
Protein-Protein Interactions of Viroporins in Coronaviruses and Paramyxoviruses: New Targets for Antivirals?


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. The Envelope (E) Protein in Coronaviruses (CoVs)
1.2. General Features of the Envelope (E) Protein in CoVs

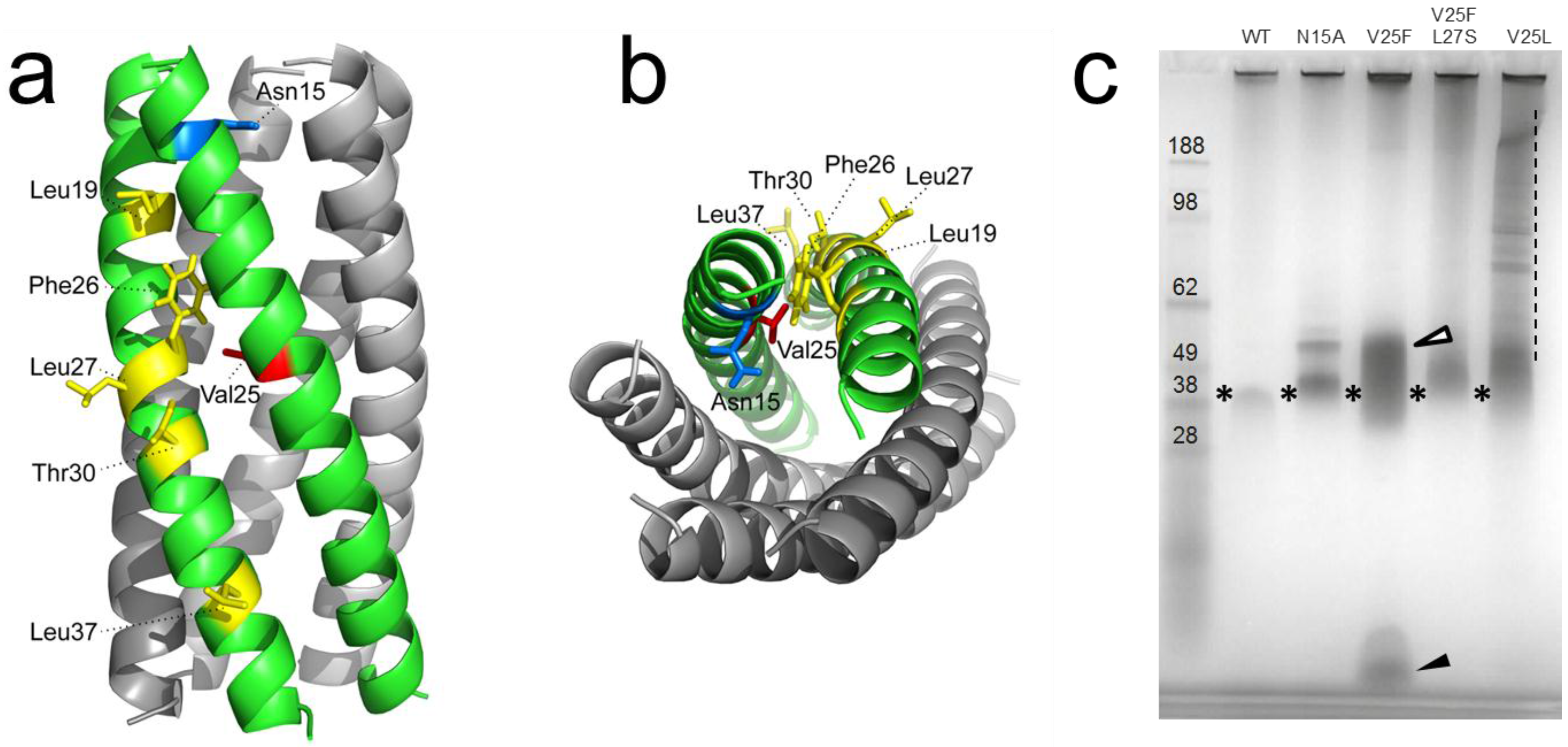
1.3. Structural Studies and Relevant Domains of CoV E Proteins


1.4. Protein-Protein Interactions
2. The Small Hydrophobic (SH) Protein in the Respiratory Syncytial Virus (RSV)
2.1. The Respiratory Syncytial Virus
2.2. The Small Hydrophobic Protein
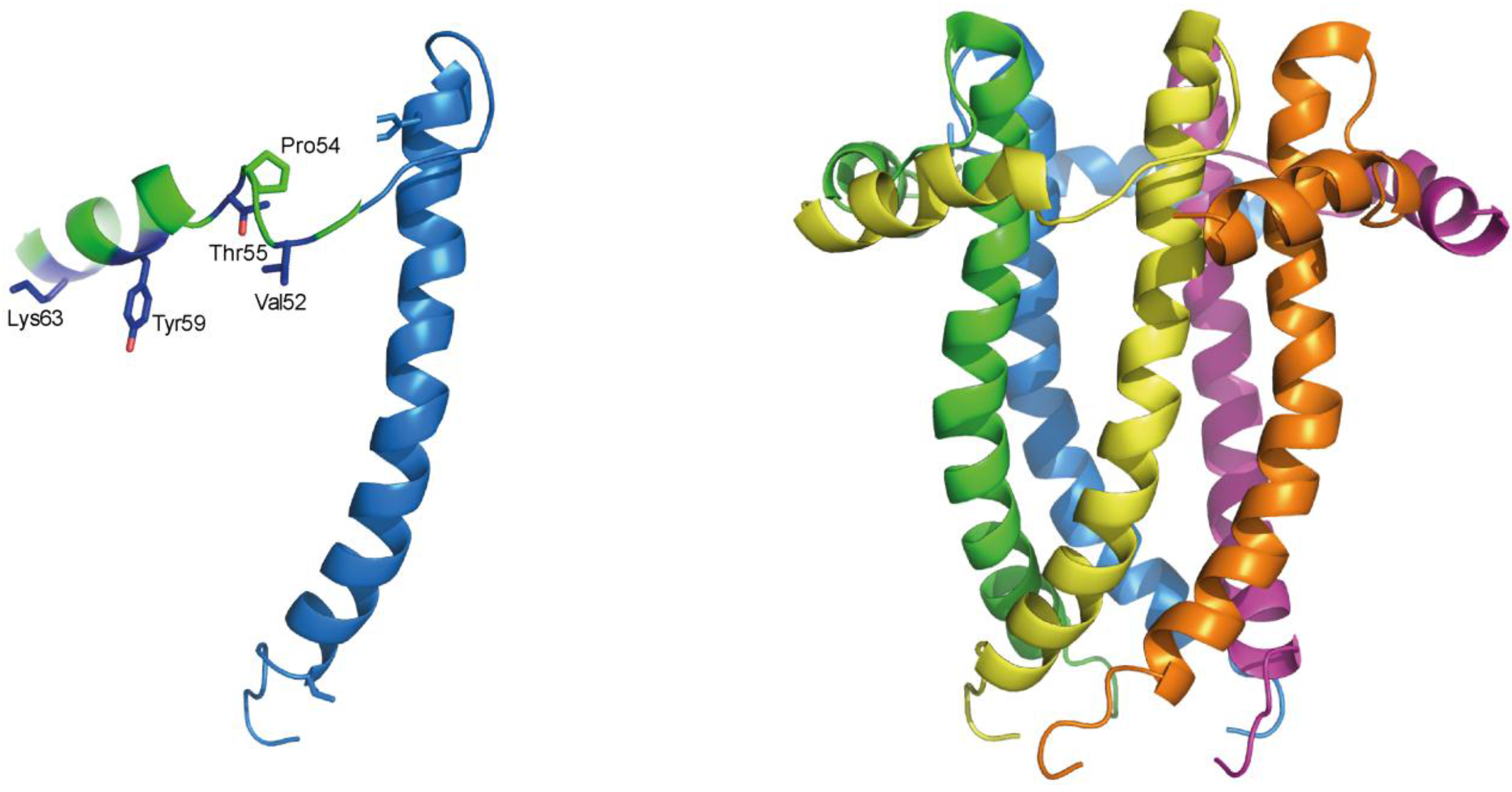
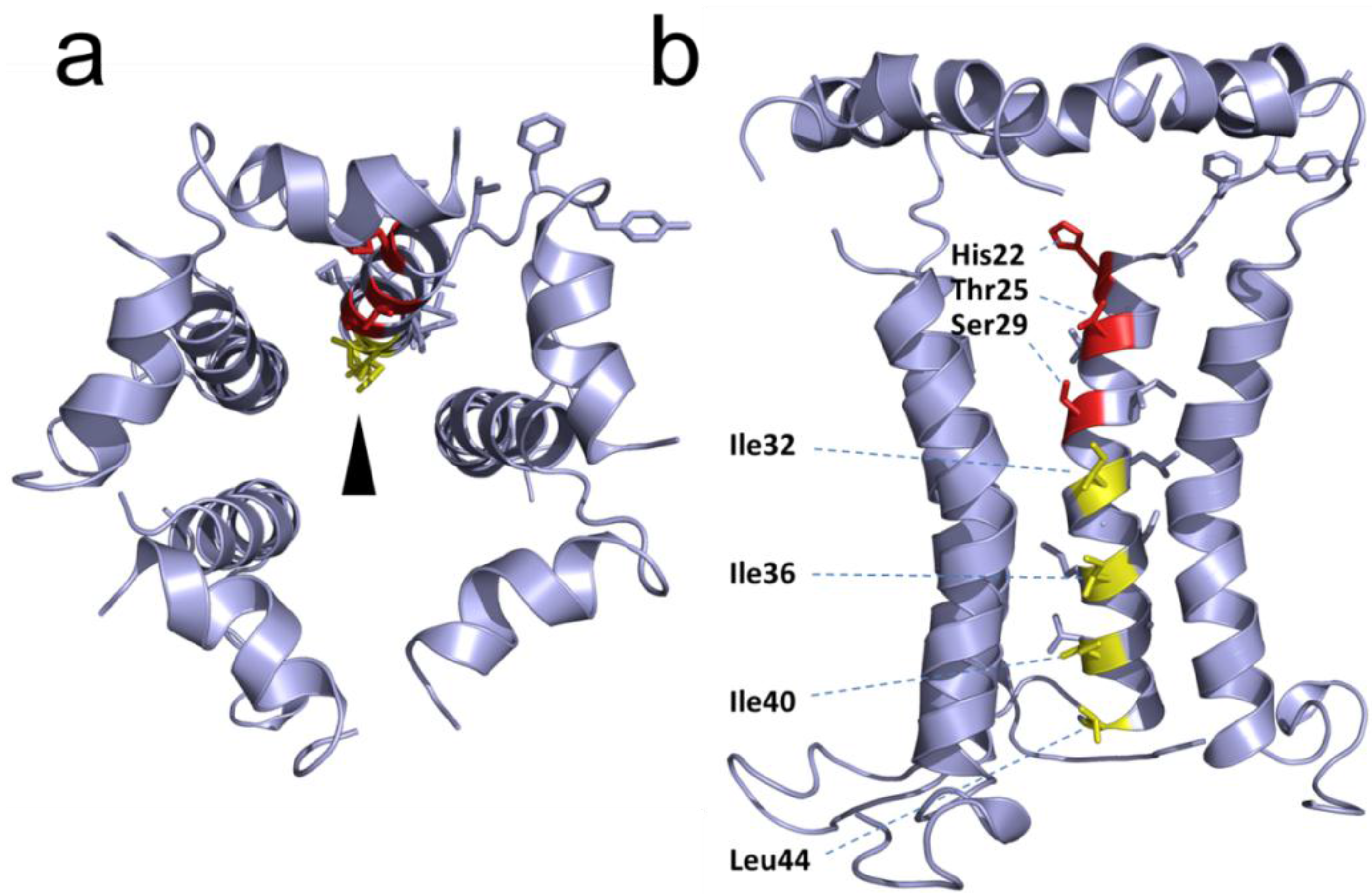
2.3. Structural Studies and Relevant Domains of RSV SH Protein


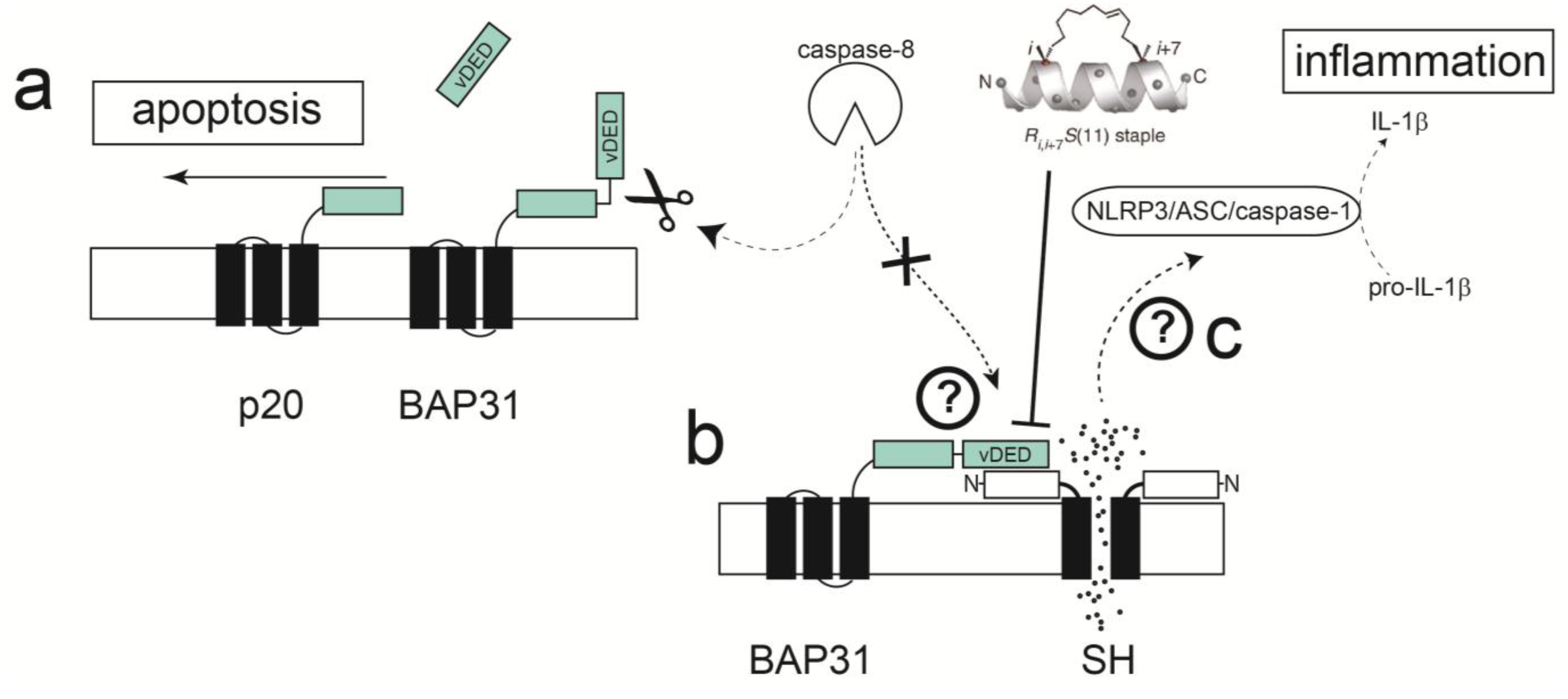
2.4. Protein-Protein Interactions Involving hRSV SH Protein

2.5. Disruption of Protein-Protein Interactions; Potential for Clinically Relevant Strategies

Acknowledgments
Author Contributions
Conflicts of Interest
References
- Enjuanes, L.; Brian, D.; Cavanagh, D.; Holmes, K.; Lai, M.M.C.; Laude, H.; Masters, P.; Rottier, P.; Siddell, S.G.; Spaan, W.J.M.; et al. Coronaviridae. In Virus taxonomy. Classification and Nomenclature of Viruses; van Regenmortel, M.H.V., Fauquet, C.M., Bishop, D.H.L., Carsten, E.B., Estes, M.K., Lemon, S.M., McGeoch, D.J., Maniloff, J., Mayo, M.A., Pringle, C.R., et al., Eds.; Academic Press: San Diego, CA, USA, 2000; pp. 835–849. [Google Scholar]
- Woo, P.C.Y.; Lau, S.K.P.; Huang, Y.; Yuen, K.Y. Coronavirus Diversity, Phylogeny and Interspecies Jumping. Exp. Biol. Med. 2009, 234, 1117–1127. [Google Scholar] [CrossRef] [PubMed]
- De Groot, R.J.; Baker, S.C.; Baric, R.; Enjuanes, L.; Gorbalenya, A.E.; Holmes, K.V.; Perlman, S.; Poon, L.; Rottier, P.J.M.; Talbot, P.J.; et al. Family Coronaviridae. In Virus Taxonomy, Ninth Report of the International Committee on Taxonomy of Viruses; King, A.M.Q., Adams, M.J., Carstens, E.B., Lefkowitz, E.J., Eds.; Elsevier: Oxford, UK, 2011; pp. 29, 806–828. [Google Scholar]
- Beaudette, F.R.; Hudson, C.B. Cultivation of the virus of infectious bronchitis. J. Am. Vet. Med. Assoc. 1937, 90, 51–60. [Google Scholar]
- Tyrrell, D.A.; Bynoe, M.L. Cultivation of a novel type of common cold virus in organ cultures. Br. Med. J. 1965, 1, 1467–1470. [Google Scholar] [CrossRef] [PubMed]
- Holmes, K.V. SARS coronavirus: A new challenge for prevention and therapy. J. Clin. Investig. 2003, 111, 1605–1609. [Google Scholar] [CrossRef] [PubMed]
- Raj, V.S.; Osterhaus, A.D.M.E.; Fouchier, R.A.M.; Haagmans, B.L. MERS: Emergence of a novel human coronavirus. Curr. Opin. Virol. 2014, 5, 58–62. [Google Scholar] [CrossRef] [PubMed]
- Lou, Z.; Sun, Y.; Rao, Z. Current progress in antiviral strategies. Trends Pharmacol. Sci. 2014, 35, 86–102. [Google Scholar] [CrossRef] [PubMed]
- Kilianski, A.; Baker, S.C. Cell-based antiviral screening against coronaviruses: Developing virus-specific and broad-spectrum inhibitors. Antivir. Res. 2014, 101, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Kilianski, A.; Mielech, A.M.; Deng, X.; Baker, S.C. Assessing activity and inhibition of Middle East respiratory syndrome coronavirus papain-like and 3C-like proteases using luciferase-based biosensors. J. Virol. 2013, 87, 11955–11962. [Google Scholar] [CrossRef] [PubMed]
- Graham, R.L.; Becker, M.M.; Eckerle, L.D.; Bolles, M.; Denison, M.R.; Baric, R.S. A live, impaired-fidelity coronavirus vaccine protects in an aged, immunocompromised mouse model of lethal disease. Nat. Med. 2012, 18, 1820–1826. [Google Scholar] [CrossRef] [PubMed]
