G.J.; Kuchroo, V.K.; Ahmed, R. Cooperation of Tim-3 and PD-1 in CD8 T-cell exhaustion during chronic viral infection. Proc. Natl. Acad. Sci. USA 2010, 107, 14733–14738. [Google Scholar] [CrossRef] [PubMed]
- Zapata, J.C.; Poonia, B.; Bryant, J.; Davis, H.; Ateh, E.; George, L.; Crasta, O.; Zhang, Y.; Slezak, T.; Jaing, C.; et al. An attenuated Lassa vaccine in SIV-infected rhesus macaques does not persist or cause arenavirus disease but does elicit Lassa virus-specific immunity. Virol. J. 2013, 10, 52. [Google Scholar] [CrossRef] [PubMed]
- Niedergang, F.; Dautry-Varsat, A.; Alcover, A. Peptide antigen or superantigen-induced down-regulation of TCRs involves both stimulated and unstimulated receptors. J. Immunol. 1997, 159, 1703–1710. [Google Scholar] [PubMed]
- Morrow, M.P.; Yan, J.; Pankhong, P.; Shedlock, D.J.; Lewis, M.G.; Talbott, K.; Toporovski, R.; Khan, A.S.; Sardesai, N.Y.; Weiner, D.B. IL-28B/IFN-lambda 3 drives granzyme B loading and significantly increases CTL killing activity in macaques. Mol. Ther. 2010, 18, 1714–1723. [Google Scholar] [CrossRef] [PubMed]
- De Souza, M.S.; Ratto-Kim, S.; Chuenarom, W.; Schuetz, A.; Chantakulkij, S.; Nuntapinit, B.; Valencia-Micolta, A.; Thelian, D.; Nitayaphan, S.; Pitisuttithum, P.; et al. The Thai phase III trial (RV144) vaccine regimen induces T cell responses that preferentially target epitopes within the V2 region of HIV-1 envelope. J. Immunol. 2012, 188, 5166–5176. [Google Scholar]
- Rerks-Ngarm, S.; Pitisuttithum, P.; Nitayaphan, S.; Kaewkungwal, J.; Chiu, J.; Paris, R.; Premsri, N.; Namwat, C.; de Souza, M.; Adams, E.; et al. Vaccination with ALVAC and AIDSVAX to prevent HIV-1 infection in Thailand. N. Engl. J. Med. 2009, 361, 2209–2220. [Google Scholar] [CrossRef] [PubMed]
- Hope, T.J. Moving ahead an HIV vaccine: To neutralize or not, a key HIV vaccine question. Nat. Med. 2011, 17, 1195–1197. [Google Scholar] [CrossRef] [PubMed]
- Padian, N.S.; McCoy, S.I.; Karim, S.S.; Hasen, N.; Kim, J.; Bartos, M.; Katabira, E.; Bertozzi, S.M.; Schwartlander, B.; Cohen, M.S. HIV prevention transformed: The new prevention research agenda. Lancet 2011, 378, 269–278. [Google Scholar] [CrossRef] [PubMed]
- Barber, D.L.; Wherry, E.J.; Masopust, D.; Zhu, B.; Allison, J.P.; Sharpe, A.H.; Freeman, G.J.; Ahmed, R. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature 2006, 439, 682–687. [Google Scholar] [CrossRef] [PubMed]
- Ying, H.; Zaks, T.Z.; Wang, R.F.; Irvine, K.R.; Kammula, U.S.; Marincola, F.M.; Leitner, W.W.; Restifo, N.P. Cancer therapy using a self-replicating RNA vaccine. Nat. Med. 1999, 5, 823–827. [Google Scholar] [CrossRef] [PubMed]
- Leitner, W.W.; Hwang, L.N.; Bergmann-Leitner, E.S.; Finkelstein, S.E.; Frank, S.; Restifo, N.P. Apoptosis is essential for the increased efficacy of alphaviral replicase-based DNA vaccines. Vaccine 2004, 22, 1537–1544. [Google Scholar] [CrossRef] [PubMed]
- Kojima, Y.; Jounai, N.; Takeshita, F.; Nakazawa, M.; Okuda, K.; Watabe, S.; Xin, K.Q.; Okuda, K. The degree of apoptosis as an immunostimulant for a DNA vaccine against HIV-1 infection. Vaccine 2007, 25, 438–445. [Google Scholar] [CrossRef] [PubMed]
