
A Piscine Birnavirus Induces Inhibition of Protein Synthesis in CHSE-214 Cells Primarily through the Induction of eIF2α Phosphorylation

Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Virus Propagation
2.3. Virus Infection and Metabolic Labeling
2.4. Western Blot Analysis
2.5. Cell Viability Assay
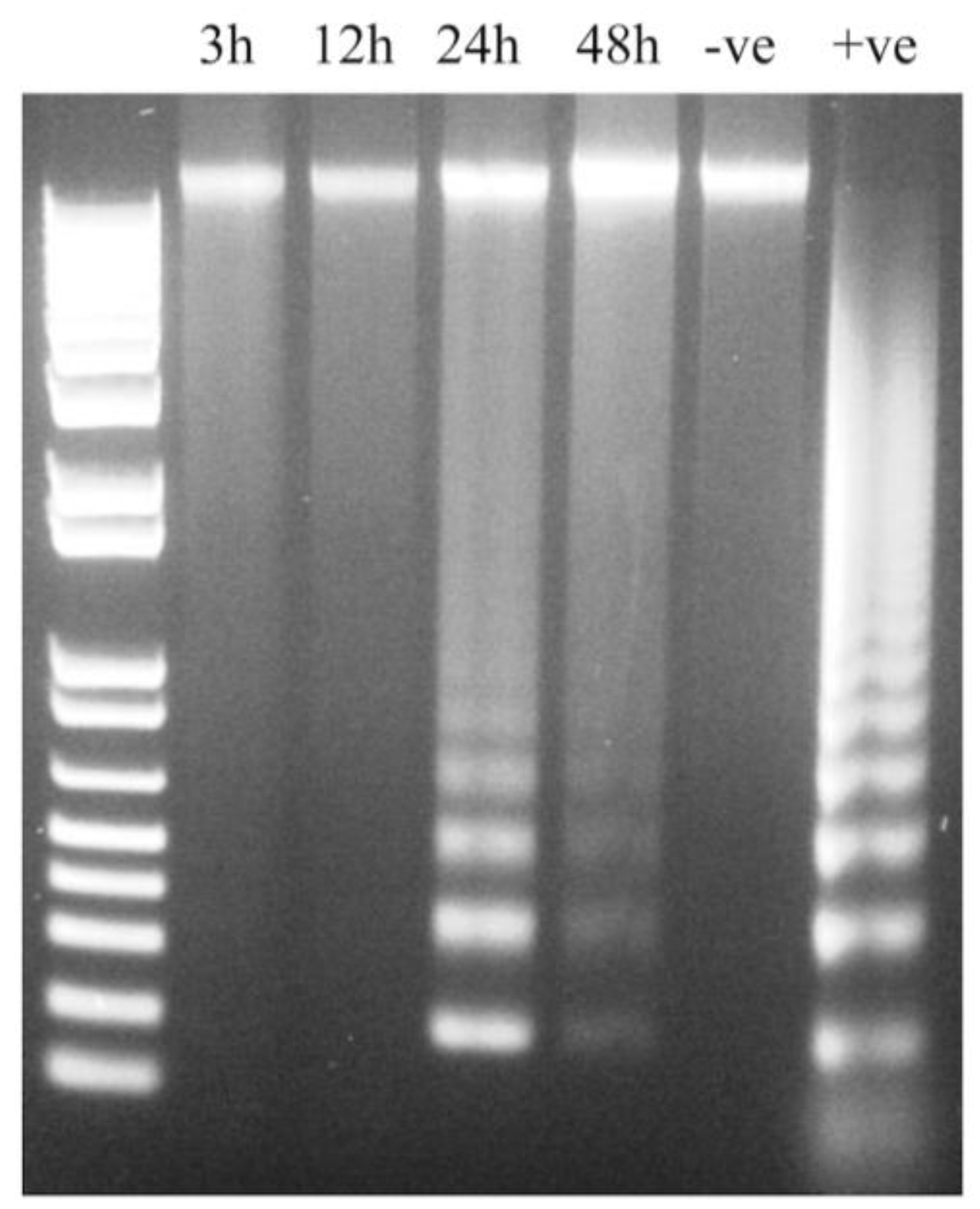
2.6. Assessment of Apoptosis by DNA Fragmentation
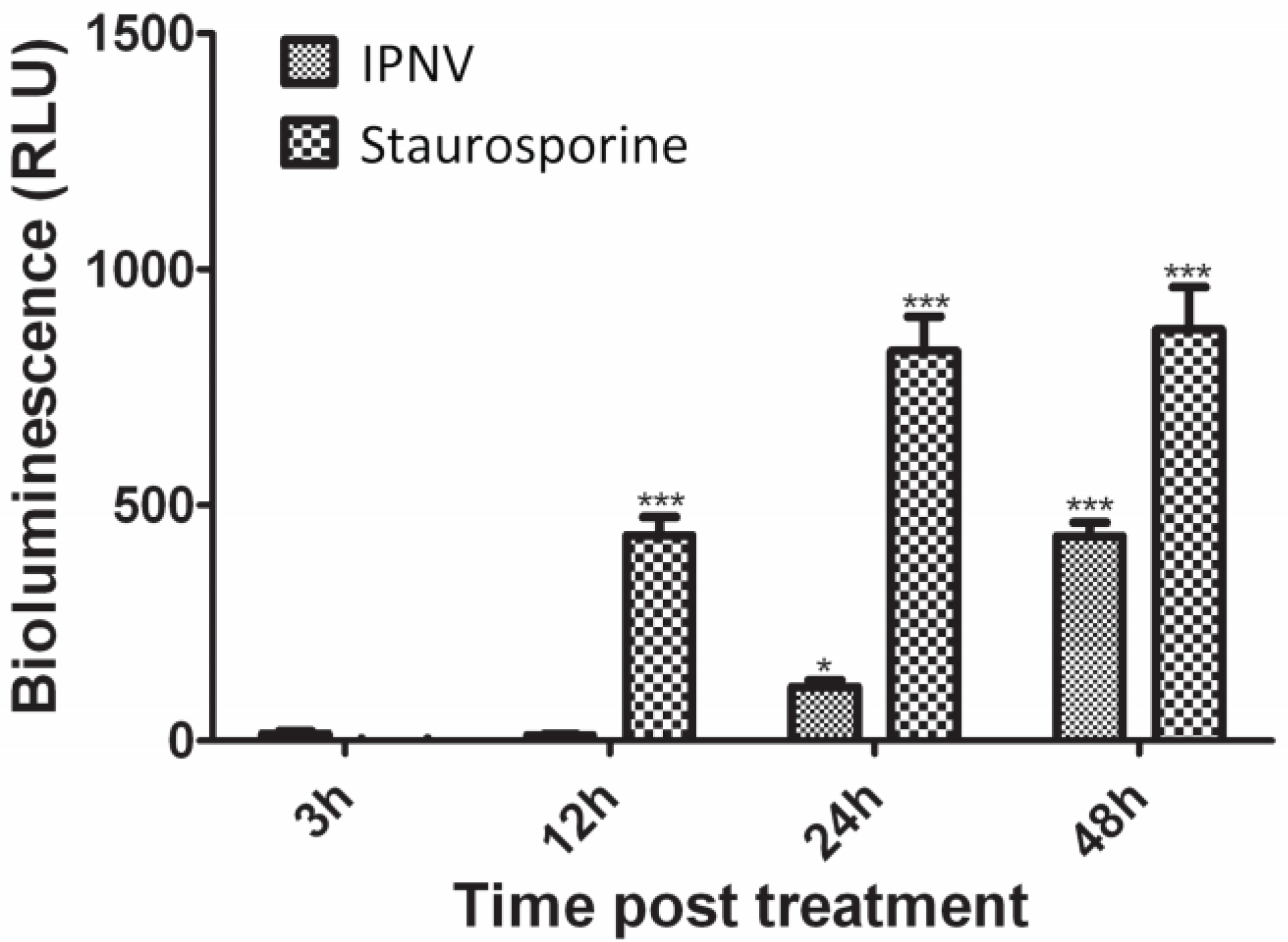
2.7. Caspase 3 Assay
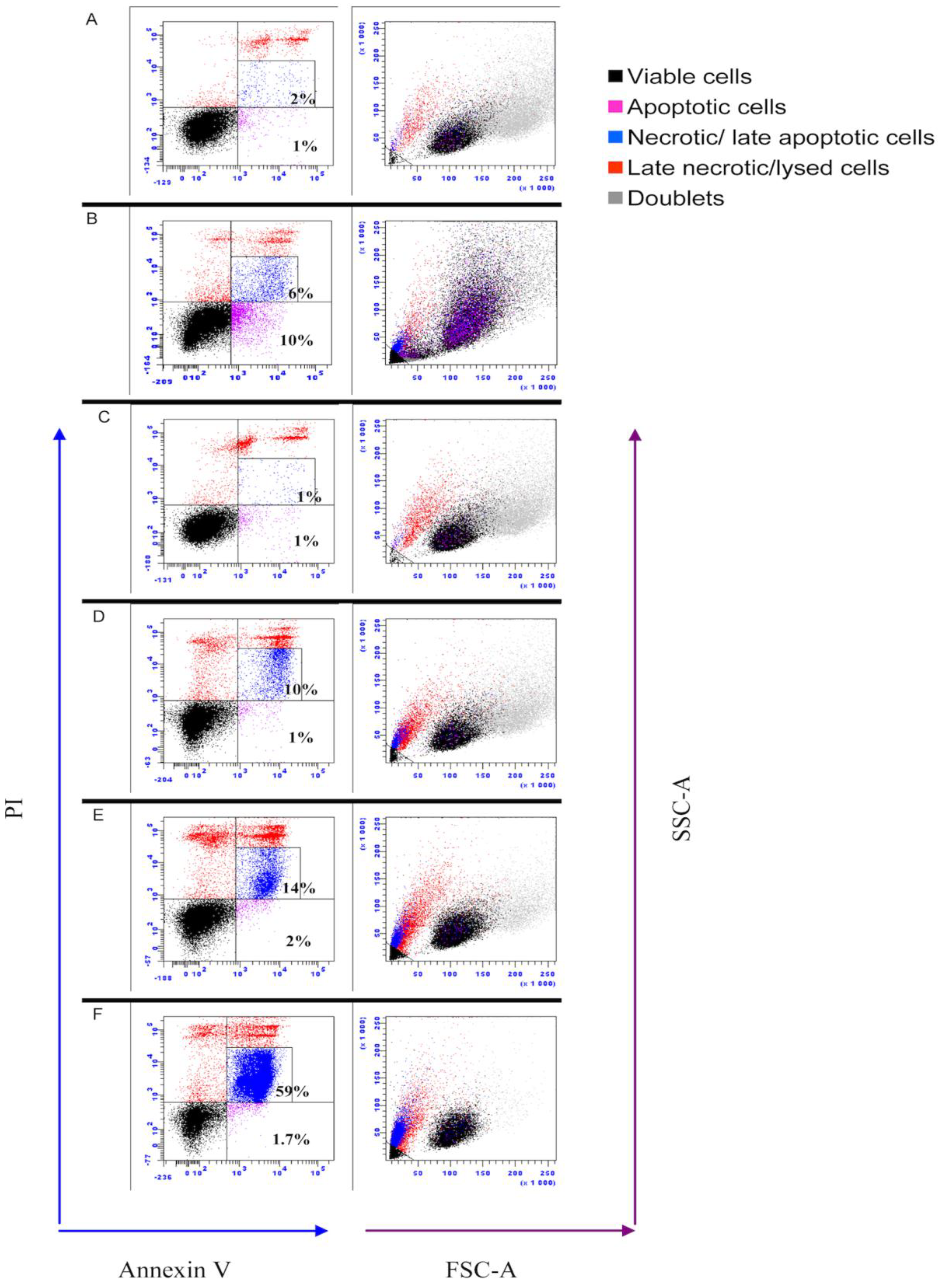
2.8. Assessment of Apoptosis by Flow Cytometry
2.9. Quantitative Real-Time PCR Analysis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Primer sequence (5’-3’) | Genbank accession no. |
|---|---|---|
| Mx-Forward Mx-Reverse | TGCAACCACAGAGGCTTTGAA GGCTTGGTCAGGATGCCTAAT | U66475 |
| B actin Forward B actin Reverse | CCAGTCCTGCTCACTGAGGC GGTCTCAAACATGATCTGGGTCA | AF012125 |
| PKR Forward PKR Reverse | TGAACACAGCCAGAAGAACAA GACTACCGCCACATAACTCCA | EF523422.1 |
| IPNv Forward IPNv Reverse | CAACAGGGTTCGACAAACCATAC TTGACGATGTCGGCGTTTC | |
| IFNα Forward IFNα Reverse | TGGGAGGAGATATCACAAAGC TCCCAGGTGACAGATTTCAT | NM_001123570.1 |
2.10. Statistical Analysis
3. Results
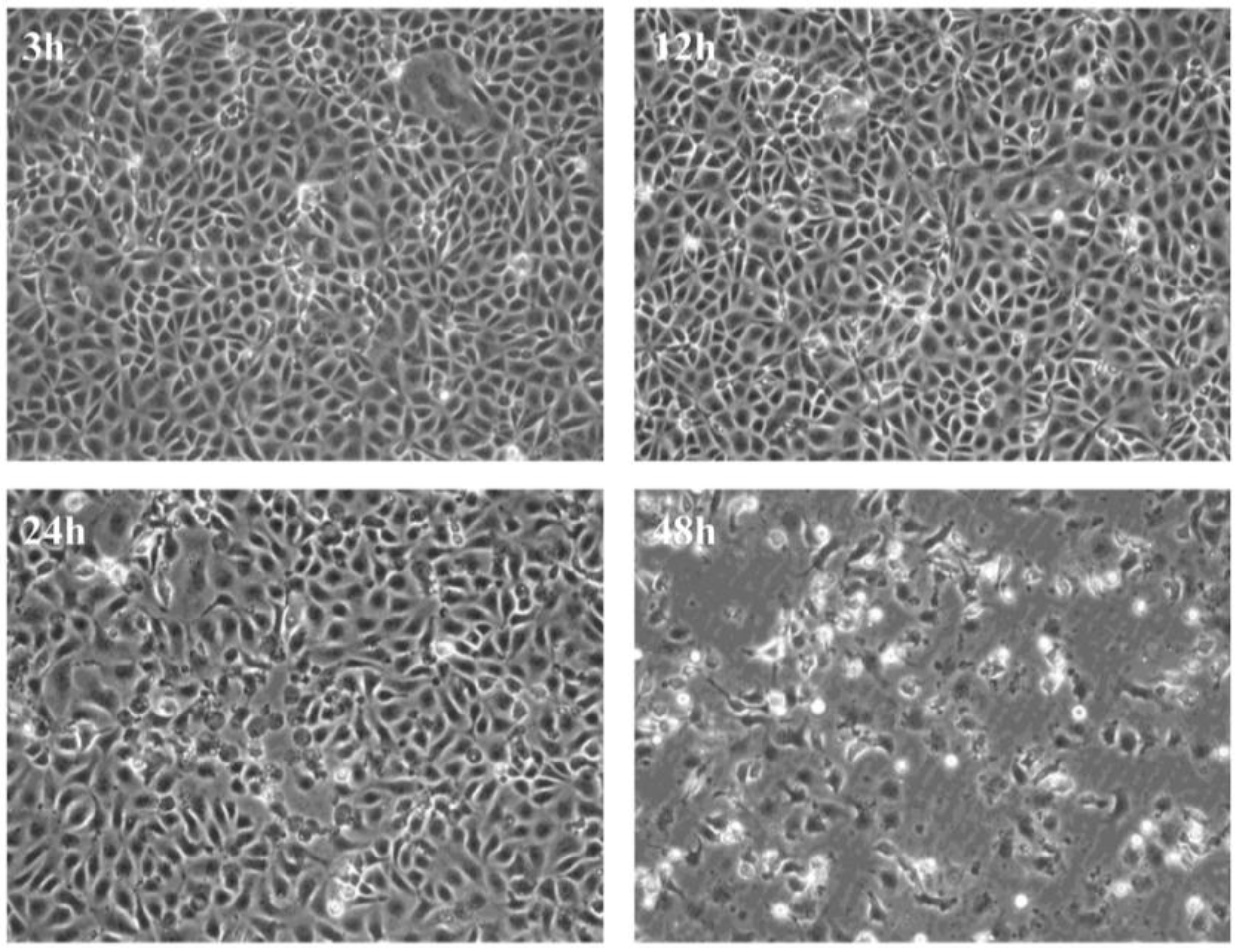
3.1. Virus Replication in CHSE-214 Cells

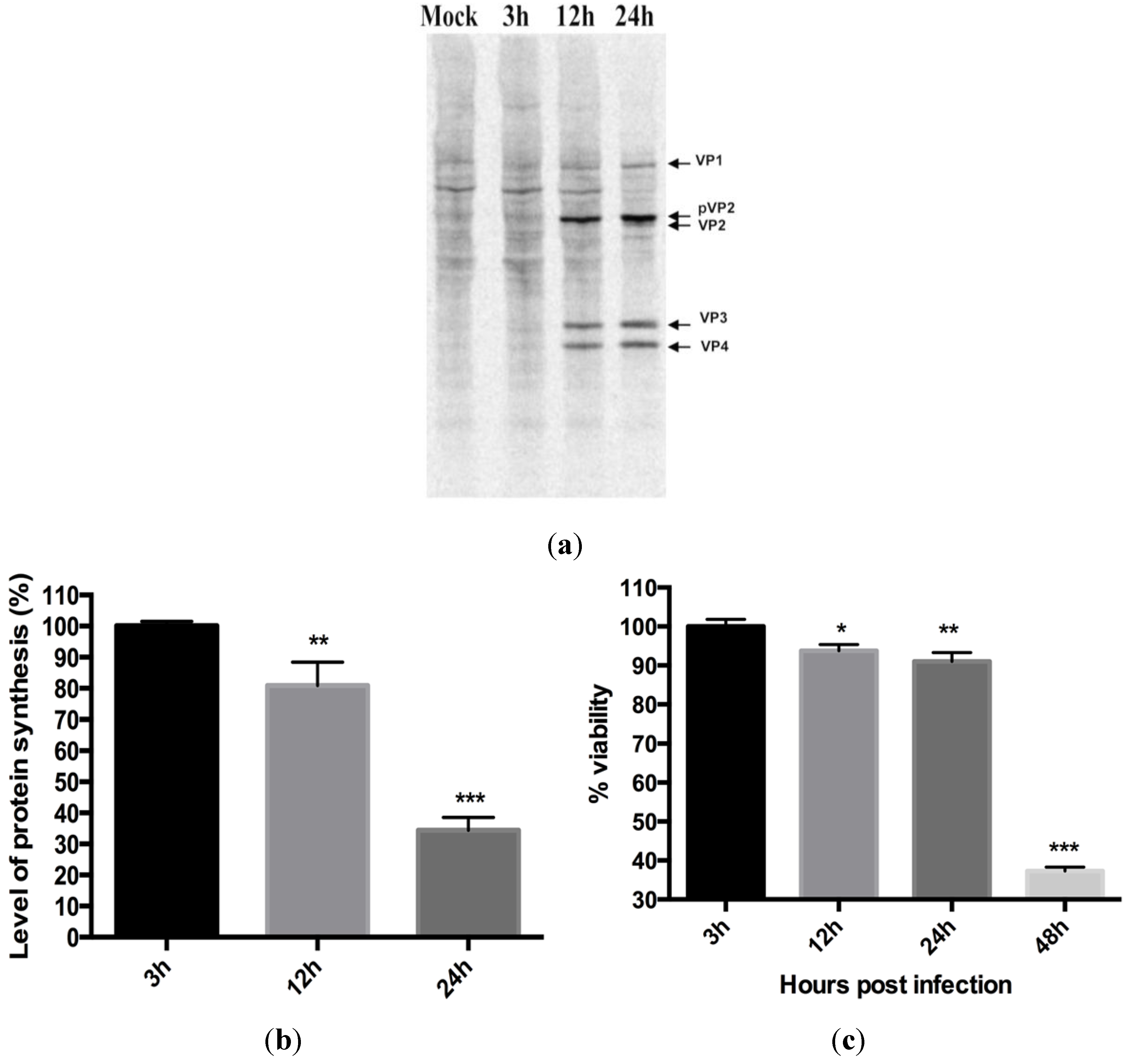
3.2. Virus Replication Results in Inhibition of Protein Synthesis
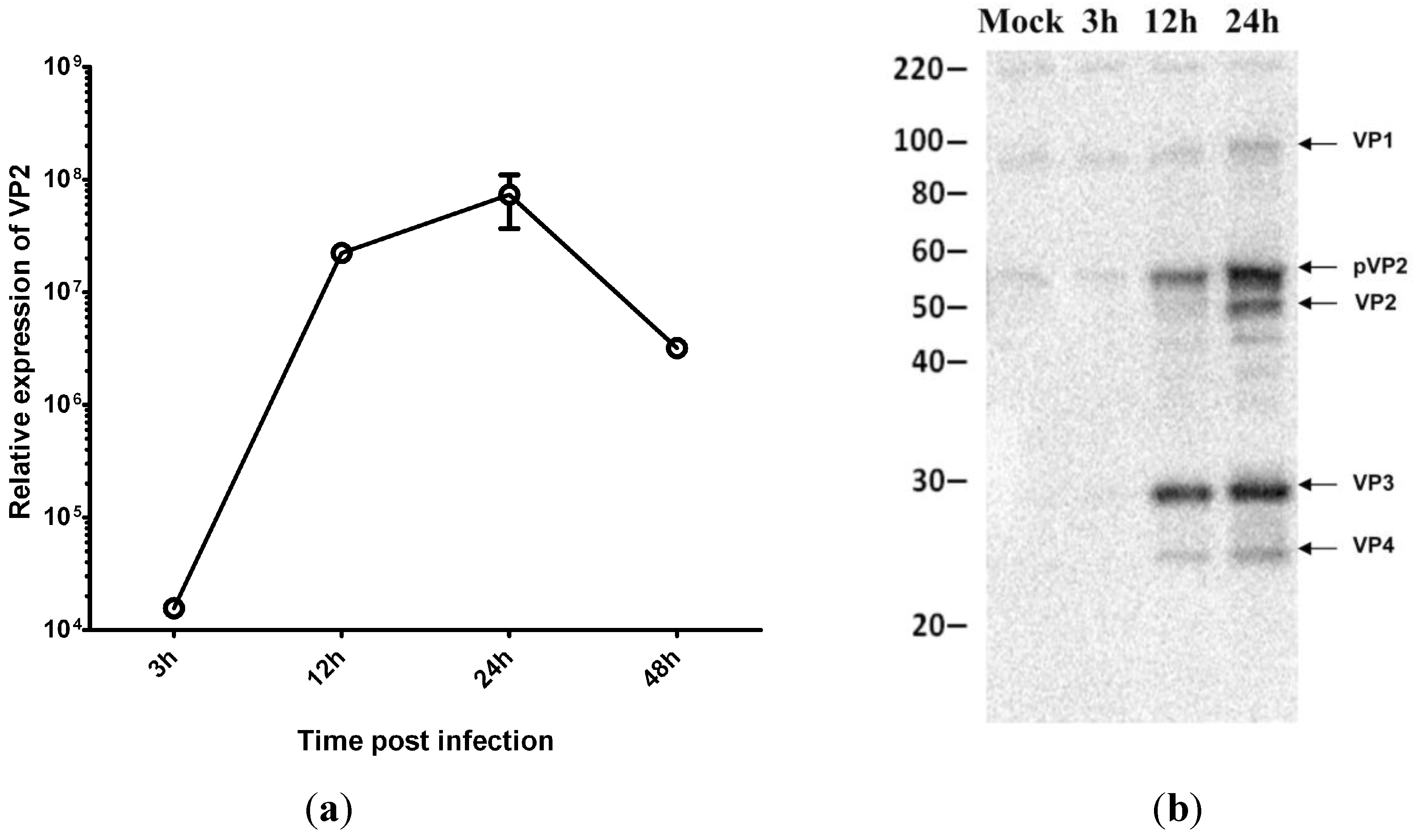
3.3. Virus Replication Increases despite Inhibition of Host Protein Synthesis at Early Time Post Infection


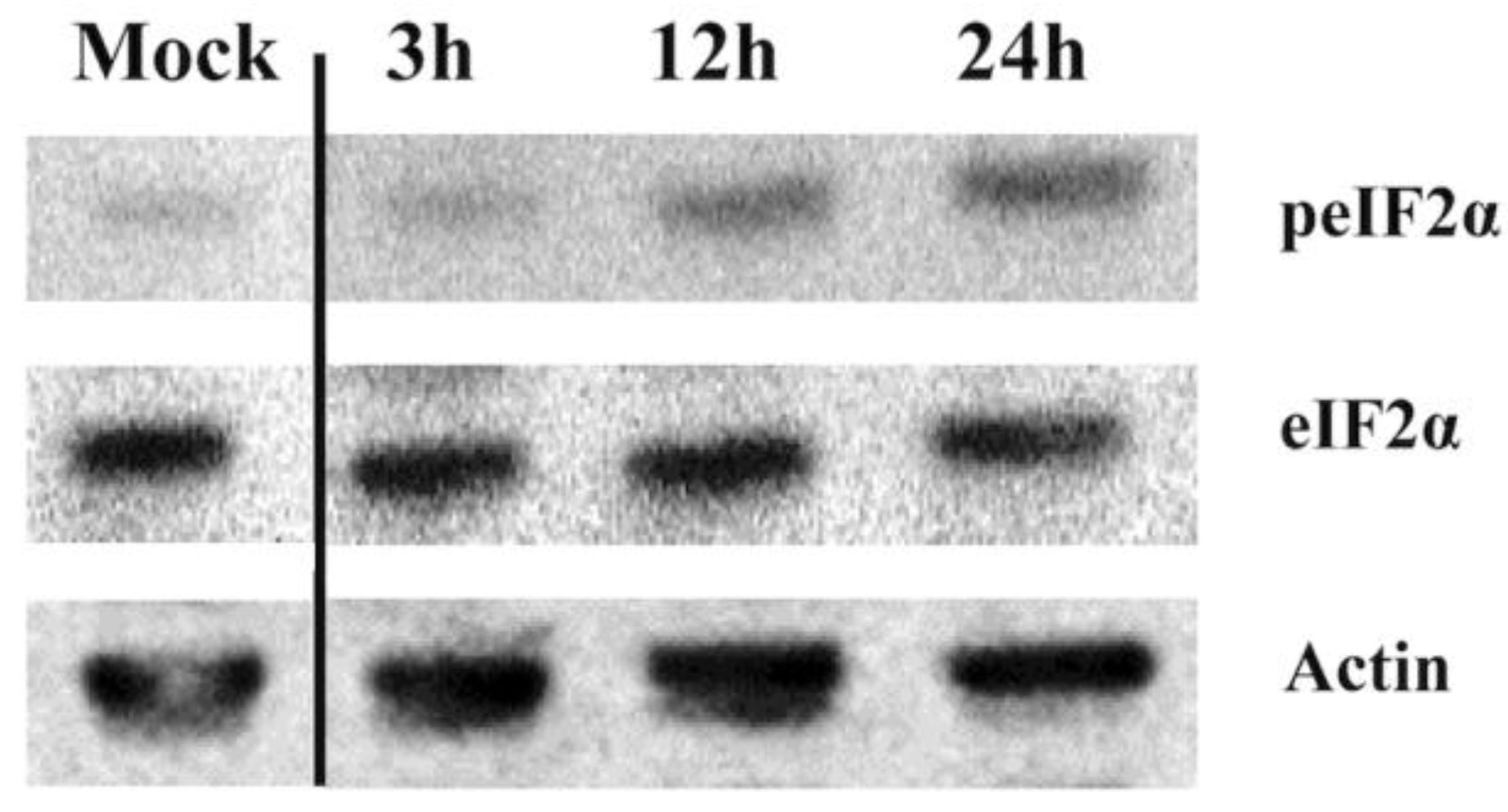
3.4. IPNV Infection of CHSE Cells Results in eIF2α Phosphorylation

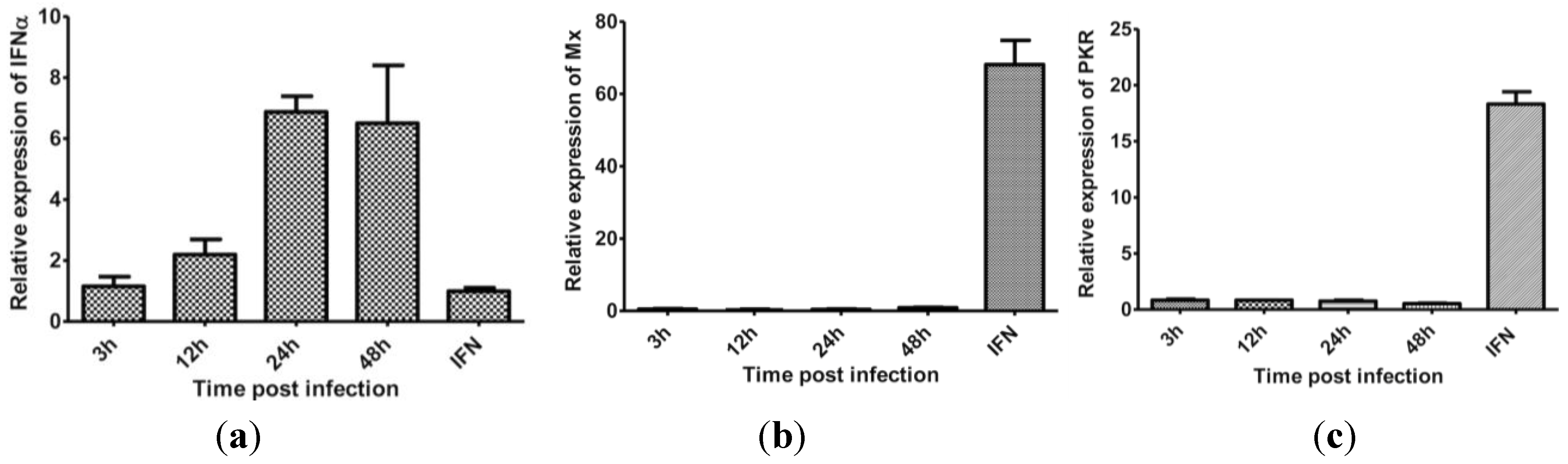
3.5. The Effect of Protein Synthesis Inhibition on Type I IFN Response

3.6. IPNV Induces More Necrosis than Apoptosis at Early Time Post Infection



4. Discussion
Acknowledgement
Author Contributions
Conflict of Interest
References