- Enjuanes, L.; Nieto-Torres, J.L.; Jimenez-Guardeno, J.M.; DeDiego, M.L. Recombinant Live Vaccines to Protect against the Severe Acute Respiratory Syndrome Coronavirus. In Replicating Vaccines, Birkhauser Advances in Infectious Diseases; Dormitzer, P., Mandl, C.W., Rappuoli, R., Eds.; Springer: Basel, Switzerland, 2011; pp. 73–97. [Google Scholar]
- Heald-Sargent, T.; Gallagher, T. Ready, set, fuse! The coronavirus spike protein and acquisition of fusion competence. Viruses 2012, 4, 557–580. [Google Scholar] [CrossRef] [PubMed]
- Masters, P.S. The molecular biology of coronaviruses. Adv. Virus Res. 2006, 66, 193–292. [Google Scholar] [PubMed]
- Liu, D.X.; Fung, T.S.; Chong, K.; Shukla, A.; Hilgenfeld, R. Accessory proteins of SARS-CoV and other coronaviruses. Antivir. Res. 2014, 109, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.X.; Cavanagh, D.; Green, P.; Inglis, S.C. A Polycistronic Messenger-RNA Specified by the Coronavirus Infectious-Bronchitis Virus. Virology 1991, 184, 531–544. [Google Scholar] [CrossRef]
- Godet, M.; L’Haridon, R.; Vautherot, J.-F.; Laude, H. TGEV corona virus ORF4 encodes a membrane protein that is incorporated into virions. Virology 1992, 188, 666–675. [Google Scholar] [CrossRef]
- Yu, X.; Bi, W.; Weiss, S.R.; Leibowitz, J.L. Mouse hepatitis virus gene 5b protein is a new virion envelope protein. Virology 1994, 202, 1018–1023. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Yuan, Q.; Torres, J.; Tam, J.P.; Liu, D.X. Biochemical and functional characterization of the membrane association and membrane permeabilizing activity of the severe acute respiratory syndrome coronavirus envelope protein. Virology 2006, 349, 264–275. [Google Scholar] [CrossRef] [PubMed]
- Corse, E.; Machamer, C.E. Infectious bronchitis virus E protein is targeted to the Golgi complex and directs release of virus-like particles. J. Virol. 2000, 74, 4319–4326. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.X.; Inglis, S.C. Association of the infectious bronchitis virus 3c protein with the virion envelope. Virology 1991, 185, 911–917. [Google Scholar] [CrossRef]
- Tung, F.Y.T.; Abraham, S.; Sethna, M.; Hung, S.-L.; Sethna, P.; Hogue, B.G.; Brian, D.A. The 9-kDa hydrophobic protein encoded at the 3′ end of the porcine transmissible gastroenteritis coronavirus genome is membrane-associated. Virology 1992, 186, 676–683. [Google Scholar] [CrossRef]
- Corse, E.; Machamer, C.E. The cytoplasmic tails of infectious bronchitis virus E and M proteins mediate their interaction. Virology 2003, 312, 25–34. [Google Scholar] [CrossRef]
- Raamsman, M.J.B.; Krijnse Locker, J.; de Hooge, A.; de Vries, A.A.F.; Griffiths, G.; Vennema, H.; Rottier, P.J.M. Characterization of the coronavirus mouse hepatitis virus strain A59 small membrane protein E. J. Virol. 2000, 74, 2333–2342. [Google Scholar] [CrossRef] [PubMed]
- Lopez, L.A.; Riffle, A.J.; Pike, S.L.; Gardner, D.; Hogue, B.G. Importance of conserved cysteine residues in the coronavirus envelope protein. J. Virol. 2008, 82, 3000–3010. [Google Scholar] [CrossRef] [PubMed]
- Nieto-Torres, J.L.; DeDiego, M.L.; Alvarez, E.; Jimenez-Guardeno, J.M.; Regla-Nava, J.A.; Llorente, M.; Kremer, L.; Shuo, S.; Enjuanes, L. Subcellular location and topology of severe acute respiratory syndrome coronavirus envelope protein. Virology 2011, 415, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.X.; Yuan, Q.; Liao, Y. Coronavirus envelope protein: A small membrane protein with multiple functions. Cell. Mol. Life Sci. 2007, 64, 2043–2048. [Google Scholar] [CrossRef] [PubMed]