- Villa-Morales, M.; Fernandez-Piqueras, J. Targeting the Fas/FasL signaling pathway in cancer therapy. Expert Opin. Ther. Targets 2012, 16, 85–101. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Mao, C.P.; Peng, S.; Hung, C.F.; Wu, T.C. RNA interference-mediated in vivo silencing of fas ligand as a strategy for the enhancement of DNA vaccine potency. Hum. Gene Ther. 2008, 19, 763–773. [Google Scholar] [CrossRef] [PubMed]
- Zuniga, E.; Motran, C.C.; Montes, C.L.; Yagita, H.; Gruppi, A. Trypanosoma cruzi infection selectively renders parasite-specific IgG+ B lymphocytes susceptible to Fas/Fas ligand-mediated fratricide. J. Immunol. 2002, 168, 3965–3973. [Google Scholar] [CrossRef] [PubMed]
- Allam, R.; Lawlor, K.E.; Yu, E.C.; Mildenhall, A.L.; Moujalled, D.M.; Lewis, R.S.; Ke, F.; Mason, K.D.; White, M.J.; Stacey, K.J.; Strasser, A.; et al. Mitochondrial apoptosis is dispensable for NLRP3 inflammasome activation but non-apoptotic caspase-8 is required for inflammasome priming. EMBO Rep. 2014, 15, 982–990. [Google Scholar] [CrossRef] [PubMed]
- Leverrier, S.; Salvesen, G.S.; Walsh, C.M. Enzymatically active single chain caspase-8 maintains T-cell survival during clonal expansion. Cell Death Differ. 2011, 18, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Hansen, S.G.; Ford, J.C.; Lewis, M.S.; Ventura, A.B.; Hughes, C.M.; Coyne-Johnson, L.; Whizin, N.; Oswald, K.; Shoemaker, R.; Swanson, T.; et al. Profound early control of highly pathogenic SIV by an effector memory T-cell vaccine. Nature 2011, 473, 523–527. [Google Scholar] [CrossRef] [PubMed]
- Elahi, S.; Dinges, W.L.; Lejarcegui, N.; Laing, K.J.; Collier, A.C.; Koelle, D.M.; McElrath, M.J.; Horton, H. Protective HIV-specific CD8+ T cells evade Treg cell suppression. Nat. Med. 2011, 17, 989–995. [Google Scholar] [CrossRef] [PubMed]
- del Pozo Balado Mdel, M.; Leal, M.; Mendez Lagares, G.; Mata, R.C.; Lopez-Cortes, L.F.; Viciana, P.; Pacheco, Y.M. Increased regulatory T cell counts in HIV-infected nonresponders to hepatitis B virus vaccine. J. Infect. Dis. 2010, 202, 362–369. [Google Scholar]
- Kursar, M.; Bonhagen, K.; Fensterle, J.; Kohler, A.; Hurwitz, R.; Kamradt, T.; Kaufmann, S.H.; Mittrucker, H.W. Regulatory CD4+CD25+ T cells restrict memory CD8+ T cell responses. J. Exp. Med. 2002, 196, 1585–1592. [Google Scholar] [CrossRef] [PubMed]
- Pandiyan, P.; Zheng, L.; Ishihara, S.; Reed, J.; Lenardo, M.J. CD4+CD25+Foxp3+ regulatory T cells induce cytokine deprivation-mediated apoptosis of effector CD4+ T cells. Nat. Immunol. 2007, 8, 1353–1362. [Google Scholar] [CrossRef] [PubMed]
- Seino, K.; Iwabuchi, K.; Kayagaki, N.; Miyata, R.; Nagaoka, I.; Matsuzawa, A.; Fukao, K.; Yagita, H.; Okumura, K. Chemotactic activity of soluble Fas ligand against phagocytes. J. Immunol. 1998, 161, 4484–4488. [Google Scholar] [PubMed]
- Coffman, R.L.; Sher, A.; Seder, R.A. Vaccine adjuvants: Putting innate immunity to work. Immunity 2010, 33, 492–503. [Google Scholar] [CrossRef] [PubMed]
- Penaloza-MacMaster, P.; Kamphorst, A.O.; Wieland, A.; Araki, K.; Iyer, S.S.; West, E.E.; O’Mara, L.; Yang, S.; Konieczny, B.T.; Sharpe, A.H.; et al. Interplay between regulatory T cells and PD-1 in modulating T cell exhaustion and viral control during chronic LCMV infection. J. Exp. Med. 2014, 211, 1905–1918. [Google Scholar] [CrossRef] [PubMed]
