
The Chikungunya Virus Capsid Protein Contains Linear B Cell Epitopes in the N- and C-Terminal Regions that are Dependent on an Intact C-Terminus for Antibody Recognition

Abstract
:1. Introduction

2. Material and Methods
2.1. Cell and Virus Culture
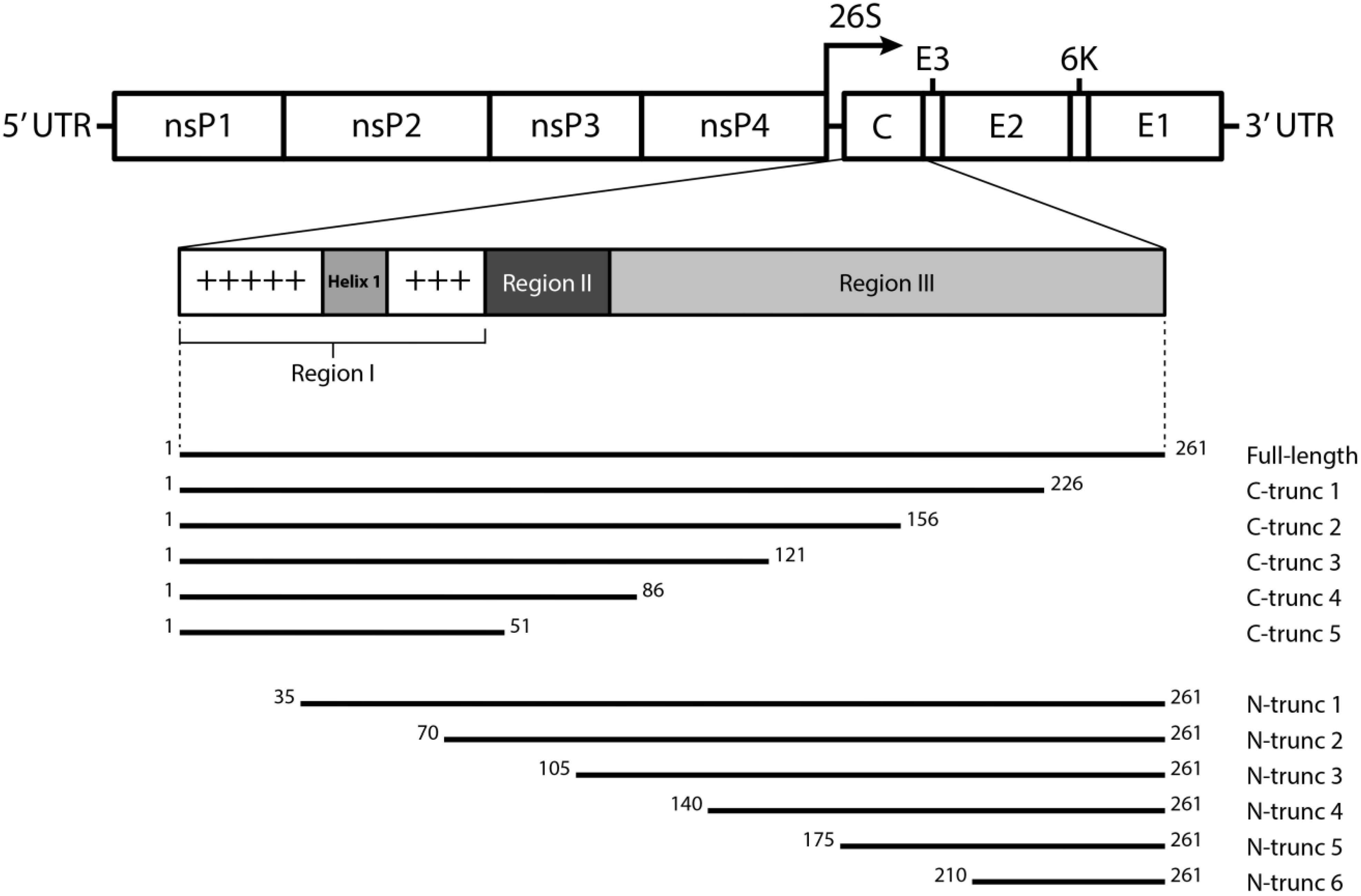
2.2. Cloning and Expression of CHIKV CP Truncations

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer code | Sequence (5′ to 3′) | Tm (°C) | Expected size (bp) |
|---|---|---|---|
| CHIKV Capsid F PCDNA | TATATAGCTAGCATGGAGTTCATCCCAACCCAA | 76.7 | - |
| CHIKV Capsid N1 R PCDNA | TATATAGGATCCGGCCACCACGCGTCCCT | 72.3 | 678 |
| CHIKV Capsid N2 R PCDNA | TATATAGGATCCGTGGTGCCAGTTGTAGTAC | 55.3 | 573 |
| CHIKV Capsid N3 R PCDNA | TATATAGGATCCCCGCTTAAAGGCCAGTTT | 61.7 | 468 |
| CHIKV Capsid N4 R PCDNA | TATATAGGATCCACCTTCGTGCTTGACTTC | 57.8 | 363 |
| CHIKV Capsid N5 R PCDNA | TATATAGGATCCCTGCTTCTTTTGATTTGTG | 55.9 | 258 |
| CHIKV Capsid N6 R PCDNA | TATATAGGATCCCAGTTTATTAACTGCTGAGATC | 55.3 | 153 |
| CHIKV Capsid R PCDNA | TATATAGGATCCACTCCACTCTTCGGCCCC | 74.8 | - |
| CHIKV Capsid C1 F PCDNA | TATATAGCTAGCATGAGGCAAGCTGGGCAAC | 67.6 | 678 |
| CHIKV Capsid C2 F PCDNA | TATATAGCTAGCATGAAGCAAAAACAACAGGC | 74.8 | 573 |
| CHIKV Capsid C3 F PCDNA | TATATAGCTAGCATGTGCATGAAAATCGAAAAT | 60.7 | 468 |
| CHIKV Capsid C4 F PCDNA | TATATAGCTAGCATGAAGGGGACCATCGATAA | 67.7 | 363 |
| CHIKV Capsid C5 F PCDNA | TATATAGCTAGCATGTCGAAGTTCACCCATGA | 65.6 | 258 |
2.3. Immunofluorescence Assay (IFA)
2.4. Western/Dot Blot
2.5. Competitive Binding Enzyme-Linked Immunosorbent Assay (ELISA)
2.6. Assessing Reactivity of mAbs to Synthetic Peptides
| No. | Sequence | Length | Protein * | Position | MIA | ELISA | Dot Blot |
|---|---|---|---|---|---|---|---|
| 1. | MEFIPTQTFYNRRYQPRPWT | 20 | CHIKV CP | 1–20 | − | − | − |
| 2. | NRRYQPRPWTPRPTIQVIRP | 20 | CHIKV CP | 11–30 | − | − | − |
| 3. | PRPTIQVIRPRPRPQRQAGQ | 20 | CHIKV CP | 21–40 | − | − | − |
| 4. | RPRPQRQAGQLAQLISAVNK | 20 | CHIKV CP | 31–50 | − | − | − |
| 5. | LAQLISAVNKLTMRAVPQQK | 20 | CHIKV CP | 41–60 | − | − | − |
| 6. | LTMRAVPQQKPRRNRKNKKQ | 20 | CHIKV CP | 51–70 | − | − | − |
| 7. | PRRNRKNKKQKQKQQAPQNN | 20 | CHIKV CP | 61–80 | − | − | − |