- DeDiego, M.L.; Nieto-Torres, J.L.; Jimenez-Guardeno, J.M.; Regla-Nava, J.A.; Alvarez, E.; Oliveros, J.C.; Zhao, J.; Fett, C.; Perlman, S.; Enjuanes, L. Severe acute respiratory syndrome coronavirus envelope protein regulates cell stress response and apoptosis. PLoS Pathog. 2011, 7, e1002315. [Google Scholar] [CrossRef] [PubMed]
- DeDiego, M.L.; Nieto-Torres, J.L.; Regla-Nava, J.A.; Jimenez-Guardeño, J.M.; Fett, C.; Fernandez-Delgado, R.; Castaño-Rodriguez, C.; Perlman, S.; Enjuanes, L. Inhibition of NF-κB-mediated inflammation in severe acute respiratory syndrome coronavirus-infected mice increases survival. J. Virol. 2014, 88, 913–924. [Google Scholar] [CrossRef] [PubMed]
- Lamirande, E.W.; DeDiego, M.L.; Roberts, A.; Jackson, J.P.; Alvarez, E.; Sheahan, T.; Shieh, W.J.; Zaki, S.R.; Baric, R.; Enjuanes, L.; et al. A live attenuated severe acute respiratory syndrome coronavirus is immunogenic and efficacious in golden Syrian hamsters. J. Virol. 2008, 82, 7721–7724. [Google Scholar] [CrossRef] [PubMed]
- Netland, J.; DeDiego, M.L.; Zhao, J.; Fett, C.; Alvarez, E.; Nieto-Torres, J.L.; Enjuanes, L.; Perlman, S. Immunization with an attenuated severe acute respiratory syndrome coronavirus deleted in E protein protects against lethal respiratory disease. Virology 2010, 399, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Almazán, F.; DeDiego, M.L.; Sola, I.; Zuñiga, S.; Nieto-Torres, J.L.; Marquez-Jurado, S.; Andrés, G.; Enjuanes, L. Engineering a Replication-Competent, Propagation-Defective Middle East Respiratory Syndrome Coronavirus as a Vaccine Candidate. MBio 2013, 4, e00650-13. [Google Scholar] [CrossRef] [PubMed]
- Regla-Nava, J.A.; Nieto-Torres, J.L.; Jimenez-Guardeño, J.M.; Fernandez-Delgado, R.; Fett, C.; Castaño-Rodríguez, C.; Perlman, S.; Enjuanes, L.; de Diego, M.L. Severe acute respiratory syndrome coronaviruses with mutations in the E protein are attenuated and promising vaccine candidates. J. Virol. 2015, 89, 3870–3887. [Google Scholar] [CrossRef] [PubMed]
- Nieto-Torres, J.L.; Dediego, M.L.; Verdia-Baguena, C.; Jimenez-Guardeno, J.M.; Regla-Nava, J.A.; Fernandez-Delgado, R.; Castano-Rodriguez, C.; Alcaraz, A.; Torres, J.; Aguilella, V.M.; et al. Severe acute respiratory syndrome coronavirus envelope protein ion channel activity promotes virus fitness and pathogenesis. PLoS Pathog. 2014, 10, e1004077. [Google Scholar] [CrossRef] [PubMed]
- Ruch, T.R.; Machamer, C.E. A single polar residue and distinct membrane topologies impact the function of the infectious bronchitis coronavirus E protein. PLoS Pathog. 2012, 8, e1002674. [Google Scholar] [CrossRef] [PubMed]
- Ruch, T.R.; Machamer, C.E. The hydrophobic domain of infectious bronchitis virus E protein alters the host secretory pathway and is important for release of infectious virus. J. Virol. 2011, 85, 675–685. [Google Scholar] [CrossRef] [PubMed]
- Lavi, E.; Wang, Q.; Weiss, S.R.; Gonatas, N.K. Syncytia formation induced by coronavirus infection is associated with fragmentation and rearrangement of the Golgi apparatus. Virology 1996, 221, 325–334. [Google Scholar] [CrossRef] [PubMed]
- Ulasli, M.; Verheije, M.H.; de Haan, C.A.M.; Reggiori, F. Qualitative and quantitative ultrastructural analysis of the membrane rearrangements induced by coronavirus. Cell. Microbiol. 2010, 12, 844–861. [Google Scholar] [CrossRef] [PubMed]
- Ruch, T.R.; Machamer, C.E. The coronavirus E protein: Assembly and beyond. Viruses 2012, 4, 363–382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torres, J. Nanyang Technological University: Singapore, Unpublished work. 2015.
