
Evaluation of Trichodysplasia Spinulosa-Associated Polyomavirus Capsid Protein as a New Carrier for Construction of Chimeric Virus-Like Particles Harboring Foreign Epitopes

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Bioinformatics Analysis of TSPyV VP1 Protein
2.2. Construction of Expression Plasmids
2.3. Generation, Purification and Electron Microscopy Analysis of Chimeric VLPs
2.4. Immunization of Mice
2.5. Generation of Monoclonal Antibodies against Intact TSPyV VP1-Derived VLPs
2.6. Indirect Enzyme-Linked Immunosorbent Assay (ELISA)
2.7. SDS-PAGE and Western Blot Analysis
2.8. Preparation of Murine Spleen Cell Cultures and Stimulation with VLPs
2.9. ELISPOT Analysis
2.10. Flow Cytometry Analysis
2.11. Statistical Analysis
3. Results
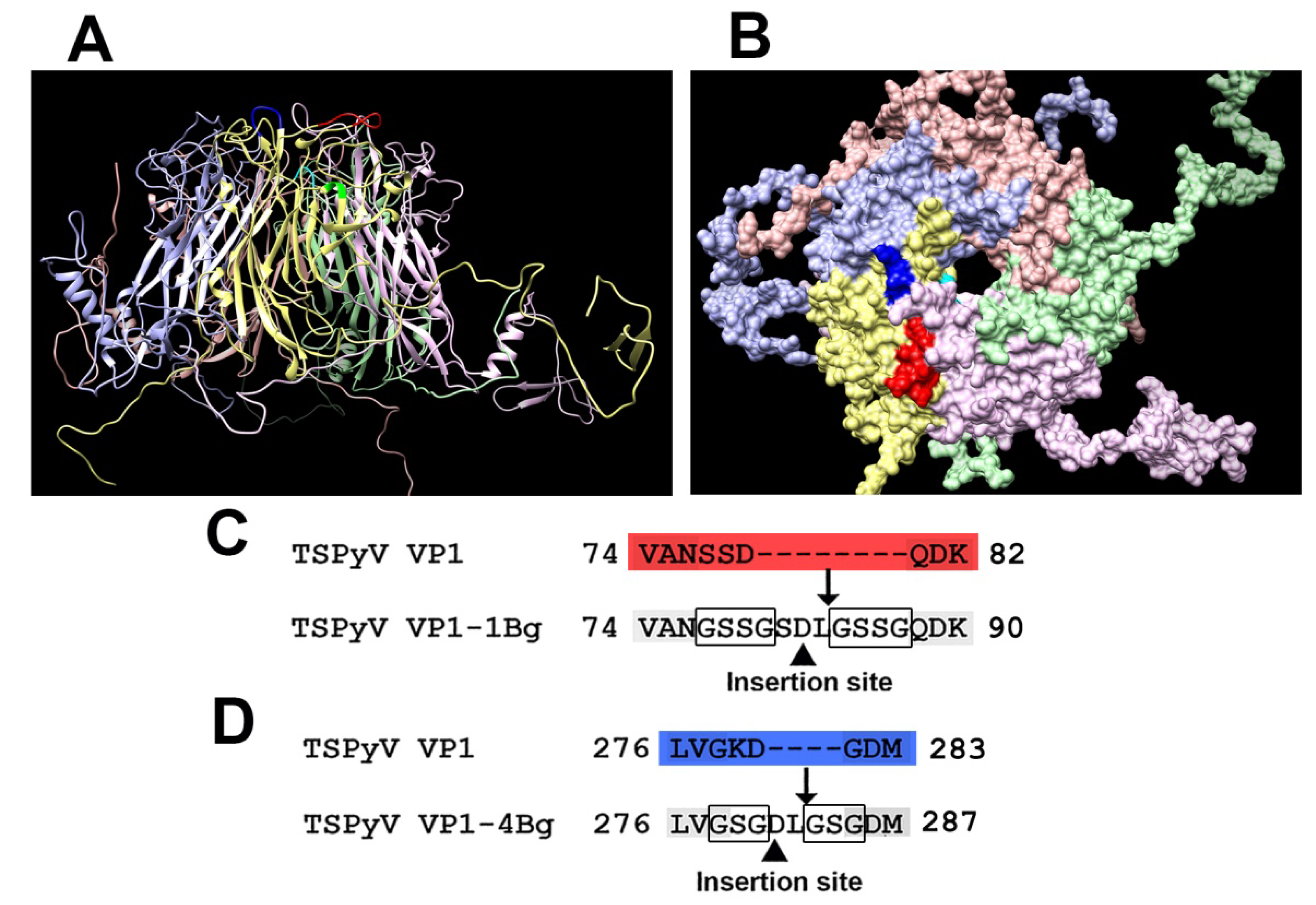
3.1. Computer Modeling of TSPyV VP1 Protein and Selection of Insert Positions




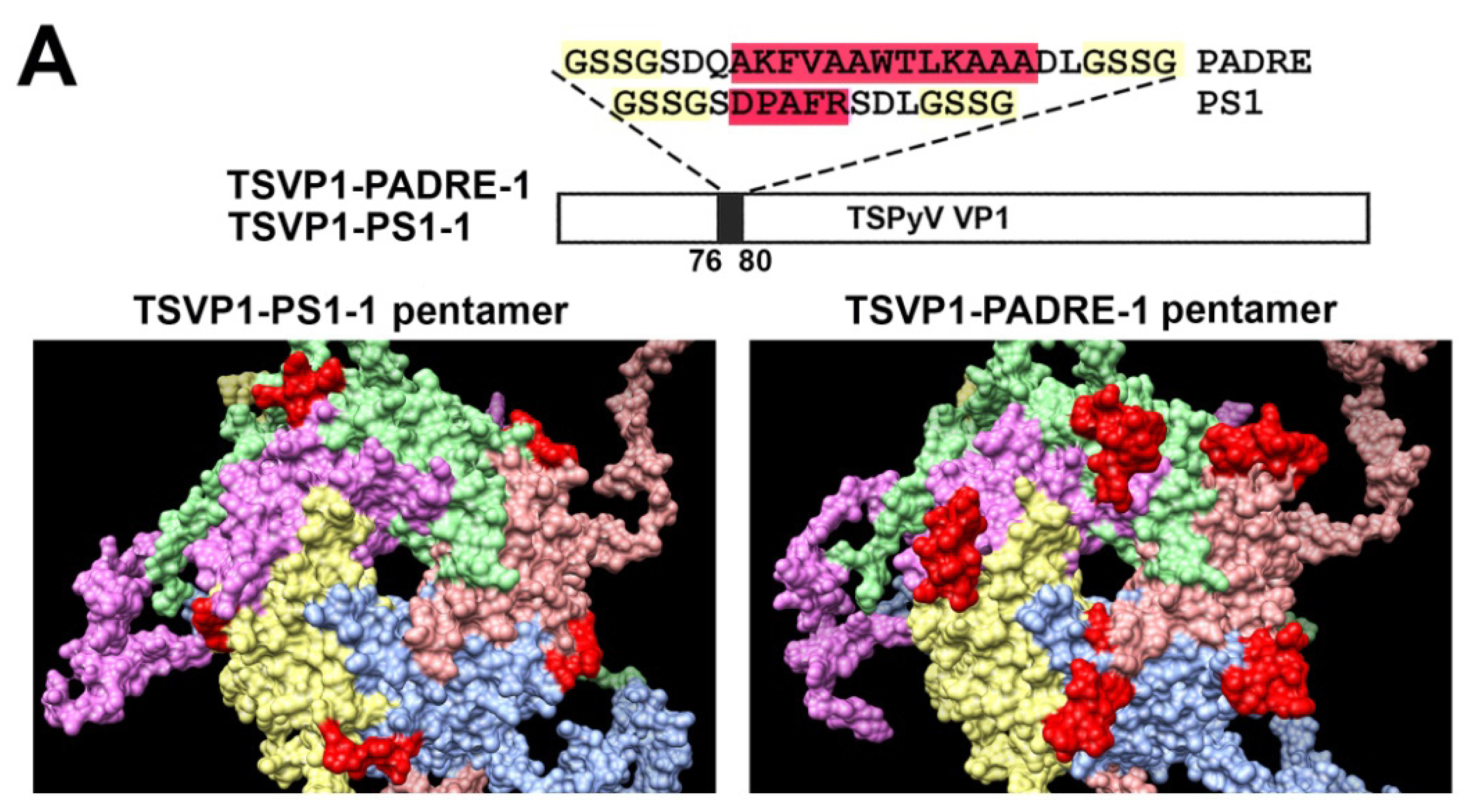
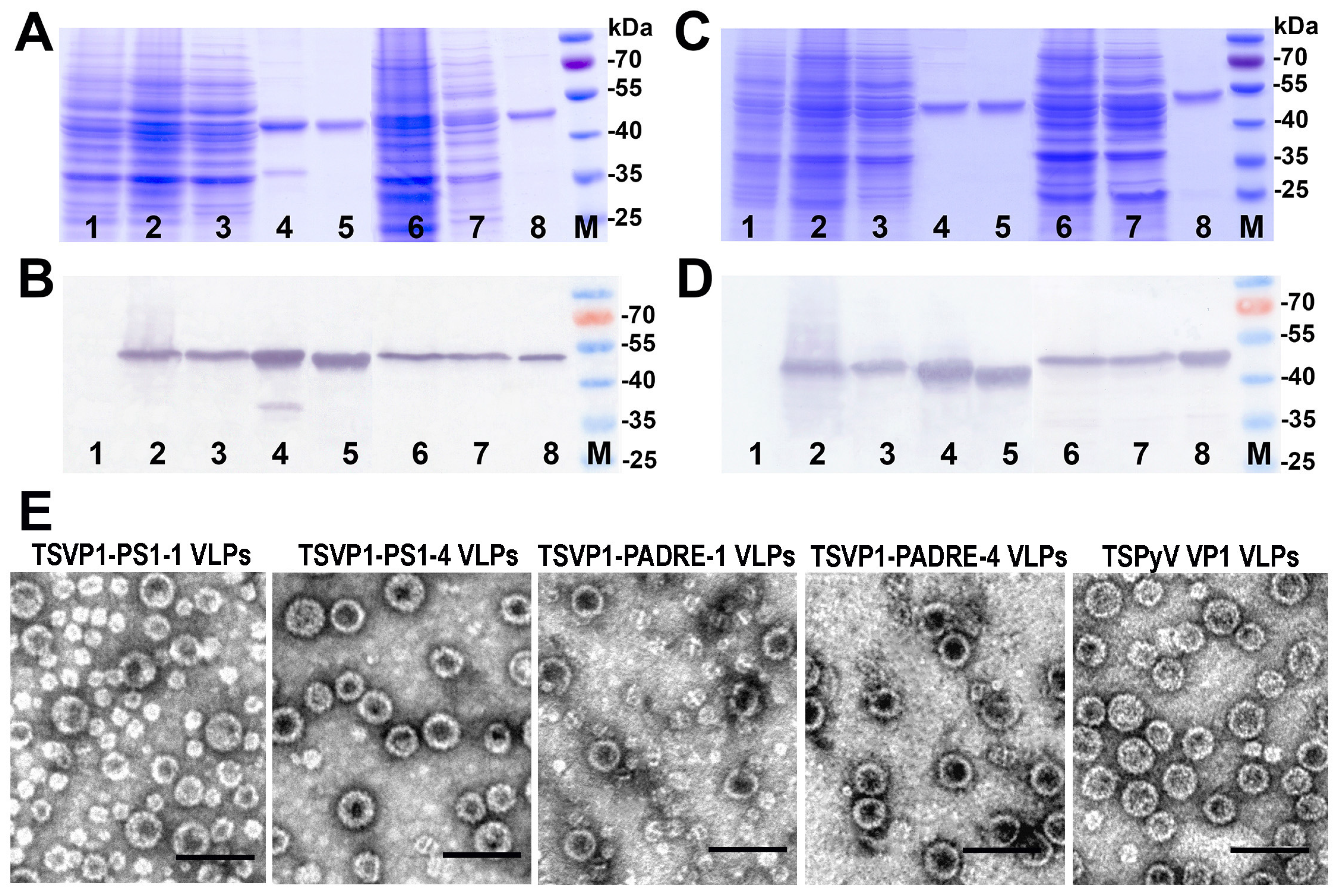
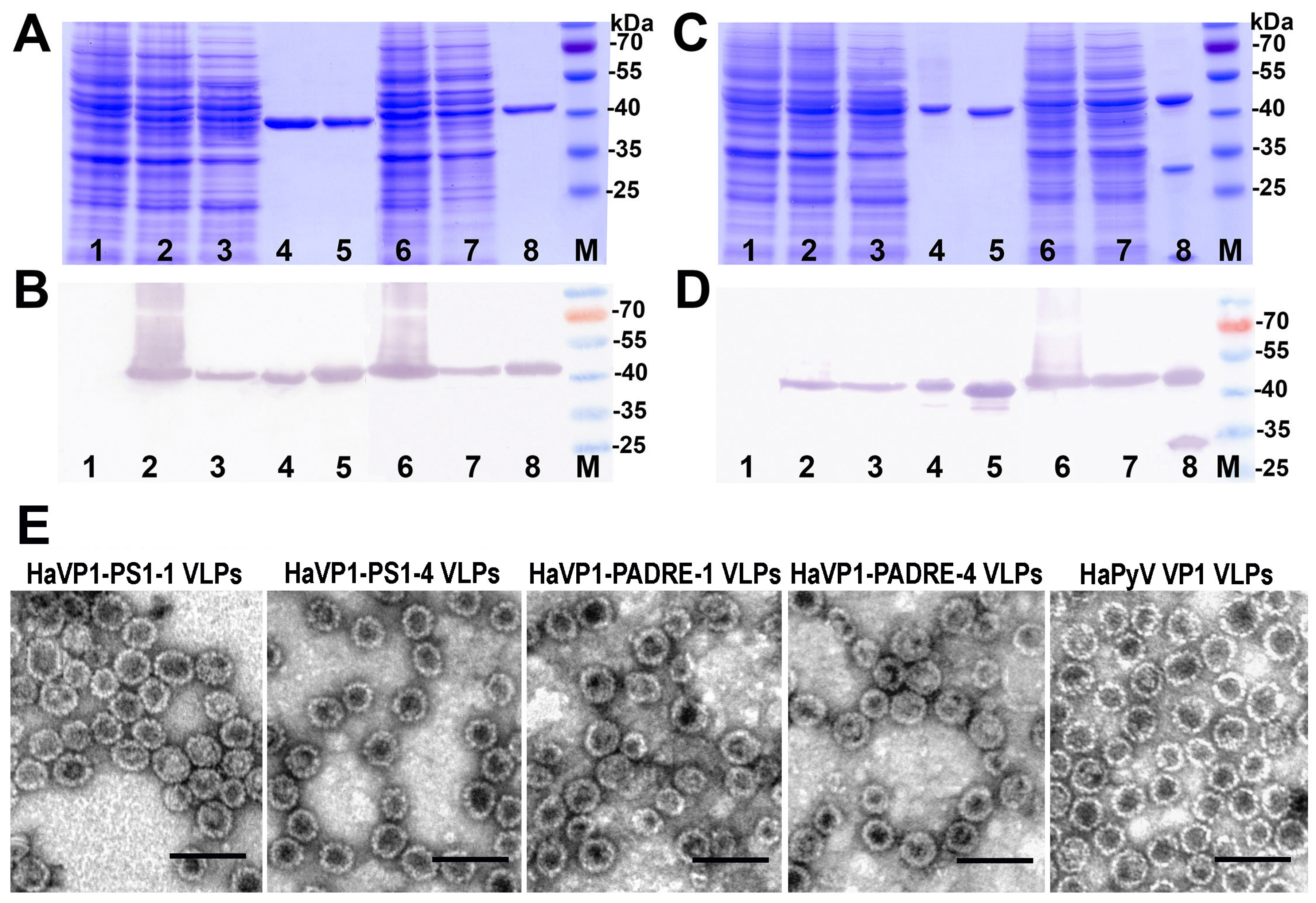
3.2. Construction and Purification of Chimeric VLPs


3.3. Generation of Monoclonal Antibodies against TSPyV VP1 Protein and Their Use to Analyze the Structure of Chimeric VLPs
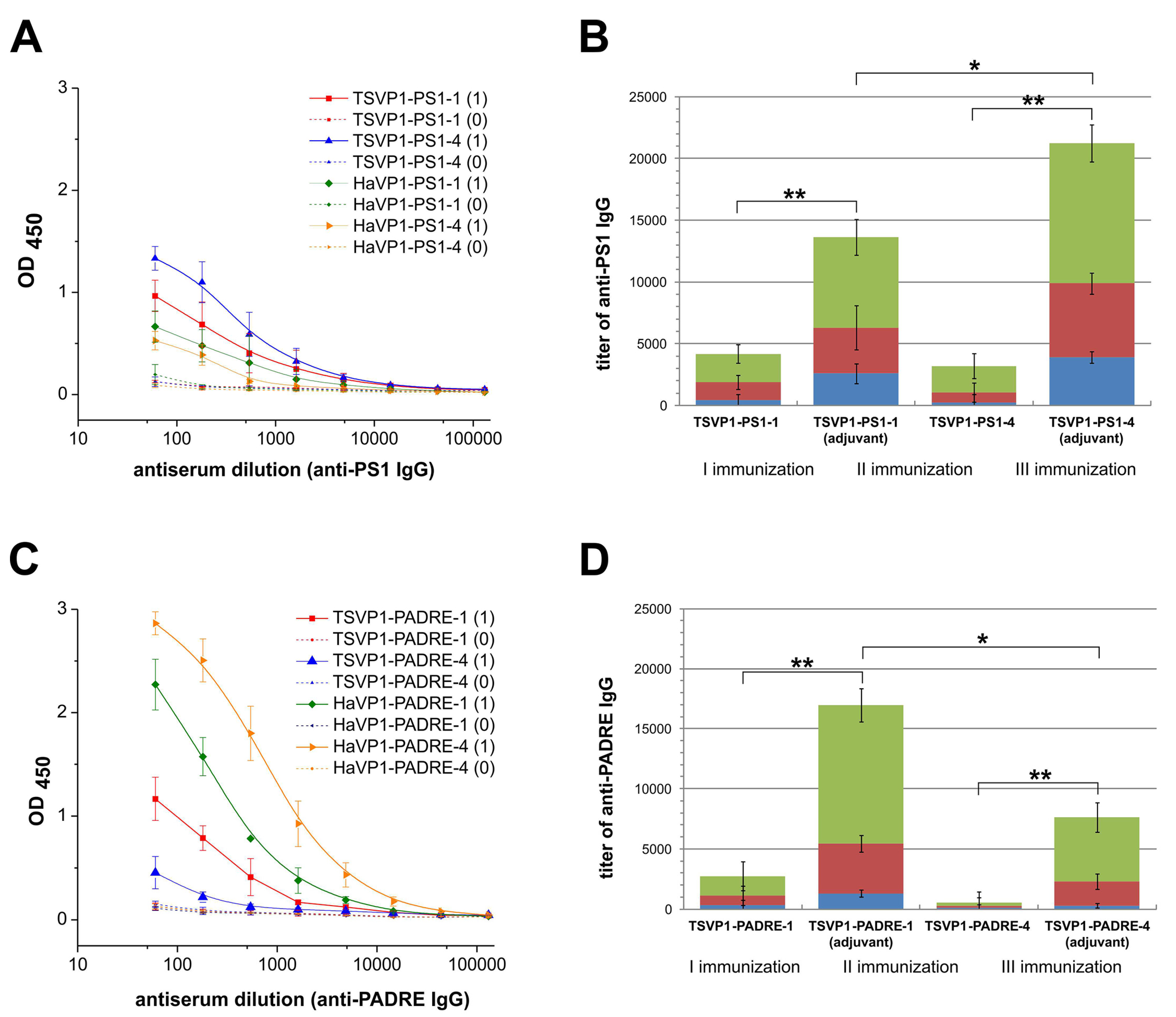