| 8. | KQKQQAPQNNTNQKKQPPKK | 20 | CHIKV CP | 71–90 | − | − | − |
| 9. | TNQKKQPPKKKPAQKKKKPG | 20 | CHIKV CP | 81–100 | − | − | − |
| 10. | KPAQKKKKPGRRERMCMKIE | 20 | CHIKV CP | 91–110 | − | − | − |
| 11. | RRERMCMKIENDCIFEVKHE | 20 | CHIKV CP | 101–120 | − | − | − |
| 12. | NDCIFEVKHEGKVTGYACLV | 20 | CHIKV CP | 111–130 | − | − | − |
| 13. | GKVTGYACLVGDKVMKPAHV | 20 | CHIKV CP | 121–140 | − | − | − |
| 14. | GDKVMKPAHVKGTIDNADLA | 20 | CHIKV CP | 131–150 | − | − | − |
| 15. | KGTIDNADLAKLAFKRSSKY | 20 | CHIKV CP | 141–160 | − | − | − |
| 16. | KLAFKRSSKYDLECAQIPVH | 20 | CHIKV CP | 151–170 | − | − | − |
| 17. | DLECAQIPVHMKSDASKFTH | 20 | CHIKV CP | 161–180 | − | − | − |
| 18. | MKSDASKFTHEKPEGYYNWH | 20 | CHIKV CP | 171–190 | − | − | − |
| 19. | EKPEGYYNWHHGAVQYSGGR | 20 | CHIKV CP | 181–200 | − | − | − |
| 20. | HGAVQYSGGRFTIPTGAGKP | 20 | CHIKV CP | 191–210 | − | − | − |
| 21. | FTIPTGAGKPGDSGRPIFDN | 20 | CHIKV CP | 201–220 | − | − | − |
| 22. | GDSGRPIFDNKGRVVAIVLG | 20 | CHIKV CP | 211–230 | − | − | − |
| 23. | KGRVVAIVLGGANEGARTAL | 20 | CHIKV CP | 221–240 | − | − | − |
| 24. | GANEGARTALSVVTWNKDIV | 20 | CHIKV CP | 231–250 | − | − | − |
| 25. | SVVTWNKDIVTKITPEGAEEW | 21 | CHIKV CP | 241–261 | − | − | − |
| 26. | SAAKHARKERNITGGHPVSR ǂ | 20 | WNVKUN NS5 | 38–57 | + | + | + |
| 27. | CTTVESHGNYSTQVGATQAG ǂ | 20 | WNVNY99 E | 146–165 | + | + | + |
| 28. | Full-length CHIKV CP | 261 | CHIKV CP | N/A | # ND | + | + |
2.7. Immunoprecipitation of CHIKV CP
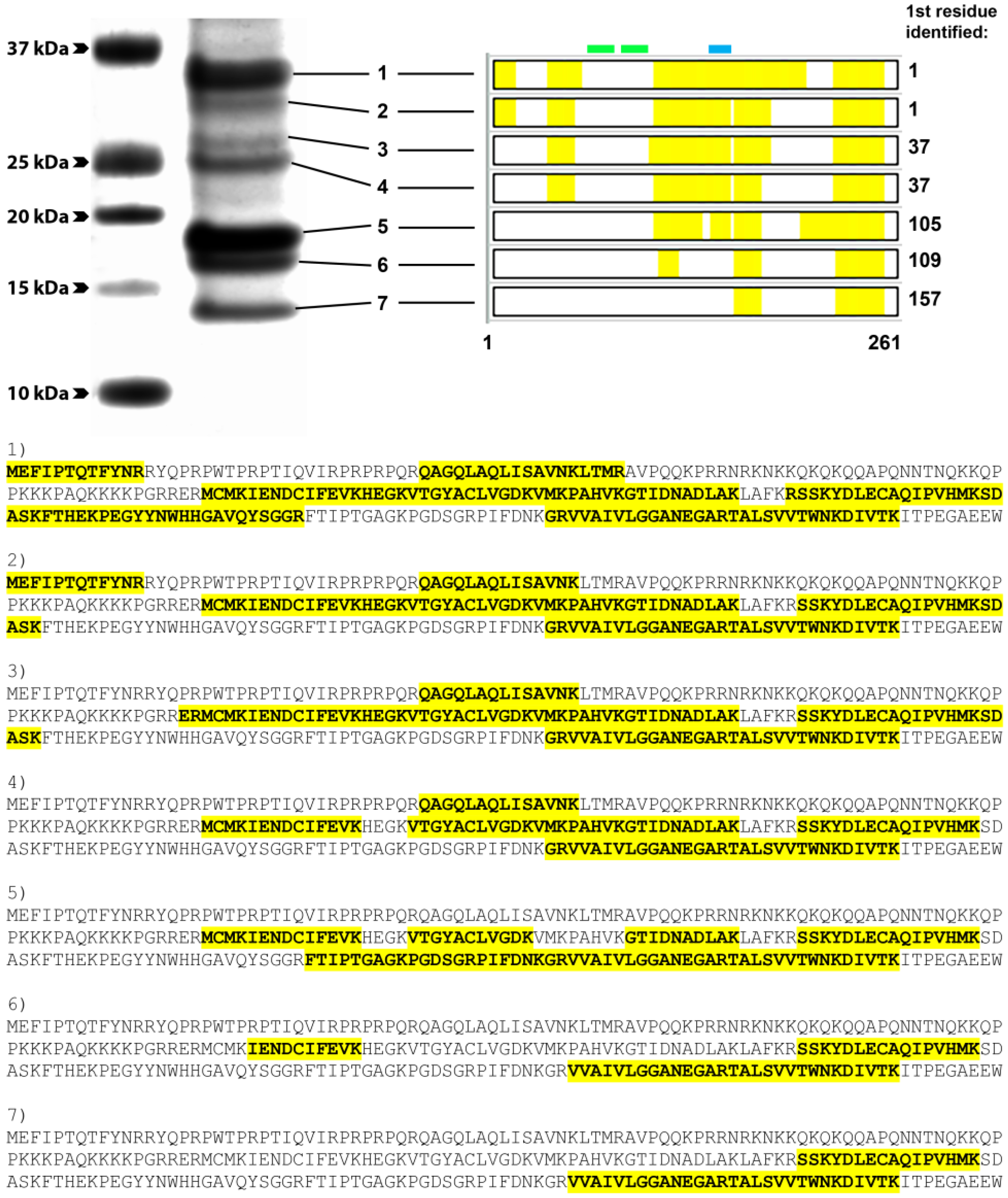
2.8. Mass Spectrometry Analysis
2.9. Data Analysis
3. Results
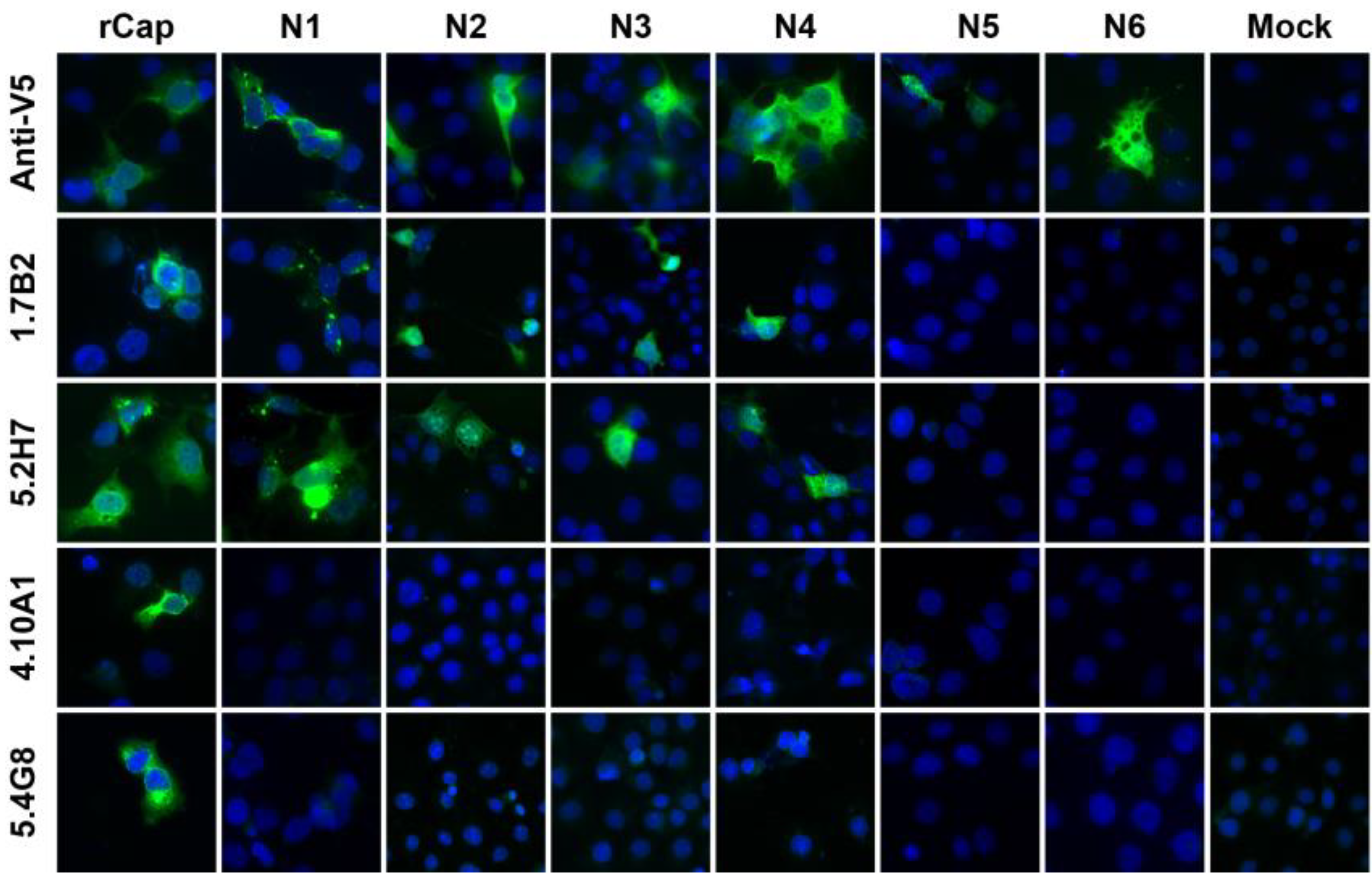
3.1. Anti-CHIKV CP mAbs Recognize Linear Epitopes


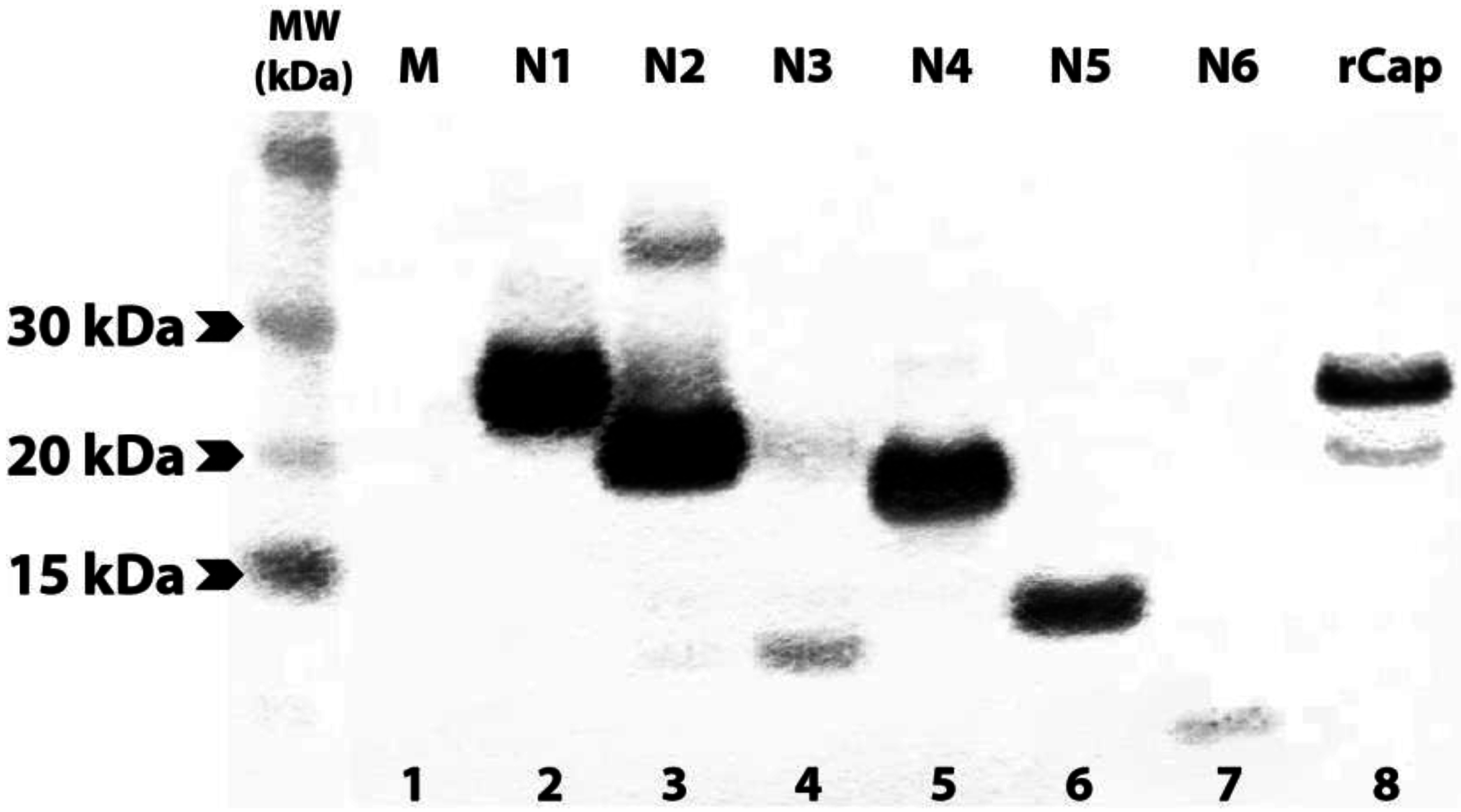
3.2. Expression of N- and C-terminally Truncated CHIKV CP


| Monoclonal antibody | Group | Reactivity in Western blot | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| FL | N1 | N2 | N3 | N4 | N5 | N6 | C1 | C2 | C3 | C4 | C5 | ||
| 1.7B2 | 1 | + | + | + | + | + | − | − | − | − | − | − | − |
| 4.1H11 | 1 | + | + | + | + | + | − | − | − | − | − | − | − |
| 5.2H7 | 1 | + | + | + | + | + | − | − | − | − | − | − | − |