- Torres, J.; Wang, J.; Parthasarathy, K.; Liu, D.X. The transmembrane oligomers of coronavirus protein E. Biophys. J. 2005, 88, 1283–1290. [Google Scholar] [CrossRef] [PubMed]
- Torres, J.; Parthasarathy, K.; Lin, X.; Saravanan, R.; Kukol, A.; Liu, D.X. Model of a putative pore: The pentameric α-helical bundle of SARS coronavirus E protein in lipid bilayers. Biophys. J. 2006, 91, 938–947. [Google Scholar] [CrossRef] [PubMed]
- Parthasarathy, K.; Ng, L.; Lin, X.; Liu, D.X.; Pervushin, K.; Gong, X.; Torres, J. Structural flexibility of the pentameric SARS coronavirus envelope protein ion channel. Biophys. J. 2008, 95, L39–L41. [Google Scholar] [CrossRef] [PubMed]
- Parthasarathy, K.; Lu, H.; Surya, W.; Vararattanavech, A.; Pervushin, K.; Torres, A. Expression and purification of coronavirus envelope proteins using a modified beta-barrel construct. Prot. Exp. Purif. 2012, 85, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Surya, W.; Claudine, S.; Torres, J. Structure of a Conserved Golgi Complex-targeting Signal in Coronavirus Envelope Proteins. J. Biol. Chem. 2014, 289, 12535–12549. [Google Scholar] [CrossRef] [PubMed]
- Pervushin, K.; Tan, E.; Parthasarathy, K.; Lin, X.; Jiang, F.L.; Yu, D.; Vararattanavech, A.; Soong, T.W.; Liu, D.X.; Torres, J. Structure and inhibition of the SARS coronavirus envelope protein ion channel. PLoS Pathog. 2009, 5, e1000511. [Google Scholar] [CrossRef] [PubMed]
- Surya, W.; Li, Y.; Verdià-Bàguena, C.; Aguilella, V.M.; Torres, J. MERS coronavirus envelope protein has a single transmembrane domain that forms pentameric ion channels. Virus Res. 2015, 201, 61–66. [Google Scholar] [CrossRef] [PubMed]
- Verdia-Baguena, C.; Nieto-Torres, J.L.; Alcaraz, A.; DeDiego, M.L.; Torres, J.; Aguilella, V.M.; Enjuanes, L. Coronavirus E protein forms ion channels with functionally and structurally-involved membrane lipids. Virology 2012, 432, 485–494. [Google Scholar] [CrossRef] [PubMed]
- Wilson, L.; Gage, P.; Ewart, G. Hexamethylene amiloride blocks E protein ion channels and inhibits coronavirus replication. Virology 2006, 353, 294–306. [Google Scholar] [CrossRef] [PubMed]
- Maeda, J.; Repass, J.F.; Maeda, A.; Makino, S. Membrane topology of coronavirus E protein. Virology 2001, 281, 163–169. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Q.; Liao, Y.; Torres, J.; Tam, J.P.; Liu, D.X. Biochemical evidence for the presence of mixed membrane topologies of the Severe Acute Respiratory Syndrome coronavirus envelope protein expressed in mammalian cells. FEBS Lett. 2006, 580, 3192–3200. [Google Scholar] [CrossRef] [PubMed]
- Torres, J.; Maheswari, U.; Parthasarathy, K.; Ng, L.F.; Liu, D.X.; Gong, X.D. Conductance and amantadine binding of a pore formed by a lysine-flanked transmembrane domain of SARS coronavirus envelope protein. Protein Sci. 2007, 16, 2065–2071. [Google Scholar] [CrossRef] [PubMed]
- Gan, S.W.; Vararattanavech, A.; Nordin, N.; Eshaghi, S.; Torres, J. A cost-effective method for simultaneous homo-oligomeric size determination and monodispersity conditions for membrane proteins. Anal. Biochem. 2011, 416, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Ramjeesingh, M.; Huan, L.J.; Garami, E.; Bear, C.E. Novel method for evaluation of the oligomeric structure of membrane proteins. Biochem. J. 1999, 342 Pt 1, 119–123. [Google Scholar] [CrossRef] [PubMed]
- Surya, W.; Samsó, M.; Torres, J. Structural and Functional Aspects of Viroporins in Human Respiratory Viruses: Respiratory Syncytial Virus and Coronaviruses. In Respiratory Disease and Infection-A New Insight; Vats, M., Ed.; InTech: Rijeka, Croatia, 2013; pp. 47–76. [Google Scholar]
- Ghosh, A.; Pithadia, A.S.; Bhat, J.; Bera, S.; Midya, A.; Fierke, C.A.; Ramamoorthy, A.; Bhunia, A. Self-Assembly of a Nine-Residue Amyloid-Forming Peptide Fragment of SARS Corona Virus E-Protein: Mechanism of Self Aggregation and Amyloid-Inhibition of hIAPP. Biochemistry 2015, 54, 2249–2261. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.R.; Lin, L.D.; Machamer, C.E. Identification of a Golgi complex-targeting signal in the cytoplasmic tail of the severe acute respiratory syndrome coronavirus envelope protein. J. Virol. 2011, 85, 5794–5803. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, L.D.; White, J.M. Mutational analysis of the candidate internal fusion peptide of the avian leukosis and sarcoma virus subgroup A envelope glycoprotein. J. Virol. 1998, 72, 3259–3267. [Google Scholar] [PubMed]
- Ito, H.; Watanabe, S.; Sanchez, A.; Whitt, M.A.; Kawaoka, Y. Mutational analysis of the putative fusion domain of Ebola virus glycoprotein. J. Virol. 1999, 73, 8907–8912. [Google Scholar] [PubMed]
- Wolfsberg, T.G.; Straight, P.D.; Gerena, R.L.; Huovila, A.P.; Primakoff, P.; Myles, D.G.; White, J.M. ADAM, a widely distributed and developmentally regulated gene family encoding membrane proteins with a disintegrin and metalloprotease domain. Dev. Biol. 1995, 169, 378–383. [Google Scholar] [CrossRef] [PubMed]
- Perry, A.C.; Gichuhi, P.M.; Jones, R.; Hall, L. Cloning and analysis of monkey fertilin reveals novel alpha subunit isoforms. Biochem. J. 1995, 307, 843–850. [Google Scholar] [PubMed]
- Whitt, M.A.; Zagouras, P.; Crise, B.; Rose, J.K. A fusion-defective mutant of the vesicular stomatitis virus glycoprotein. J. Virol. 1990, 64, 4907–4913. [Google Scholar] [PubMed]
- Kielian, M.; Rey, F.A. Virus membrane-fusion proteins: More than one way to make a hairpin. Nat. Rev. 2006, 4, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Rey, F.A.; Heinz, F.X.; Mandl, C.; Kunz, C.; Harrison, S.C. The envelope glycoprotein from tick-borne encephalitis virus at 2 A resolution. Nature 1995, 375, 291–298. [Google Scholar] [CrossRef] [PubMed]
- Root, M.J.; Steger, H.K. HIV-1 gp41 as a target for viral entry inhibition. Curr. Pharm. Des. 2004, 10, 1805–1825. [Google Scholar] [CrossRef] [PubMed]
- Langosch, D.; Hofmann, M.; Ungermann, C. The role of transmembrane domains in membrane fusion. Cell. Mol. Life Sci. 2007, 64, 850–864. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, R.T.; Kushnir, A.S.; White, J.M. The transmembrane domain of influenza hemagglutinin exhibits a stringent length requirement to support the hemifusion to fusion transition. J. Cell Biol. 2000, 151, 425–437. [Google Scholar] [CrossRef] [PubMed]
- Odell, D.; Wanas, E.; Yan, J.; Ghosh, H.P. Influence of membrane anchoring and cytoplasmic domains on the fusogenic activity of vesicular stomatitis virus glycoprotein G. J. Virol. 1997, 71, 7996–8000. [Google Scholar] [PubMed]