- Su, Q.Z.; Wang, S.; Baltzis, D.; Qu, L.K.; Raven, J.F.; Li, S.Y.; Wong, A.H.T.; Koromilas, A.E. Interferons induce tyrosine phosphorylation of the eIF2 alpha kinase PKR through activation of Jak1 and Tyk2. EMBO Rep. 2007, 8, 265–270. [Google Scholar] [CrossRef] [PubMed]
- Asano, K.; Clayton, J.; Shalev, A.; Hinnebusch, A.G. A multifactor complex of eukaryotic initiation factors, eIE1, eIF2, eIF3, eIF5, and initiator tRNA(Met) is an important translation initiation intermediate in vivo. Genes Dev. 2000, 14, 2534–2546. [Google Scholar] [CrossRef] [PubMed]
- Medigeshi, G.R.; Lancaster, A.M.; Hirsch, A.J.; Briese, T.; Lipkin, W.I.; Defilippis, V.; Fruh, K.; Mason, P.W.; Nikolich-Zugich, J.; Nelson, J.A. West Nile virus infection activates the unfolded protein response, leading to CHOP induction and apoptosis. J Virol. 2007, 81, 10849–10860. [Google Scholar] [CrossRef] [PubMed]
- Barber, G.N. Host defense, viruses and apoptosis. Cell Death Differ. 2001, 8, 113–126. [Google Scholar] [CrossRef] [PubMed]
- Marissen, W.E.; Lloyd, R.E. Eukaryotic translation initiation factor 4G is targeted for proteolytic cleavage by caspase 3 during inhibition of translation in apoptotic cells. Mol. Cell. Biol. 1998, 18, 7565–7574. [Google Scholar] [PubMed]
- Gradi, A.; Svitkin, Y.V.; Imataka, H.; Sonenberg, N. Proteolysis of human eukaryotic translation initiation factor eIF4GII, but not eIF4GI, coincides with the shutoff of host protein synthesis after poliovirus infection. Proc. Natl. Acad. Sci. USA 1998, 95, 11089–11094. [Google Scholar] [CrossRef] [PubMed]
- Joachims, M.; van Breugel, P.C.; Lloyd, R.E. Cleavage of poly(A)-binding protein by enterovirus proteases concurrent with inhibition of translation in vitro. J. Virol. 1999, 73, 718–727. [Google Scholar] [PubMed]
- Vende, P.; Piron, M.; Castagne, N.; Poncet, D. Efficient translation of rotavirus mRNA requires simultaneous interaction of NSP3 with the eukaryotic translation initiation factor eIF4G and the mRNA 3' end. J. Virol. 2000, 74, 7064–7071. [Google Scholar] [CrossRef] [PubMed]