| 5.5D11 | 1 | + | + | + | + | + | − | − | − | − | − | − | − |
| 5.5G9 | 1 | + | + | + | + | + | − | − | − | − | − | − | − |
| 5.1B12 | 2 | + | − | − | − | − | − | − | − | − | − | − | − |
| 5.5A11 | 2 | + | − | − | − | − | − | − | − | − | − | − | − |
| 4.8E2 | 2 | + | − | − | − | − | − | − | − | − | − | − | − |
| 4.10A11 | 2 | + | − | − | − | − | − | − | − | − | − | − | − |
| 5.2F8 | 2 | + | − | − | − | − | − | − | − | − | − | − | − |
| 5.4G8 | 2 | + | − | − | − | − | − | − | − | − | − | − | − |
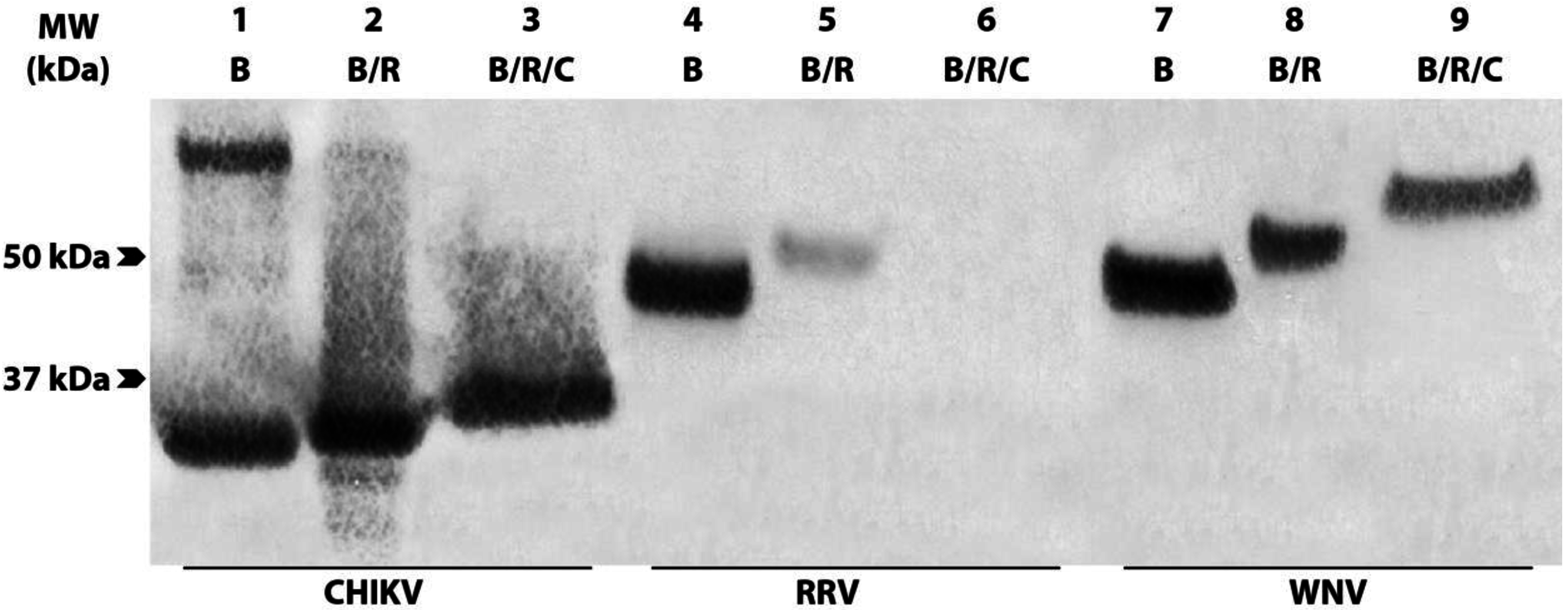
3.3. Group 1 mAbs Bind Native N-terminally Truncated CP in CHIKV Lysates


3.4. Group 1 mAbs Target a Series of Overlapping Epitopes on CP


4. Discussion

5. Conclusions
Supplementary Files
Supplementary File 1Acknowledgements
Author Contributions
Conflicts of Interest
References
- Suhrbier, A.; Jaffar-Bandjee, M.C.; Gasque, P. Arthritogenic alphaviruses—An overview. Nat. Rev. Rheumatol. 2012, 8, 420–429. [Google Scholar] [CrossRef] [PubMed]
- Tesh, R.B. Arthritides caused by mosquito-borne viruses. Annu. Rev. Med. 1982, 33, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Staples, J.E.; Breiman, R.F.; Powers, A.M. Chikungunya fever: An epidemiological review of a re-emerging infectious disease. Clin. Infect. Dis. 2009, 49, 942–948. [Google Scholar] [CrossRef] [PubMed]
- Borgherini, G.; Poubeau, P.; Staikowsky, F.; Lory, M.; Le Moullec, N.; Becquart, J.P.; Wengling, C.; Michault, A.; Paganin, F. Outbreak of chikungunya on Reunion Island: Early clinical and laboratory features in 157 adult patients. Clin. Infect. Dis. 2007, 44, 1401–1407. [Google Scholar] [CrossRef] [PubMed]
- Renault, P.; Solet, J.L.; Sissoko, D.; Balleydier, E.; Larrieu, S.; Filleul, L.; Lassalle, C.; Thiria, J.; Rachou, E.; de Valk, H.; et al. A major epidemic of chikungunya virus infection on Reunion Island, France, 2005–2006. Am. J. Trop. Med. Hyg. 2007, 77, 727–731. [Google Scholar] [PubMed]
- Grandadam, M.; Caro, V.; Plumet, S.; Thiberge, J.M.; Souares, Y.; Failloux, A.B.; Tolou, H.J.; Budelot, M.; Cosserat, D.; Leparc-Goffart, I.; et al. Chikungunya virus, southeastern France. Emerg. Infect. Dis. 2011, 17, 910–913. [Google Scholar] [CrossRef] [PubMed]