- Giraudo, C.G.; Hu, C.; You, D.; Slovic, A.M.; Mosharov, E.V.; Sulzer, D.; Melia, T.J.; Rothman, J.E. SNAREs can promote complete fusion and hemifusion as alternative outcomes. J. Cell Biol. 2005, 170, 249–260. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, M.W.; Peplowska, K.; Rohde, J.; Poschner, B.C.; Ungermann, C.; Langosch, D. Self-interaction of a SNARE transmembrane domain promotes the hemifusion-to-fusion transition. J. Mol. Biol. 2006, 364, 1048–1060. [Google Scholar] [CrossRef] [PubMed]
- Roy, R.; Peplowska, K.; Rohde, J.; Ungermann, C.; Langosch, D. Role of the Vam3p transmembrane segment in homodimerization and SNARE complex formation. Biochemistry 2006, 45, 7654–7660. [Google Scholar] [CrossRef] [PubMed]
- Langosch, D.; Crane, J.M.; Brosig, B.; Hellwig, A.; Tamm, L.K.; Reed, J. Peptide mimics of SNARE transmembrane segments drive membrane fusion depending on their conformational plasticity. J. Mol. Biol. 2001, 311, 709–721. [Google Scholar] [CrossRef] [PubMed]
- Dennison, S.M.; Greenfield, N.; Lenard, J.; Lentz, B.R. VSV transmembrane domain (TMD) peptide promotes PEG-mediated fusion of liposomes in a conformationally sensitive fashion. Biochemistry 2002, 41, 14925–14934. [Google Scholar] [CrossRef] [PubMed]
- Cleverley, D.Z.; Lenard, J. The transmembrane domain in viral fusion: Essential role for a conserved glycine residue in vesicular stomatitis virus G protein. Proc. Nat. Acad. Sci. USA 1998, 95, 3425–3430. [Google Scholar] [CrossRef] [PubMed]
- Chellgren, B.W.; Creamer, T.P. Side-chain entropy effects on protein secondary structure formation. Proteins 2006, 62, 411–420. [Google Scholar] [CrossRef] [PubMed]
- Fischer, F.; Stegen, C.F.; Masters, P.S.; Samsonoff, W.A. Analysis of constructed E gene mutants of mouse hepatitis virus confirms a pivotal role for E protein in coronavirus assembly. J. Virol. 1998, 72, 7885–7894. [Google Scholar] [PubMed]
- Corse, E.; Machamer, C.E. The cytoplasmic tail of infectious bronchitis virus E protein directs Golgi targeting. J. Virol. 2002, 76, 1273–1284. [Google Scholar] [CrossRef] [PubMed]
- Lamirande, E.W.; Dediego, M.L.; Roberts, A.; Jackson, J.P.; Alvarez, E.; Sheahan, T.; Shieh, W.J.; Zaki, S.R.; Baric, R.; Enjuanes, L.; et al. A live attenuated SARS coronavirus is immunogenic and efficacious in golden Syrian hamsters. J. Virol. 2008, 82, 7721–7724. [Google Scholar] [CrossRef] [PubMed]
- Siu, Y.L.; Teoh, K.T.; Lo, J.; Chan, C.M.; Kien, F.; Escriou, N.; Tsao, S.W.; Nicholls, J.M.; Altmeyer, R.; Peiris, J.S.; et al. The M, E, and N structural proteins of the severe acute respiratory syndrome coronavirus are required for efficient assembly, trafficking, and release of virus-like particles. J. Virol. 2008, 82, 11318–11330. [Google Scholar] [CrossRef] [PubMed]
- Vennema, H.; Godeke, G.J.; Rossen, J.W.; Voorhout, W.F.; Horzinek, M.C.; Opstelten, D.J.; Rottier, P.J. Nucleocapsid-independent assembly of coronavirus-like particles by co-expression of viral envelope protein genes. EMBO J. 1996, 15, 2020–2028. [Google Scholar] [PubMed]
- Ho, Y.; Lin, P.H.; Liu, C.Y.Y.; Lee, S.P.; Chao, Y.C. Assembly of human severe acute respiratory syndrome coronavirus-like particles. Biochem. Biophys. Res. Commun. 2004, 318, 833–838. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.P.; Liu, D.X. The missing link in coronavirus assembly. Retention of the avian coronavirus infectious bronchitis virus envelope protein in the pre-Golgi compartments and physical interaction between the envelope and membrane proteins. J. Biol. Chem. 2001, 276, 17515–17523. [Google Scholar] [CrossRef] [PubMed]
- Hogue, B.G.; Machamer, C.E. Coronavirus structural proteins and virus assembly. In Nidoviruses; Perlman, S., Gallagher, T., Snijder, E.J., Eds.; ASM Press: Washington, DC, USA, 2008; pp. 179–200. [Google Scholar]
- De Haan, C.A.; Vennema, H.; Rottier, P.J. Assembly of the coronavirus envelope: Homotypic interactions between the M proteins. J. Virol. 2000, 74, 4967–4978. [Google Scholar] [CrossRef] [PubMed]
- Rottier, P.J.M. The Coronaviridae; Siddell, S.G., Ed.; Plenum: New York, NY, USA, 1995; pp. 115–139. [Google Scholar]
- Baudoux, P.; Carrat, C.; Besnardeau, L.; Charley, B.; Laude, H. Coronavirus pseudoparticles formed with recombinant M and E proteins induce alpha interferon synthesis by leukocytes. J. Virol. 1998, 72, 8636–8643. [Google Scholar] [PubMed]
- Kuo, L.; Masters, P.S. Evolved variants of the membrane protein can partially replace the envelope protein in murine coronavirus assembly. J. Virol. 2010, 84, 12872–12885. [Google Scholar] [CrossRef] [PubMed]
- Teoh, K.T.; Siu, Y.L.; Chan, W.L.; Schluter, M.A.; Liu, C.J.; Peiris, J.S.; Bruzzone, R.; Margolis, B.; Nal, B. The SARS coronavirus E protein interacts with PALS1 and alters tight junction formation and epithelial morphogenesis. Mol. Biol. Cell 2010, 21, 3838–3852. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Guardeno, J.M.; Nieto-Torres, J.L.; DeDiego, M.L.; Regla-Nava, J.A.; Enjuanes, L.; Fernandez-Delgado, R.; Castano-Rodriguez, C. The PDZ-Binding Motif of Severe Acute Respiratory Syndrome Coronavirus Envelope Protein Is a Determinant of Viral Pathogenesis. PLoS Pathog. 2014, 10, e1004320. [Google Scholar] [CrossRef] [PubMed]
- Ye, F.; Zhang, M.J. Structures and target recognition modes of PDZ domains: Recurring themes and emerging pictures. Biochem. J. 2013, 455, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.K.; Pan, L.F.; Chen, J.; Zhang, M.J. Extensions of PDZ domains as important structural and functional elements. Protein Cell 2010, 1, 737–751. [Google Scholar] [CrossRef] [PubMed]
- Penkert, R.R.; DiVittorio, H.M.; Prehoda, K.E. Internal recognition through PDZ domain plasticity in the Par-6-Pals1 complex. Nat. Struct. Mol. Biol. 2004, 11, 1122–1127. [Google Scholar] [CrossRef] [PubMed]
- Hillier, B.J.; Christopherson, K.S.; Prehoda, K.E.; Bredt, D.S.; Lim, W.A. Unexpected modes of PDZ domain scaffolding revealed by structure of nNOS-syntrophin complex. Science 1999, 284, 812–815. [Google Scholar] [CrossRef] [PubMed]
- DiVittorio, H.; Penkert, R.; Prehoda, K.E. Mechanism of PDZ internal peptide recognition: Bypassing the requirement for a C-terminus. Protein Sci. 2004, 13, 138–138. [Google Scholar]
- Kumar, S.; Boehm, J.; Lee, J.C. p38 map kinases: Key signalling molecules as therapeutic targets for inflammatory diseases. Nat. Rev. Drug Discov. 2003, 2, 717–726. [Google Scholar] [CrossRef] [PubMed]
- Smits, S.L.; de Lang, A.; van den Brand, J.M.A.; Leijten, L.M.; van IJcken, W.F.; Eijkemans, M.J.C.; van Amerongen, G.; Kuiken, T.; Andeweg, A.C.; Osterhaus, A.D.M.E.; et al. Exacerbated Innate Host Response to SARS-CoV in Aged Non-Human Primates. PLoS Pathog. 2010, 6, e1000756. [Google Scholar] [CrossRef] [PubMed]
- Javier, R.T.; Rice, A.P. Emerging Theme: Cellular PDZ Proteins as Common Targets of Pathogenic Viruses. J. Virol. 2011, 85, 11544–11556. [Google Scholar] [CrossRef] [PubMed]