- Piron, M.; Vende, P.; Cohen, J.; Poncet, D. Rotavirus RNA-binding protein NSP3 interacts with eIF4GI and evicts the poly(A) binding protein from eIF4F. EMBO J. 1998, 17, 5811–5821. [Google Scholar] [CrossRef] [PubMed]
- Etchison, D.; Milburn, S.C.; Edery, I.; Sonenberg, N.; Hershey, J.W.B. Inhibition of Hela-Cell Protein-Synthesis Following Poliovirus Infection Correlates with the Proteolysis of A 220,000-Dalton Polypeptide Associated with Eukaryotic Initiation Factor-Iii and A Cap Binding-Protein Complex. J. Biol. Chem. 1982, 257, 4806–4810. [Google Scholar] [PubMed]
- Khoo, D.; Perez, C.; Mohr, I. Characterization of RNA determinants recognized by the arginine- and proline-rich region of Us11, a herpes simplex virus type 1-encoded double-stranded RNA binding protein that prevents PKR activation. J. Virol. 2002, 76, 11971–11981. [Google Scholar] [CrossRef] [PubMed]
- Spurgeon, M.E.; Ornelles, D.A. The Adenovirus E1B 55-Kilodalton and E4 Open Reading Frame 6 Proteins Limit Phosphorylation of eIF2 alpha during the Late Phase of Infection. J. Virol. 2009, 83, 9970–9982. [Google Scholar] [CrossRef] [PubMed]
- Dobos, P. The molecular biology of infectious pancreatic necrosis virus (IPNV). Annu. Rev. Fish Dis. 1995, 5, 25–54. [Google Scholar] [CrossRef]
- Havarstein, L.S.; Kalland, K.H.; Christie, K.E.; Endresen, C. Sequence of the Large Double-Stranded-RNA Segment of the N1 Strain of Infectious Pancreatic Necrosis Virus—A Comparison with Other Birnaviridae. J. Gen. Virol. 1990, 71, 299–308. [Google Scholar] [CrossRef] [PubMed]
- Magyar, G.; Dobos, P. Evidence for the Detection of the Infectious Pancreatic Necrosis Virus Polyprotein and the 17-Kda Polypeptide in Infected-Cells and of the Ns Protease in Purified Virus. Virology 1994, 204, 580–589. [Google Scholar] [CrossRef] [PubMed]
- Duncan, R.; Nagy, E.; Krell, P.J.; Dobos, P. Synthesis of the Infectious Pancreatic Necrosis Virus Polyprotein, Detection of A Virus-Encoded Protease, and Fine-Structure Mapping of Genome Segment-A Coding Regions. J. Virol. 1987, 61, 3655–3664. [Google Scholar] [PubMed]