- Rezza, G.; Nicoletti, L.; Angelini, R.; Romi, R.; Finarelli, A.C.; Panning, M.; Cordioli, P.; Fortuna, C.; Boros, S.; Magurano, F.; et al. Infection with Chikungunya virus in Italy: An outbreak in a temperate region. Lancet 2007, 370, 1840–1846. [Google Scholar] [CrossRef] [PubMed]
- Lam, S.K.; Chua, K.B.; Hooi, P.S.; Rahimah, M.A.; Kumari, S.; Tharmaratnam, M.; Chuah, S.K.; Smith, D.W.; Sampson, I.A. Chikungunya infection—An emerging disease in Malaysia. Southeast Asian J. Trop. Med. Public Health 2001, 32, 447–451. [Google Scholar] [PubMed]
- Munasinghe, D.R.; Amarasekera, P.J.; Fernando, C.F. An epidemic of dengue-like fever in Ceylon (chikungunya—A clinical and haematological study. Ceylon Med. J. 1966, 11, 129–142. [Google Scholar] [PubMed]
- Horwood, P.; Bande, G.; Dagina, R.; Guillaumot, L.; Aaskov, J.; Pavlin, B. The threat of chikungunya in Oceania. West. Pac. Surveill. Response J. 2013, 4, 8–10. [Google Scholar] [CrossRef] [PubMed]
- Van Bortel, W.; Dorleans, F.; Rosine, J.; Blateau, A.; Rousset, D.; Matheus, S.; Leparc-Goffart, I.; Flusin, O.; Prat, C.; Cesaire, R.; et al. Chikungunya outbreak in the Caribbean region, December 2013 to March 2014, and the significance for Europe. Euro Surveill. 2014, 19. [Google Scholar] [CrossRef]
- Jaffar-Bandjee, M.C.; Ramful, D.; Gauzere, B.A.; Hoarau, J.J.; Krejbich-Trotot, P.; Robin, S.; Ribera, A.; Selambarom, J.; Gasque, P. Emergence and clinical insights into the pathology of Chikungunya virus infection. Expert Rev. Anti Infect. Ther. 2010, 8, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Tandale, B.V.; Sathe, P.S.; Arankalle, V.A.; Wadia, R.S.; Kulkarni, R.; Shah, S.V.; Shah, S.K.; Sheth, J.K.; Sudeep, A.B.; Tripathy, A.S.; et al. Systemic involvements and fatalities during Chikungunya epidemic in India, 2006. J. Clin. Virol. 2009, 46, 145–149. [Google Scholar] [CrossRef] [PubMed]
- Economopoulou, A.; Dominguez, M.; Helynck, B.; Sissoko, D.; Wichmann, O.; Quenel, P.; Germonneau, P.; Quatresous, I. Atypical Chikungunya virus infections: Clinical manifestations, mortality and risk factors for severe disease during the 2005–2006 outbreak on Reunion. Epidemiol. Infect. 2009, 137, 534–541. [Google Scholar] [CrossRef] [PubMed]