- Ji, H.L.; Song, W.; Gao, Z.; Su, X.F.; Nie, H.G.; Jiang, Y.; Peng, J.B.; He, Y.X.; Liao, Y.; Zhou, Y.J.; et al. SARS-CoV proteins decrease levels and activity of human ENaC via activation of distinct PKC isoforms. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2009, 296, L372–L383. [Google Scholar] [CrossRef] [PubMed]
- Wetherill, L.F.; Holmes, K.K.; Verow, M.; Muller, M.; Howell, G.; Harris, M.; Fishwick, C.; Stonehouse, N.; Foster, R.; Blair, G.E.; et al. High-Risk Human Papillomavirus E5 Oncoprotein Displays Channel-Forming Activity Sensitive to Small-Molecule Inhibitors. J. Virol. 2012, 86, 5341–5351. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, D.J.; Finbow, M.E.; Andresson, T.; Mclean, P.; Smith, K.; Bubb, V.; Schlegel, R. Bovine Papillomavirus-E5 Oncoprotein Binds to the 16k Component of Vacuolar H+-Atpases. Nature 1991, 352, 347–349. [Google Scholar] [CrossRef] [PubMed]
- Schapiro, F.; Sparkowski, J.; Adduci, A.; Suprynowicz, F.; Schlegel, R.; Grinstein, S. Golgi alkalinization by the papillomavirus E5 oncoprotein. J. Cell Biol. 2000, 148, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Blount, R.E., Jr.; Morris, J.A.; Savage, R.E. Recovery of cytopathogenic agent from chimpanzees with coryza. Proc. Soc. Exp. Biol. Med. 1956, 92, 544–549. [Google Scholar] [PubMed]
- Dowell, S.F.; Anderson, L.J.; Gary, H.E., Jr.; Erdman, D.D.; Plouffe, J.F.; File, T.M., Jr.; Marston, B.J.; Breiman, R.F. Respiratory syncytial virus is an important cause of community-acquired lower respiratory infection among hospitalized adults. J. Infect. Dis. 1996, 174, 456–462. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Thompson, W.W.; Viboud, C.G.; Ringholz, C.M.; Cheng, P.Y.; Steiner, C.; Abedi, G.R.; Anderson, L.J.; Brammer, L.; Shay, D.K. Hospitalizations Associated With Influenza and Respiratory Syncytial Virus in the United States, 1993–2008. Clin. Infect. Dis. 2012, 54, 1427–1436. [Google Scholar] [CrossRef] [PubMed]
- Nair, H.; Nokes, D.J.; Gessner, B.D.; Dherani, M.; Madhi, S.A.; Singleton, R.J.; O’Brien, K.L.; Roca, A.; Wright, P.F.; Bruce, N.; et al. Global burden of acute lower respiratory infections due to respiratory syncytial virus in young children: A systematic review and meta-analysis. Lancet 2010, 375, 1545–1555. [Google Scholar] [CrossRef]
- Zhao, X.; Singh, M.; Malashkevich, V.N.; Kim, P.S. Structural characterization of the human respiratory syncytial virus fusion protein core 2. Proc. Nat. Acad. Sci. USA 2000, 97, 14172–14177. [Google Scholar] [CrossRef] [PubMed]
- Lambert, D.M.; Barney, S.; Lambert, A.L.; Guthrie, K.; Medinas, R.; Davis, D.E.; Bucy, T.; Erickson, J.; Merutka, G.; Petteway, S.R. Peptides from conserved regions of paramyxovirus fusion (F) proteins are potent inhibitors of viral fusion. Proc. Nat. Acad. Sci. USA 1996, 93, 2186–2191. [Google Scholar] [CrossRef] [PubMed]
- Shepherd, N.E.; Hoang, H.N.; Desai, V.S.; Letouze, E.; Young, P.R.; Fairlie, D.P. Modular alpha-helical mimetics with antiviral activity against respiratory syncitial virus. J. Am. Chem. Soc. 2006, 128, 13284–13289. [Google Scholar] [CrossRef] [PubMed]
- Pastey, M.K.; Gower, T.L.; Spearman, P.W.; Crowe, J.E.; Graham, B.S. A RhoA-derived peptide inhibits syncytium formation induced by respiratory syncytial virus and parainfluenza virus type 3. Nat. Med. 2000, 6, 35–40. [Google Scholar] [PubMed]
- Razinkov, V.; Gazumyan, A.; Nikitenko, A.; Ellestad, G.; Krishnamurthy, G. RFI-641 inhibits entry of respiratory syncytial virus via interactions with fusion protein. Chem. Biol. 2001, 8, 645–659. [Google Scholar] [CrossRef]
- Douglas, J.L.; Panis, M.L.; Ho, E.; Lin, K.Y.; Krawczyk, S.H.; Grant, D.M.; Cai, R.; Swaminathan, S.; Chen, X.; Cihlar, T. Small molecules VP-14637 and JNJ-2408068 inhibit respiratory syncytial virus fusion by similar mechanisms. Antimicrob. Agents Chemother. 2005, 49, 2460–2466. [Google Scholar] [CrossRef] [PubMed]
- Roymans, D.; de Bondt, H.L.; Arnoult, E.; Geluykens, P.; Gevers, T.; van Ginderen, M.; Verheyen, N.; Kim, H.D.; Willebrords, R.; Bonfanti, J.F.; et al. Binding of a potent small-molecule inhibitor of six-helix bundle formation requires interactions with both heptad-repeats of the RSV fusion protein. Proc. Nat. Acad. Sci. USA 2010, 107, 308–313. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Behera, A.K.; Lockey, R.F.; Zhang, J.; Bhullar, G.; de La Cruz, C.P.; Chen, L.C.; Leong, K.W.; Huang, S.K.; Mohapatra, S.S. Intranasal gene transfer by chitosan-DNA nanospheres protects BALB/c mice against acute respiratory syncytial virus infection. Hum. Gene Ther. 2002, 13, 1415–1425. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Yang, H.; Kong, X.; Mohapatra, S.; san Juan-Vergara, H.; Hellermann, G.; Behera, S.; Singam, R.; Lockey, R.F.; Mohapatra, S.S. Inhibition of respiratory syncytial virus infection with intranasal siRNA nanoparticles targeting the viral NS1 gene. Nat. Med. 2005, 11, 233–233. [Google Scholar] [CrossRef]
- McLellan, J.S.; Chen, M.; Joyce, M.G.; Sastry, M.; Stewart-Jones, G.B.E.; Yang, Y.P.; Zhang, B.S.; Chen, L.; Srivatsan, S.; Zheng, A.Q.; et al. Structure-Based Design of a Fusion Glycoprotein Vaccine for Respiratory Syncytial Virus. Science 2013, 342, 592–598. [Google Scholar] [CrossRef] [PubMed]
- McLellan, J.S.; Chen, M.; Leung, S.; Graepel, K.W.; Du, X.L.; Yang, Y.P.; Zhou, T.Q.; Baxa, U.; Yasuda, E.; Beaumont, T.; et al. Structure of RSV Fusion Glycoprotein Trimer Bound to a Prefusion-Specific Neutralizing Antibody. Science 2013, 340, 1113–1117. [Google Scholar] [CrossRef] [PubMed]
- Schepens, B.; Sedeyn, K.; Vande Ginste, L.; de Baets, S.; Schotsaert, M.; Roose, K.; Houspie, L.; van Ranst, M.; Gilbert, V.; van Rooijen, N.; et al. Protection and mechanism of action of a novel human respiratory syncytial virus vaccine candidate based on the extracellular domain of small hydrophobic protein. EMBO Mol. Med. 2014, 6, 1436–1454. [Google Scholar] [CrossRef] [PubMed]
- Bird, G.H.; Boyapalle, S.; Wong, T.; Opoku-Nsiah, K.; Bedi, R.; Crannell, W.C.; Perry, A.F.; Nguyen, H.; Sampayo, V.; Devareddy, A.; et al. Mucosal delivery of a double-stapled RSV peptide prevents nasopulmonary infection. J. Clin. Investig. 2014, 124, 2113–2124. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention. Respiratory syncytial virus activity: United States, 1999–2000 season. Morb. Mortal. Wkly. Rep. 2000, 49, 1091–1093. [Google Scholar]
- Weiner, L.B.; Polak, M.J.; Masaquel, A.; Mahadevia, P.J. Cost-Effectiveness of Respiratory Syncytial Virus Prophylaxis with Palivizumab among Preterm Infants Covered by Medicaid in the United States. Value Health 2011, 14, A118–A118. [Google Scholar] [CrossRef]
- Hall, C.B.; Powell, K.R.; Macdonald, N.E.; Gala, C.L.; Menegus, M.E.; Suffin, S.C.; Cohen, H.J. Respiratory Syncytial Viral-Infection in Children with Compromised Immune Function. N. Engl. J. Med. 1986, 315, 77–81. [Google Scholar] [CrossRef] [PubMed]