- Macdonald, R.D.; Dobos, P. Identification of the Proteins Encoded by Each Genome Segment of Infectious Pancreatic Necrosis Virus. Virology 1981, 114, 414–422. [Google Scholar] [CrossRef] [PubMed]
- Duncan, R.; Mason, C.L.; Nagy, E.; Leong, J.A.; Dobos, P. Sequence-Analysis of Infectious Pancreatic Necrosis Virus Genome Segment-B and Its Encoded VP1 Protein—A Putative RNA-Dependent RNA-Polymerase Lacking the Gly Asp Asp Motif. Virology 1991, 181, 541–552. [Google Scholar] [CrossRef] [PubMed]
- Lothrop, D.; Nicholson, B.L. Inhibition of Cellular DNA-Synthesis in Cells Infected with Infectious Pancreatic Necrosis Virus. J. Virol. 1974, 14, 485–492. [Google Scholar] [PubMed]
- Nicholson, B.L. Macromolecule Synthesis in RTG-2 Cells Following Infection with Infectiouus Pancreatic Necrosis (IPN) Virus. J. Gen. Virol. 1971, 13, 369–372. [Google Scholar] [CrossRef] [PubMed]
- Garner, J.N.; Joshi, B.; Jagus, R. Characterization of rainbow trout and zebrafish eukaryotic initiation factor 2 alpha and its response to endoplasmic reticulum stress and IPNV infection. Dev. Comp. Immunol. 2003, 27, 217–231. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Evensen, Ø.; Mutoloki, S. Delayed protein shut down and cytopathic changes lead to high yields of infectious pancreatic necrosis virus cultured in Asian Grouper cells. J. Virol. Methods 2014, 195, 228–235. [Google Scholar] [CrossRef] [PubMed]
- Lannan, C.N.; Winton, J.R.; Fryer, J.L. Fish cell lines: Establishment and characterization of nine cell lines from salmonids. In Vitro 1984, 20, 671–676. [Google Scholar] [CrossRef] [PubMed]
- Munang'andu, H.M.; Fredriksen, B.N.; Mutoloki, S.; Brudeseth, B.; Kuo, T.Y.; Marjara, I.S.; Dalmo, R.A.; Evensen, O. Comparison of vaccine efficacy for different antigen delivery systems for infectious pancreatic necrosis virus vaccines in Atlantic salmon (Salmo salar L.) in a cohabitation challenge model. Vaccine 2012, 30, 4007–4016. [Google Scholar] [CrossRef] [PubMed]