- Mavalankar, D.; Shastri, P.; Bandyopadhyay, T.; Parmar, J.; Ramani, K.V. Increased mortality rate associated with Chikungunya epidemic, Ahmedabad, India. Emerg. Infect. Dis. 2008, 14, 412–415. [Google Scholar] [CrossRef] [PubMed]
- Strauss, J.H.; Strauss, E.G. The alphaviruses: Gene expression, replication, and evolution. Microbiol. Rev. 1994, 58, 491–562. [Google Scholar] [PubMed]
- Choi, H.K.; Tong, L.; Minor, W.; Dumas, P.; Boege, U.; Rossmann, M.G.; Wengler, G. Structure of Sindbis virus core protein reveals a chymotrypsin-like serine proteinase and the organization of the virion. Nature 1991, 354, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Aguilar, P.V.; Weaver, S.C.; Basler, C.F. Capsid protein of eastern equine encephalitis virus inhibits host cell gene expression. J. Virol. 2007, 81, 3866–3876. [Google Scholar] [CrossRef] [PubMed]
- Elgizoli, M.; Dai, Y.; Kempf, C.; Koblet, H.; Michel, M.R. Semliki Forest virus capsid protein acts as a pleiotropic regulator of host cellular protein synthesis. J. Virol. 1989, 63, 2921–2928. [Google Scholar] [PubMed]
- Hong, E.M.; Perera, R.; Kuhn, R.J. Alphavirus capsid protein helix I controls a checkpoint in nucleocapsid core assembly. J. Virol. 2006, 80, 8848–8855. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.; Rai, J.; John, L.; Schaefer, S.; Putzer, B.M.; Herchenroder, O. Chikungunya virus capsid protein contains nuclear import and export signals. Virol. J. 2013, 10, 269. [Google Scholar] [CrossRef] [PubMed]
- Favre, D.; Studer, E.; Michel, M.R. Two nucleolar targeting signals present in the N-terminal part of Semliki Forest virus capsid protein. Arch. Virol. 1994, 137, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Jakob, R. Nucleolar accumulation of Semliki Forest virus nucleocapsid C protein: Influence of metabolic status, cytoskeleton and receptors. J. Med. Microbiol. 1994, 40, 389–392. [Google Scholar] [CrossRef] [PubMed]
- Nigg, E.A.; Baeuerle, P.A.; Luhrmann, R. Nuclear import-export: In search of signals and mechanisms. Cell 1991, 66, 15–22. [Google Scholar] [CrossRef]