3.4. Immunogenicity of Chimeric VLPs: Induction of Antibody Response

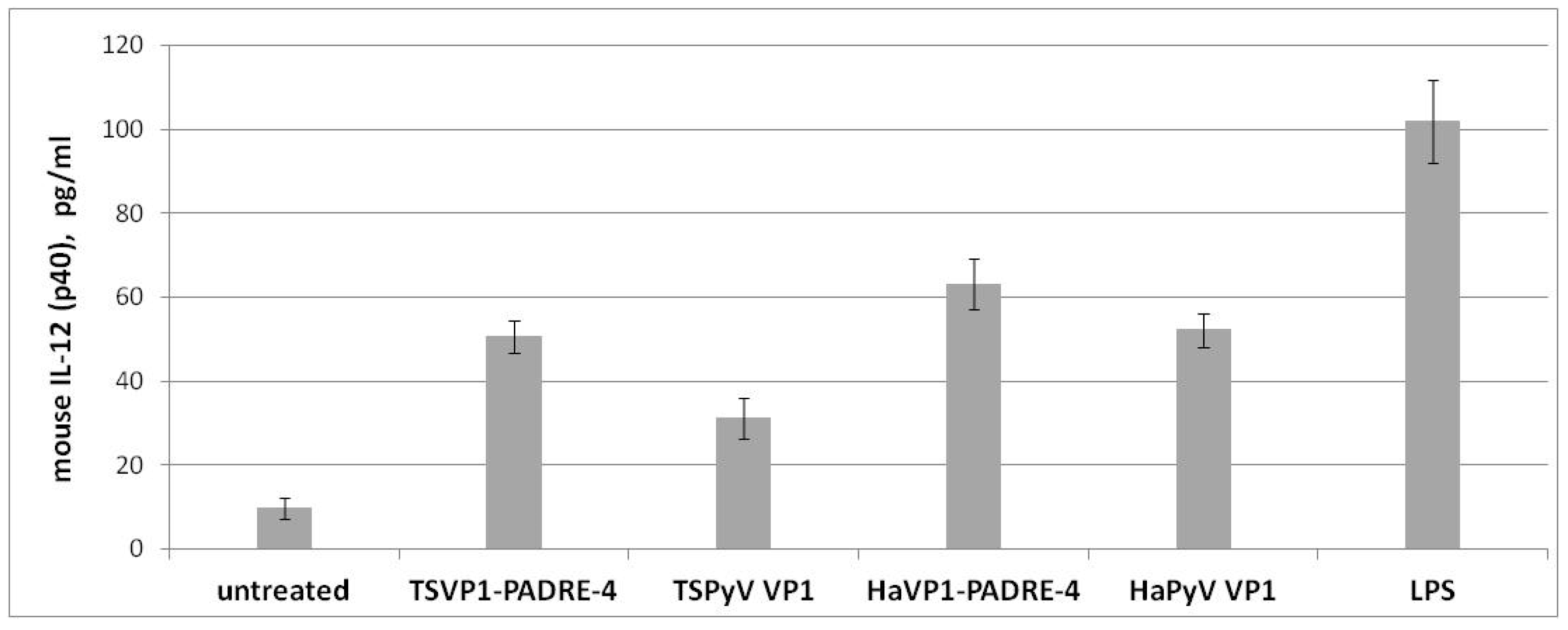
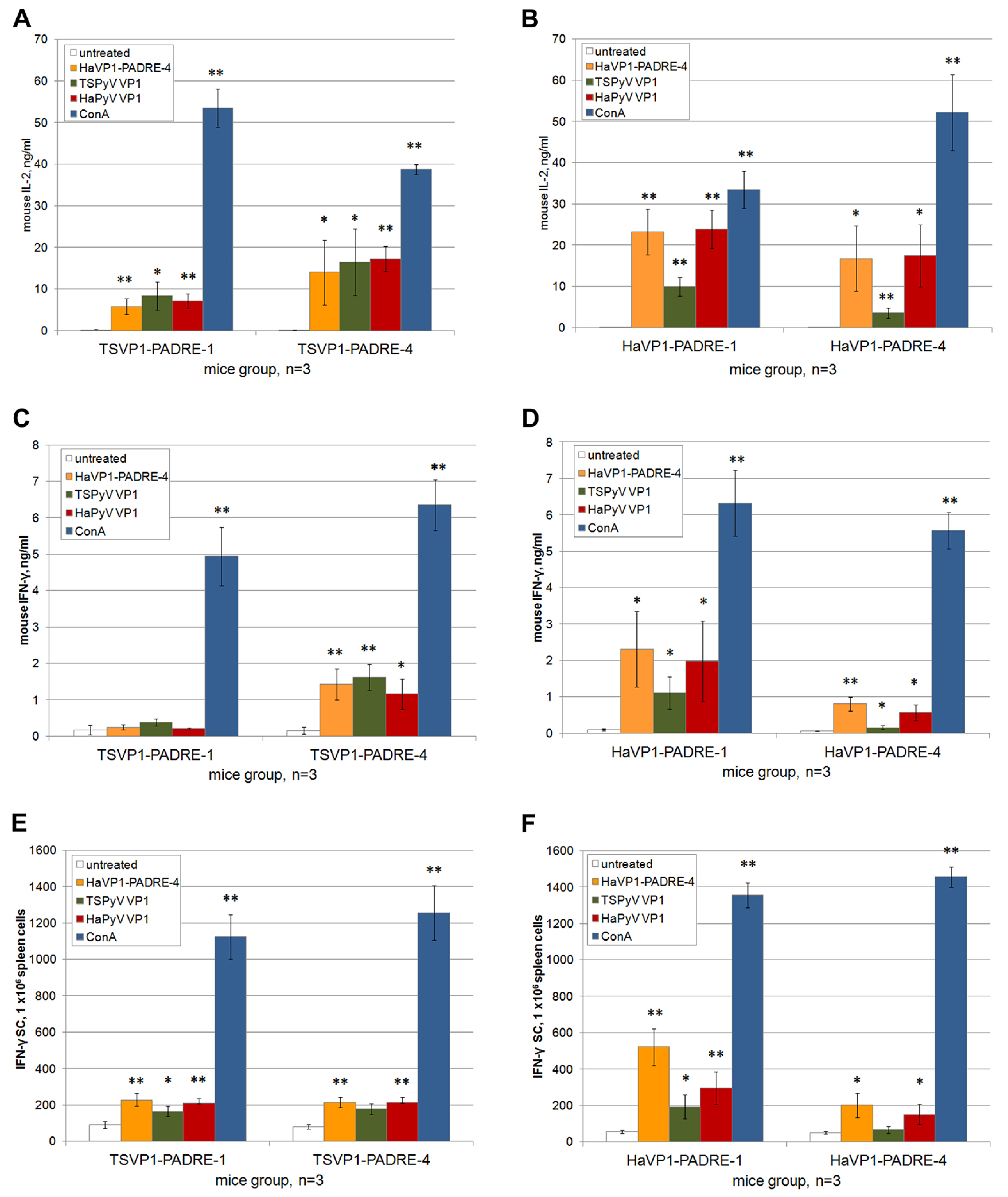
3.5. Activation of Murine Spleen Cells by Chimeric VLPs



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| % of CD4+ CD69+ Cells within CD4+ T Cells Population | ||||||
| Antigen Used for Stimulation | Untreated | HaVP1-PADRE-4 | TSPyV VP1 | HaPyV VP1 | ConA | |
| Antigen Used for Immunization | ||||||
| TSVP1-PADRE-1 | 0.10 ±0.005 | 0.64 ± 0.05 | 0.58 ± 0.05 | 0.53 ± 0.09 | 24.42 ± 1.96 | |
| TSVP1-PADRE-4 | 0.11 ± 0.004 | 0.93 ± 0.1 | 0.57 ± 0.01 | 0.73 ± 0.05 | 21.64 ± 1.73 | |