- Song, H.C.; Santi, N.; Evensen, O.; Vakharia, V.N. Molecular determinants of infectious pancreatic necrosis virus virulence and cell culture adaptation. J. Virol. 2005, 79, 10289–10299. [Google Scholar] [CrossRef] [PubMed]
- Evensen, O.; Rimstad, E. Immunohistochemical identification of infectious pancreatic necrosis virus in paraffin-embedded tissues of Atlantic salmon (Salmo salar). J. Vet. Diagn. Invest 1990, 2, 288–293. [Google Scholar] [CrossRef]
- Xu, C.; Guo, T.C.; Mutoloki, S.; Haugland, O.; Marjara, I.S.; Evensen, O. Alpha Interferon and Not Gamma Interferon Inhibits Salmonid Alphavirus Subtype 3 Replication In Vitro. J. Virol. 2010, 84, 8903–8912. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.R.; Lin, T.L.; Hsu, Y.L.; Wu, J.L. Apoptosis precedes necrosis of fish cell line with infectious pancreatic necrosis virus infection. Virology 1998, 250, 76–84. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.; Saikumar, P.; Weinberg, J.M.; Venkatachalam, M.A. Internucleosomal DNA cleavage triggered by plasma membrane damage during necrotic cell death—Involvement of serine but not cysteine proteases. Am. J. Pathol. 1997, 151, 1205–1213. [Google Scholar] [PubMed]
- Didenko, V.V.; Ngo, H.; Baskin, D.S. Early necrotic DNA degradation—Presence of blunt-ended DNA breaks, 3' and 5' overhangs in apoptosis, but only 5' overhangs in early necrosis. Am. J. Pathol. 2003, 162, 1571–1578. [Google Scholar] [CrossRef] [PubMed]
- Honda, O.; Kuroda, M.; Joja, I.; Asaumi, J.; Takeda, Y.; Akaki, S.; Togami, I.; Kanazawa, S.; Kawasaki, S.; Hiraki, Y. Assessment of secondary necrosis of Jurkat cells using a new microscopic system and double staining method with annexin V and propidium iodide. Int. J. Oncol. 2000, 16, 283–288. [Google Scholar] [PubMed]