- Clark, D.C.; Lobigs, M.; Lee, E.; Howard, M.J.; Clark, K.; Blitvich, B.J.; Hall, R.A. In situ reactions of monoclonal antibodies with a viable mutant of Murray Valley encephalitis virus reveal an absence of dimeric NS1 protein. J. Gen. Virol. 2007, 88, 1175–1183. [Google Scholar] [CrossRef] [PubMed]
- Hobson-Peters, J.; Toye, P.; Sanchez, M.D.; Bossart, K.N.; Wang, L.F.; Clark, D.C.; Cheah, W.Y.; Hall, R.A. A glycosylated peptide in the West Nile virus envelope protein is immunogenic during equine infection. J. Gen. Virol. 2008, 89, 3063–3072. [Google Scholar] [CrossRef] [PubMed]
- Setoh, Y.X.; Hobson-Peters, J.; Prow, N.A.; Young, P.R.; Hall, R.A. Expression of recombinant West Nile virus prM protein fused to an affinity tag for use as a diagnostic antigen. J. Virol. Methods 2011, 175, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Goh, L.Y.; Hobson-Peters, J.; Prow, N.A.; Gardner, J.; Bielefeldt-Ohmann, H.; Pyke, A.T.; Suhrbier, A.; Hall, R.A. Neutralizing monoclonal antibodies to the E2 protein of chikungunya virus protects against disease in a mouse model. Clin. Immunol. 2013, 149, 487–497. [Google Scholar] [CrossRef] [PubMed]
- Hall, R.A.; Tan, S.E.; Selisko, B.; Slade, R.; Hobson-Peters, J.; Canard, B.; Hughes, M.; Leung, J.Y.; Balmori-Melian, E.; Hall-Mendelin, S.; et al. Monoclonal antibodies to the West Nile virus NS5 protein map to linear and conformational epitopes in the methyltransferase and polymerase domains. J. Gen. Virol. 2009, 90, 2912–2922. [Google Scholar] [CrossRef] [PubMed]
- Hobson-Peters, J.; Toye, P. A whole-blood homogeneous assay for the multiplex detection of the factor V G1691A and the prothrombin G20210A mutations. Mol. Cell. Probes 2005, 19, 290–297. [Google Scholar] [CrossRef] [PubMed]
- Hastie, M.L.; Headlam, M.J.; Patel, N.B.; Bukreyev, A.A.; Buchholz, U.J.; Dave, K.A.; Norris, E.L.; Wright, C.L.; Spann, K.M.; Collins, P.L.; et al. The human respiratory syncytial virus nonstructural protein 1 regulates type I and type II interferon pathways. Mol. Cell. Proteomics 2012, 11, 108–127. [Google Scholar] [CrossRef] [PubMed]