| HaVP1-PADRE-1 | 0.10 ± 0.03 | 1.13 ± 0.15 | 0.65 ± 0.05 | 0.84 ± 0.09 | 18.4 ± 2.83 | |
| HaVP1-PADRE-4 | 0.12 ± 0.006 | 0.72 ±0.08 | 0.50 ±0.03 | 0.62 ±0.08 | 22.07 ±1.67 | |
| % of CD8+ CD69+ Cells within CD8+ T Cells Population | ||||||
| Antigen Used for Stimulation | Untreated | HaVP1-PADRE-4 | TSPyV VP1 | HaPyV VP1 | ConA | |
| Antigen Used for Immunization | ||||||
| TSVP1-PADRE-1 | 0.67 ± 0.02 | 1.03 ± 0.09 | 0.77 ± 0.05 | 0.81 ± 0.09 | 32.44 ± 2.62 | |
| TSVP1-PADRE-4 | 0.64 ± 0.04 | 0.99 ± 0.07 | 0.86 ± 0.07 | 0.79 ± 0.15 | 33.10 ± 0.34 | |
| HaVP1-PADRE-1 | 0.65 ± 0.03 | 2.46 ± 0.41 | 1.03 ± 0.01 | 2.52 ± 0.25 | 27.70 ± 0.72 | |
| HaVP1-PADRE-4 | 0.68 ± 0.03 | 1.17 ± 0.11 | 0.74 ± 0.01 | 0.78 ± 0.09 | 26.11 ± 1.15 | |
4. Discussion
5. Conclusions
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Zeltins, A. Construction and characterization of virus-like particles: A review. Mol. Biotechnol. 2013, 53, 92–107. [Google Scholar] [CrossRef] [PubMed]
- Bachmann, M.F.; Jennings, G.T. Vaccine delivery: A matter of size, geometry, kinetics and molecular patterns. Nat. Rev. Immunol. 2010, 10, 787–796. [Google Scholar] [CrossRef] [PubMed]