- Garcia, M.A.; Meurs, E.F.; Esteban, M. The dsRNA protein kinase PKR: Virus and cell control. Biochimie 2007, 89, 799–811. [Google Scholar] [CrossRef] [PubMed]
- Bergan, V.; Jagus, R.; Lauksund, S.; Kileng, O.; Robertsen, B. The Atlantic salmon Z-DNA binding protein kinase phosphorylates translation initiation factor 2 alpha and constitutes a unique orthologue to the mammalian dsRNA-activated protein kinase R. FEBS J. 2008, 275, 184–197. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.L.; Wu, J.L.; Chen, M.H.; Hong, J.R. Aquatic birnavirus-induced ER stress-mediated death signaling contribute to downregulation of Bcl-2 family proteins in salmon embryo cells. PLOS ONE 2011, 6, e22935. [Google Scholar] [CrossRef] [PubMed]
- He, B. Viruses, endoplasmic reticulum stress, and interferon responses. Cell Death Differ. 2006, 13, 393–403. [Google Scholar] [CrossRef] [PubMed]
- Espinoza, J.C.; Kuznar, J. Visualization of the infectious pancreatic necrosis virus replication cycle by labeling viral intermediates with a TUNEL assay. Vet. Microbiol. 2010, 143, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Espinoza, J.C.; Cortes-Gutierrez, M.; Kuznar, J. Necrosis of infectious pancreatic necrosis virus (IPNV) infected cells rarely is preceded by apoptosis. Virus Res. 2005, 109, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.R.; Lin, T.L.; Hsu, Y.L.; Wu, J.L. Induction of apoptosis and secondary necrosis by infectious pancreatic necrosis virus in fish embryonic cells. J. Fish Dis. 1998, 21, 463–467. [Google Scholar] [CrossRef]
- Hong, J.R.; Gong, H.Y.; Wu, J.L. IPNVVP5, a novel anti-apoptosis gene of the Bcl-2 family, regulates Mcl-1 and viral protein expression. Virology 2002, 295, 217–229. [Google Scholar] [CrossRef] [PubMed]