- Searle, B.C. Scaffold: A bioinformatic tool for validating MS/MS-based proteomic studies. Proteomics 2010, 10, 1265–1269. [Google Scholar] [CrossRef] [PubMed]
- Keller, A.; Nesvizhskii, A.I.; Kolker, E.; Aebersold, R. Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal. Chem. 2002, 74, 5383–5392. [Google Scholar] [CrossRef] [PubMed]
- Goh, L.Y.; Hobson-Peters, J.; Prow, N.A.; Gardner, J.; Bielefeldt-Ohmann, H.; Suhrbier, A.; Hall, R.A. Monoclonal antibodies specific for the capsid protein of chikungunya virus suitable for multiple applications. J. Gen. Virol. 2015, 96, 507–512. [Google Scholar] [CrossRef] [PubMed]
- Blom, N.; Gammeltoft, S.; Brunak, S. Sequence- and structure-based prediction of eukaryotic protein phosphorylation sites. J. Mol. Biol. 1999, 294, 1351–1362. [Google Scholar] [CrossRef] [PubMed]
- Brandler, S.; Ruffie, C.; Combredet, C.; Brault, J.B.; Najburg, V.; Prevost, M.C.; Habel, A.; Tauber, E.; Despres, P.; Tangy, F. A recombinant measles vaccine expressing chikungunya virus-like particles is strongly immunogenic and protects mice from lethal challenge with chikungunya virus. Vaccine 2013, 31, 3718–3725. [Google Scholar] [CrossRef] [PubMed]
- Cho, B.; Kim, J.; Cho, J.E.; Jeon, B.Y.; Park, S. Expression of the capsid protein of Chikungunya virus in a baculovirus for serodiagnosis of Chikungunya disease. J. Virol. Methods 2008, 154, 154–159. [Google Scholar] [CrossRef] [PubMed]
- Bell, J.R.; Bond, M.W.; Hunkapiller, M.W.; Strauss, E.G.; Strauss, J.H.; Yamamoto, K.; Simizu, B. Structural proteins of Western equine encephalitis virus: Amino acid compositions and N-terminal sequences. J. Virol. 1983, 45, 708–714. [Google Scholar] [PubMed]
- Choi, H.K.; Lee, S.; Zhang, Y.P.; McKinney, B.R.; Wengler, G.; Rossmann, M.G.; Kuhn, R.J. Structural analysis of Sindbis virus capsid mutants involving assembly and catalysis. J. Mol. Biol. 1996, 262, 151–167. [Google Scholar] [CrossRef] [PubMed]
- Ishida, I.; Simizu, B. Evidence for the presence of the minor capsid protein of Western equine encephalitis virus. Arch. Virol. 1981, 67, 159–164. [Google Scholar] [CrossRef] [PubMed]