- Salvato, M.; Borrow, P.; Shimomaye, E.; Oldstone, M.B. Molecular basis of viral persistence: A single amino acid change in the glycoprotein of lymphocytic choriomeningitis virus is associated with suppression of the antiviral cytotoxic T-lymphocyte response and establishment of persistence. J. Virol. 1991, 65, 1863–1869. [Google Scholar] [PubMed]
- Teijaro, J.R.; Ng, C.; Lee, A.M.; Sullivan, B.M.; Sheehan, K.C.; Welch, M.; Schreiber, R.D.; de la Torre, J.C.; Oldstone, M.B. Persistent LCMV infection is controlled by blockade of type I interferon signaling. Science 2013, 340, 207–211. [Google Scholar] [CrossRef] [PubMed]
- Wilson, E.B.; Yamada, D.H.; Elsaesser, H.; Herskovitz, J.; Deng, J.; Cheng, G.; Aronow, B.J.; Karp, C.L.; Brooks, D.G. Blockade of chronic type I interferon signaling to control persistent LCMV infection. Science 2013, 340, 202–207. [Google Scholar] [CrossRef] [PubMed]
- Kutzler, M.A.; Weiner, D.B. DNA vaccines: Ready for prime time? Nat. Rev. Genet. 2008, 9, 776–788. [Google Scholar] [CrossRef] [PubMed]
- Tourneur, L.; Chiocchia, G. FADD: A regulator of life and death. Trends Immunol. 2010, 31, 260–269. [Google Scholar] [CrossRef] [PubMed]
- Dillon, C.P.; Oberst, A.; Weinlich, R.; Janke, L.J.; Kang, T.B.; Ben-Moshe, T.; Mak, T.W.; Wallach, D.; Green, D.R. Survival function of the FADD-CASPASE-8-cFLIP(L) complex. Cell Rep. 2012, 1, 401–407. [Google Scholar] [CrossRef] [PubMed]
- Grunert, M.; Gottschalk, K.; Kapahnke, J.; Gundisch, S.; Kieser, A.; Jeremias, I. The adaptor protein FADD and the initiator caspase-8 mediate activation of NF-kappaB by TRAIL. Cell Death Dis. 2012, 3, e414. [Google Scholar] [CrossRef] [PubMed]
- Kreuz, S.; Siegmund, D.; Rumpf, J.J.; Samel, D.; Leverkus, M.; Janssen, O.; Hacker, G.; Dittrich-Breiholz, O.; Kracht, M.; Scheurich, P.; et al. NFkappaB activation by Fas is mediated through FADD, caspase-8, and RIP and is inhibited by FLIP. J. Cell Biol. 2004, 166, 369–380. [Google Scholar] [CrossRef] [PubMed]
- Kalams, S.A.; Parker, S.; Jin, X.; Elizaga, M.; Metch, B.; Wang, M.; Hural, J.; Lubeck, M.; Eldridge, J.; Cardinali, M.; et al. Safety and immunogenicity of an HIV-1 gag DNA vaccine with or without IL-12 and/or IL-15 plasmid cytokine adjuvant in healthy, HIV-1 uninfected adults. PLoS One 2012, 7, e29231. [Google Scholar] [CrossRef] [PubMed]
- Zou, W. Regulatory T cells, tumour immunity and immunotherapy. Nat. Rev. Immunol. 2006, 6, 295–307. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yan, J.; Zapata, J.C.; Pauza, C.D.; Salvato, M.S. Modulation of SIV and HIV DNA Vaccine Immunity by Fas-FasL Signaling. Viruses 2015, 7, 1429-1453. https://0-doi-org.brum.beds.ac.uk/10.3390/v7031429
Yan J, Zapata JC, Pauza CD, Salvato MS. Modulation of SIV and HIV DNA Vaccine Immunity by Fas-FasL Signaling. Viruses. 2015; 7(3):1429-1453. https://0-doi-org.brum.beds.ac.uk/10.3390/v7031429
Chicago/Turabian StyleYan, Jiabin, Juan Carlos Zapata, Charles David Pauza, and Maria S. Salvato. 2015. "Modulation of SIV and HIV DNA Vaccine Immunity by Fas-FasL Signaling" Viruses 7, no. 3: 1429-1453. https://0-doi-org.brum.beds.ac.uk/10.3390/v7031429