- Santi, N.; Sandtro, A.; Sindre, H.; Song, H.C.; Hong, J.R.; Thu, B.; Wu, J.L.; Vakharia, V.N.; Evensen, O. Infectious pancreatic necrosis virus induces apoptosis in vitro and in vivo independent of VP5 expression. Virology 2005, 342, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Jeffrey, I.W.; Bushell, M.; Tilleray, V.J.; Morley, S.; Clemens, M.J. Inhibition of protein synthesis in apoptosis: Differential requirements by the tumor necrosis factor alpha family and a DNA-damaging agent for caspases and the double-stranded RNA-dependent protein kinase. Cancer Res. 2002, 62, 2272–2280. [Google Scholar] [PubMed]
- Pasternak, C.A. How Viruses Damage Cells—Alterations in Plasma-Membrane Function. J. Biosci. 1984, 6, 569–583. [Google Scholar] [CrossRef]
- Galloux, M.; Libersou, S.; Morellet, N.; Bouaziz, S.; Da Costa, B.; Ouldali, M.; Lepault, J.; Delmas, B. Infectious bursal disease virus, a non-enveloped virus, possesses a capsid-associated peptide that deforms and perforates biological membranes. J. Biol. Chem. 2007, 282, 20774–20784. [Google Scholar] [CrossRef] [PubMed]
- Kang, K.H.; Park, K.S.; Yoon, J.H.; Lee, C.H. Calcium response of CHSE following infection with infectious pancreatic necrosis virus (IPNV). Korean J. Microbiol. 1993, 31, 79–84. [Google Scholar]
- Pelletier, J.; Kaplan, G.; Racaniello, V.R.; Sonenberg, N. Cap-independent translation of poliovirus mRNA is conferred by sequence elements within the 5' noncoding region. Mol. Cell Biol. 1988, 8, 1103–1112. [Google Scholar] [PubMed]
- Mir, M.A.; Panganiban, A.T. A protein that replaces the entire cellular eIF4F complex. EMBO J. 2008, 27, 3129–3139. [Google Scholar] [CrossRef] [PubMed]
- Boot, H.J.; Pritz-Verschuren, S.B. Modifications of the 3'-UTR stem-loop of infectious bursal disease virus are allowed without influencing replication or virulence. Nucleic Acids Res. 2004, 32, 211–222. [Google Scholar] [CrossRef] [PubMed]
- Barry, G.; Breakwell, L.; Fragkoudis, R.; Attarzadeh-Yazdi, G.; Rodriguez-Andres, J.; Kohl, A.; Fazakerley, J.K. PKR acts early in infection to suppress Semliki Forest virus production and strongly enhances the type I interferon response. J. Gen. Virol. 2009, 90, 1382–1391. [Google Scholar] [CrossRef] [PubMed]