- Ventoso, I.; Sanz, M.A.; Molina, S.; Berlanga, J.J.; Carrasco, L.; Esteban, M. Translational resistance of late alphavirus mRNA to eIF2alpha phosphorylation: A strategy to overcome the antiviral effect of protein kinase PKR. Genes Dev. 2006, 20, 87–100. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, R. The Togaviruses (Chapter 23). In Fields Virology, 6th ed.; Lippencott Williams and Wilkins: Philadelphia, PA, USA, 2013. [Google Scholar]
- Firth, A.E.; Brierley, I. Non-canonical translation in RNA viruses. J. Gen. Virol. 2012, 93, 1385–1409. [Google Scholar] [CrossRef] [PubMed]
- Glenney, J.R., Jr.; Zokas, L.; Kamps, M.P. Monoclonal antibodies to phosphotyrosine. J. Immunol. Methods 1988, 109, 277–285. [Google Scholar] [CrossRef]
- Kehoe, J.W.; Velappan, N.; Walbolt, M.; Rasmussen, J.; King, D.; Lou, J.; Knopp, K.; Pavlik, P.; Marks, J.D.; Bertozzi, C.R.; et al. Using phage display to select antibodies recognizing post-translational modifications independently of sequence context. Mol. Cell. Proteomics 2006, 5, 2350–2363. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Goh, L.Y.H.; Hobson-Peters, J.; Prow, N.A.; Baker, K.; Piyasena, T.B.H.; Taylor, C.T.; Rana, A.; Hastie, M.L.; Gorman, J.J.; Hall, R.A. The Chikungunya Virus Capsid Protein Contains Linear B Cell Epitopes in the N- and C-Terminal Regions that are Dependent on an Intact C-Terminus for Antibody Recognition. Viruses 2015, 7, 2943-2964. https://0-doi-org.brum.beds.ac.uk/10.3390/v7062754
Goh LYH, Hobson-Peters J, Prow NA, Baker K, Piyasena TBH, Taylor CT, Rana A, Hastie ML, Gorman JJ, Hall RA. The Chikungunya Virus Capsid Protein Contains Linear B Cell Epitopes in the N- and C-Terminal Regions that are Dependent on an Intact C-Terminus for Antibody Recognition. Viruses. 2015; 7(6):2943-2964. https://0-doi-org.brum.beds.ac.uk/10.3390/v7062754
Chicago/Turabian StyleGoh, Lucas Y. H., Jody Hobson-Peters, Natalie A. Prow, Kelly Baker, Thisun B. H. Piyasena, Carmel T. Taylor, Ashok Rana, Marcus L. Hastie, Jeff J. Gorman, and Roy A. Hall. 2015. "The Chikungunya Virus Capsid Protein Contains Linear B Cell Epitopes in the N- and C-Terminal Regions that are Dependent on an Intact C-Terminus for Antibody Recognition" Viruses 7, no. 6: 2943-2964. https://0-doi-org.brum.beds.ac.uk/10.3390/v7062754