- Collins, P.L.; Melero, J.A. Progress in understanding and controlling respiratory syncytial virus: Still crazy after all these years. Virus Res. 2011, 162, 80–99. [Google Scholar] [CrossRef] [PubMed]
- Krusat, T.; Streckert, H.J. Heparin-dependent attachment of respiratory syncytial virus (RSV) to host cells. Arch. Virol. 1997, 142, 1247–1254. [Google Scholar] [CrossRef] [PubMed]
- Lamb, R.A. Paramyxovirus fusion: A hypothesis for changes. Virology 1993, 197, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Collins, P.L.; Mottet, G. Membrane orientation and oligomerization of the small hydrophobic protein of human respiratory syncytial virus. J. Gen. Virol. 1993, 74, 1445–1450. [Google Scholar] [CrossRef] [PubMed]
- Gan, S.W.; Tan, E.; Lin, X.; Yu, D.; Wang, J.; Tan, G.M.-Y.; Vararattanavech, A.; Yeo, C.Y.; Soon, C.H.; Soong, T.W.; et al. The small hydrophobic protein of the human respiratory syncytial virus forms pentameric ion channels. J. Biol. Chem. 2012, 287, 24671–24689. [Google Scholar] [CrossRef] [PubMed]
- Taylor, C.E.; Morrow, S.; Scott, M.; Young, B.; Toms, G.L. Comparative Virulence of Respiratory Syncytial Virus Subgroup-A and Subgroup-B. Lancet 1989, 1, 777–778. [Google Scholar] [CrossRef]
- Mcconnochie, K.M.; Hall, C.B.; Walsh, E.E.; Roghmann, K.J. Variation in Severity of Respiratory Syncytial Virus-Infections with Subtype. J. Pediatr. 1990, 117, 52–62. [Google Scholar] [CrossRef]
- Hall, C.B.; Walsh, E.E.; Schnabel, K.C.; Long, C.E.; Mcconnochie, K.M.; Hildreth, S.W.; Anderson, L.J. Occurrence of Group-A and Group-B of Respiratory Syncytial Virus over 15 Years-Associated Epidemiologic and Clinical Characteristics in Hospitalized and Ambulatory Children. J. Infect. Dis. 1990, 162, 1283–1290. [Google Scholar] [CrossRef] [PubMed]
- Rixon, H.W.; Brown, G.; Aitken, J.; McDonald, T.; Graham, S.; Sugrue, R.J. The small hydrophobic (SH) protein accumulates within lipid-raft structures of the Golgi complex during respiratory syncytial virus infection. J. Gen. Virol. 2004, 85, 1153–1165. [Google Scholar] [CrossRef] [PubMed]
- Techaarpornkul, S.; Barretto, N.; Peeples, M.E. Functional analysis of recombinant respiratory syncytial virus deletion mutants lacking the small hydrophobic and/or attachment glycoprotein gene. J. Virol. 2001, 75, 6825–6834. [Google Scholar] [CrossRef] [PubMed]
- Bukreyev, A.; Whitehead, S.S.; Murphy, B.R.; Collins, P.L. Recombinant respiratory syncytial virus from which the entire SH gene has been deleted grows efficiently in cell culture and exhibits site-specific attenuation in the respiratory tract of the mouse. J. Virol. 1997, 71, 8973–8982. [Google Scholar] [PubMed]
- Karron, R.A.; Buonagurio, D.A.; Georgiu, A.F.; Whitehead, S.S.; Adamus, J.E.; Harris, D.O.; Clements-Mann, M.L.; Randolph, V.B.; Udem, S.A.; Murphy, B.R.; et al. Respiratory syncytial virus (RSV) SH and G proteins are not essential for viral replication in vitro: Clinical evaluation and molecular characterization of a cold-passaged, attenuated RSV subgroup B mutant. Proc. Natl. Acad. Sci. USA 1997, 94, 13961–13966. [Google Scholar] [CrossRef] [PubMed]
- Whitehead, S.S.; Bukreyev, A.; Teng, M.N.; Firestone, C.Y.; St Claire, M.; Elkins, W.R.; Collins, P.L.; Murphy, B.R. Recombinant respiratory syncytial virus bearing a deletion of either the NS2 or SH gene is attenuated in chimpanzees. J. Virol. 1999, 73, 3438–3442. [Google Scholar] [PubMed]
- Jin, H.; Zhou, H.; Cheng, X.; Tang, R.; Munoz, M.; Nguyen, N. Recombinant respiratory syncytial viruses with deletions in the NS1, NS2, SH, and M2–2 genes are attenuated in vitro and in vivo. Virology 2000, 273, 210–218. [Google Scholar] [CrossRef] [PubMed]
- Taylor, G.; Wyld, S.; Valarcher, J.F.; Guzman, E.; Thom, M.; Widdison, S.; Buchholz, U.J. Recombinant bovine respiratory syncytial virus with deletion of the SH gene induces increased apoptosis and pro-inflammatory cytokines in vitro, and is attenuated and induces protective immunity in calves. J. Gen. Virol. 2014, 95, 1244–1254. [Google Scholar] [CrossRef] [PubMed]
- Fuentes, S.; Tran, K.C.; Luthra, P.; Teng, M.N.; He, B. Function of the respiratory syncytial virus small hydrophobic protein. J. Virol. 2007, 81, 8361–8366. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Xu, J.; Patel, J.; Fuentes, S.; Lin, Y.A.; Anderson, D.; Sakamoto, K.; Wang, L.F.; He, B.A. Function of the Small Hydrophobic Protein of J Paramyxovirus. J. Virol. 2011, 85, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Jack, P.J.M.; Boyle, D.B.; Eaton, B.T.; Wang, L.F. The complete genome sequence of J virus reveals a unique genome structure in the family Paramyxoviridae. J. Virol. 2005, 79, 10690–10700. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Bright, A.C.; Rothermel, T.A.; He, B. Induction of apoptosis by paramyxovirus simian virus 5 lacking a small hydrophobic gene. J. Virol. 2003, 77, 3371–3383. [Google Scholar] [CrossRef] [PubMed]
- Wilson, R.L.; Fuentes, S.M.; Wang, P.; Taddeo, E.C.; Klatt, A.; Henderson, A.J.; He, B. Function of small hydrophobic proteins of paramyxovirus. J. Virol. 2006, 80, 1700–1709. [Google Scholar] [CrossRef] [PubMed]
- Ichinohe, T.; Pang, I.K.; Iwasaki, A. Influenza virus activates inflammasomes via its intracellular M2 ion channel. Nat. Immunol. 2010, 11, 404–410. [Google Scholar] [CrossRef] [PubMed]
- Ito, M.; Yanagi, Y.; Ichinohe, T. Encephalomyocarditis virus viroporin 2B activates NLRP3 inflammasome. PLoS Pathog. 2012, 8, e1002857. [Google Scholar] [CrossRef] [PubMed]
- Segovia, J.; Sabbah, A.; Mgbemena, V.; Tsai, S.Y.; Chang, T.H.; Berton, M.T.; Morris, I.R.; Allen, I.C.; Ting, J.P.Y.; Bose, S. TLR2/MyD88/NF-kappa B Pathway, Reactive Oxygen Species, Potassium Efflux Activates NLRP3/ASC Inflammasome during Respiratory Syncytial Virus Infection. PLoS ONE 2012, 7, e29695. [Google Scholar] [CrossRef] [PubMed]
- Triantafilou, K.; Kar, S.; Vakakis, E.; Kotecha, S.; Triantafilou, M. Human respiratory syncytial virus viroporin SH: A viral recognition pathway used by the host to signal inflammasome activation. Immunology 2013, 140, 87–88. [Google Scholar] [CrossRef] [PubMed]
- Gan, S.W.; Ng, L.; Xin, L.; Gong, X.; Torres, J. Structure and ion channel activity of the human respiratory syncytial virus (hRSV) small hydrophobic protein transmembrane domain. Protein Sci. 2008, 17, 813–820. [Google Scholar] [CrossRef] [PubMed]
- Gan, S.W.; Torres, J. Structural and functional aspects of the small hydrophobic (SH) protein in the human respiratory syncytial virus. In Human Respiratory Syncytial Virus Infection; Resch, B., Ed.; Intech: Janeza Trdine, Croatia, 2011; pp. 75–96. [Google Scholar]