- Wertz, G.W.; Youngner, J.S. Interferon Production and Inhibition of Host Synthesis in Cells Infected with Vesicular Stomatitis Virus. J. Virol. 1970, 6, 476–484. [Google Scholar] [PubMed]
- Collet, B.; Munro, E.S.; Gahlawat, S.; Acosta, F.; Garcia, J.; Roemelt, C.; Zou, J.; Secombes, C.J.; Ellis, A.E. Infectious pancreatic necrosis virus suppresses type I interferon signalling in rainbow trout gonad cell line but not in Atlantic salmon macrophages. Fish Shellfish Immunol. 2007, 22, 44–56. [Google Scholar] [CrossRef] [PubMed]
- Skjesol, A.; Aamo, T.; Hegseth, M.N.; Robertsen, B.; Jorgensen, J.B. The interplay between infectious pancreatic necrosis virus (IPNV) and the IFN system: IFN signaling is inhibited by IPNV infection. Virus Res. 2009, 143, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Dobos, P. Virus-Specific Protein-Synthesis in Cells Infected by Infectious Pancreatic Necrosis Virus. J. Virol. 1977, 21, 242–258. [Google Scholar] [PubMed]
- Hill, B. J.; Way, K. Serological classification of infectious pancreatic necrosis (IPN) virus and other aquatic birnaviruses. Annu. Rev. Fish Dis. 1995, 5, 55–77. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gamil, A.A.A.; Mutoloki, S.; Evensen, Ø. A Piscine Birnavirus Induces Inhibition of Protein Synthesis in CHSE-214 Cells Primarily through the Induction of eIF2α Phosphorylation. Viruses 2015, 7, 1987-2005. https://0-doi-org.brum.beds.ac.uk/10.3390/v7041987
Gamil AAA, Mutoloki S, Evensen Ø. A Piscine Birnavirus Induces Inhibition of Protein Synthesis in CHSE-214 Cells Primarily through the Induction of eIF2α Phosphorylation. Viruses. 2015; 7(4):1987-2005. https://0-doi-org.brum.beds.ac.uk/10.3390/v7041987
Chicago/Turabian StyleGamil, Amr A.A., Stephen Mutoloki, and Øystein Evensen. 2015. "A Piscine Birnavirus Induces Inhibition of Protein Synthesis in CHSE-214 Cells Primarily through the Induction of eIF2α Phosphorylation" Viruses 7, no. 4: 1987-2005. https://0-doi-org.brum.beds.ac.uk/10.3390/v7041987