- Li, Y.; To, J.; Verdià-Baguena, C.; Dossena, S.; Surya, W.; Huang, M.; Paulmichl, M.; Liu, D.X.; Aguilella, V.M.; Torres, J. Inhibition of the Human Respiratory Syncytial Virus Small Hydrophobic Protein and Structural variations in a bicelle environment. J. Virol. 2014, 88, 11899–11914. [Google Scholar] [CrossRef] [PubMed]
- Schnell, J.R.; Chou, J.J. Structure and mechanism of the M2 proton channel of influenza A virus. Nature 2008, 451, 591–595. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, B.; Xie, S.; Berardi, M.J.; Zhao, X.; Dev, J.; Yu, W.; Sun, B.; Chou, J.J. Unusual architecture of the p7 channel from hepatitis C virus. Nature 2013, 498, 521–525. [Google Scholar] [CrossRef] [PubMed]
- Tapia, L.I.; Shaw, C.A.; Aideyan, L.O.; Jewell, A.M.; Dawson, B.C.; Haq, T.R.; Piedra, P.A. Gene Sequence Variability of the Three Surface Proteins of Human Respiratory Syncytial Virus (HRSV) in Texas. PLoS ONE 2014, 9, e90786. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.D.; Vazquez, M.; Buonocore, L.; Kahn, J.S. Conservation of the respiratory syncytial virus SH gene. J. Infect. Dis. 2000, 182, 1228–1233. [Google Scholar] [CrossRef] [PubMed]
- Collins, P.L.; Olmsted, R.A.; Johnson, P.R. The small hydrophobic protein of human respiratory syncytial virus: Comparison between antigenic subgroups A and B. J. Gen. Virol. 1990, 71, 1571–1576. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, K.; Tanabayashi, K.; Hishiyama, M.; Yamada, A. The mumps virus SH protein is a membrane protein and not essential for virus growth. Virology 1996, 225, 156–162. [Google Scholar] [CrossRef] [PubMed]
- Elango, N.; Kovamees, J.; Varsanyi, T.M.; Norrby, E. Messenger-RNA Sequence and Deduced Amino-Acid Sequence of the Mumps-Virus Small Hydrophobic Protein Gene. J. Virol. 1989, 63, 1413–1415. [Google Scholar] [PubMed]
- Low, K.W.; Tan, T.; Ng, K.; Tan, B.H.; Sugrue, R.J. The RSV F and G glycoproteins interact to form a complex on the surface of infected cells. Biochem. Biophys. Res. Commun. 2008, 366, 308–313. [Google Scholar] [CrossRef] [PubMed]
- Rixon, H.W.; Brown, G.; Murray, J.T.; Sugrue, R.J. The respiratory syncytial virus small hydrophobic protein is phosphorylated via a mitogen-activated protein kinase p38-dependent tyrosine kinase activity during virus infection. J. Gen. Virol. 2005, 86, 375–384. [Google Scholar] [CrossRef] [PubMed]
- Schlender, J.; Zimmer, G.; Herrler, G.; Conzelmann, K.K. Respiratory syncytial virus (RSV) fusion protein subunit F2, not attachment protein G, determines the specificity of RSV infection. J. Virol. 2003, 77, 4609–4616. [Google Scholar] [CrossRef] [PubMed]
- Karger, A.; Schmidt, U.; Buchholz, U.J. Recombinant bovine respiratory syncytial virus with deletions of the G or SH genes: G and F proteins bind heparin. J. Gen. Virol. 2001, 82, 631–640. [Google Scholar] [PubMed]
- Li, Y.; Jain, N.; Limpanawat, S.; To, J.; Quistgaard, E.M.; Nordlund, P.; Thanabalu, T.; Torres, J. Interaction between human BAP31 and Respiratory Syncytial Virus Small Hydrophobic (SH) protein. Virology 2015, 482, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Heath-Engel, H.; Zhang, D.; Nguyen, N.; Thomas, D.Y.; Hanrahan, J.W.; Shore, G.C. BAP31 Interacts with Sec61 Translocons and Promotes Retrotranslocation of CFTRΔF508 via the Derlin-1 Complex. Cell 2008, 133, 1080–1092. [Google Scholar] [CrossRef] [PubMed]
- Adachi, T.; Schamel, W.W.; Kim, K.M.; Watanabe, T.; Becker, B.; Nielsen, P.J.; Reth, M. The specificity of association of the IgD molecule with the accessory proteins BAP31/BAP29 lies in the IgD transmembrane sequence. EMBO J. 1996, 15, 1534–1541. [Google Scholar] [PubMed]
- Quistgaard, E.M.; Low, C.; Moberg, P.; Guettou, F.; Maddi, K.; Nordlund, P. Structural and biophysical characterization of the cytoplasmic domains of human BAP29 and BAP31. PLoS ONE 2013, 8, e71111. [Google Scholar] [CrossRef] [PubMed]
- Ng, F.W.H.; Nguyen, M.; Kwan, T.; Branton, P.E.; Nicholson, D.W.; Cromlish, J.A.; Shore, G.C. p28 Bap31, a Bcl-2/Bcl-X-L- and procaspase-8-associated protein in the endoplasmic reticulum. J. Cell Biol. 1997, 139, 327–338. [Google Scholar] [CrossRef] [PubMed]
- Breckenridge, D.G.; Nguyen, M.; Kuppig, S.; Reth, M.; Shore, G.C. The procaspase-8 isoform, procaspase-8L, recruited to the BAP31 complex at the endoplasmic reticulum. Proc. Nat. Acad. Sci. USA 2002, 99, 4331–4336. [Google Scholar] [CrossRef] [PubMed]
- Breckenridge, D.G.; Stojanovic, M.; Marcellus, R.C.; Shore, G.C. Caspase cleavage product of BAP31 induces mitochondrial fission through endoplasmic reticulum calcium signals, enhancing cytochrome c release to the cytosol. J. Cell Biol. 2003, 160, 1115–1127. [Google Scholar] [CrossRef] [PubMed]
- Rosati, E.; Sabatini, R.; Rampino, G.; De Falco, F.; Di Ianni, M.; Falzetti, F.; Fettucciari, K.; Bartoli, A.; Screpanti, I.; Marconi, P. Novel targets for endoplasmic reticulum stress-induced apoptosis in B-CLL. Blood 2010, 116, 2713–2723. [Google Scholar] [CrossRef] [PubMed]
- Iwasawa, R.; Mahul-Mellier, A.L.; Datler, C.; Pazarentzos, E.; Grimm, S. Fis1 and Bap31 bridge the mitochondria-ER interface to establish a platform for apoptosis induction. EMBO J. 2011, 30, 556–568. [Google Scholar] [CrossRef] [PubMed]
- Kabsch, K.; Mossadegh, N.; Kohl, A.; Komposch, G.; Schenkel, J.; Alonso, A.; Tomakidi, P. The HPV-16 E5 protein inhibits TRAIL- and FasL-mediated apoptosis in human keratinocyte raft cultures. Intervirology 2004, 47, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Regan, J.A.; Laimins, L.A. Bap31 is a novel target of the human papillomavirus E5 protein. J. Virol. 2008, 82, 10042–10051. [Google Scholar] [CrossRef] [PubMed]
- Walensky, L.D.; Bird, G.H. Hydrocarbon-Stapled Peptides: Principles, Practice, and Progress. J. Med. Chem. 2014, 57, 6275–6288. [Google Scholar] [CrossRef] [PubMed]
- Bellmann-Sickert, K.; Stone, T.A.; Poulsen, B.E.; Deber, C.M. Efflux by Small Multidrug Resistance Proteins is Inhibited by Membrane-Interactive Helix-Stapled Peptides. J. Biol. Chem. 2014, 290, 1752–1759. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Torres, J.; Surya, W.; Li, Y.; Liu, D.X. Protein-Protein Interactions of Viroporins in Coronaviruses and Paramyxoviruses: New Targets for Antivirals? Viruses 2015, 7, 2858-2883. https://0-doi-org.brum.beds.ac.uk/10.3390/v7062750
Torres J, Surya W, Li Y, Liu DX. Protein-Protein Interactions of Viroporins in Coronaviruses and Paramyxoviruses: New Targets for Antivirals? Viruses. 2015; 7(6):2858-2883. https://0-doi-org.brum.beds.ac.uk/10.3390/v7062750
Chicago/Turabian StyleTorres, Jaume, Wahyu Surya, Yan Li, and Ding Xiang Liu. 2015. "Protein-Protein Interactions of Viroporins in Coronaviruses and Paramyxoviruses: New Targets for Antivirals?" Viruses 7, no. 6: 2858-2883. https://0-doi-org.brum.beds.ac.uk/10.3390/v7062750