- Pushko, P.; Pumpens, P.; Grens, E. Development of virus-like particle technology from small highly symmetric to large complex virus-like particle structures. Intervirology 2013, 56, 141–165. [Google Scholar] [CrossRef] [PubMed]
- Clarke, B.E.; Newton, S.E.; Carroll, A.R.; Francis, M.J.; Appleyard, G.; Syred, A.D.; Highfield, P.E.; Rowlands, D.J.; Brown, F. Improved immunogenicity of a peptide epitope after fusion to hepatitis B core protein. Nature 1987, 330, 381–384. [Google Scholar] [CrossRef] [PubMed]
- Pumpens, P.; Borisova, G.P.; Crowther, R.A.; Grens, E. Hepatitis B virus core particles as epitope carriers. Intervirology 1995, 38, 63–74. [Google Scholar] [PubMed]
- Schödel, F.; Peterson, D.; Hughes, J.; Wirtz, R.; Milich, D. Hybrid hepatitis B virus core antigen as a vaccine carrier moiety: I. presentation of foreign epitopes. J. Biotechnol. 1996, 44, 91–96. [Google Scholar] [CrossRef]
- Sominskaya, I.; Skrastina, D.; Dislers, A.; Vasiljev, D.; Mihailova, M.; Ose, V.; Dreilina, D.; Pumpens, P. Construction and immunological evaluation of multivalent hepatitis B virus (HBV) core virus-like particles carrying HBV and HCV epitopes. Clin. Vaccine Immunol. 2010, 17, 1027–1033. [Google Scholar] [CrossRef] [PubMed]
- Borisova, G.; Borschukova, O.; Skrastina, D.; Dishlers, A.; Ose, V.; Pumpens, P.; Grens, E. Behavior of a short preS1 epitope on the surface of hepatitis B core particles. Biol. Chem. 1999, 380, 315–324. [Google Scholar] [CrossRef] [PubMed]
- Schödel, F.; Wirtz, R.; Peterson, D.; Hughes, J.; Warren, R.; Sadoff, J.; Milich, D. Immunity to malaria elicited by hybrid hepatitis B virus core particles carrying circumsporozoite protein epitopes. J. Exp. Med. 1994, 180, 1037–1046. [Google Scholar] [CrossRef] [PubMed]
- Ulrich, R.; Koletzki, D.; Lachmann, S.; Lundkvist, A.; Zankl, A.; Kazaks, A.; Kurth, A.; Gelderblom, H.R.; Borisova, G.; Meisel, H.; et al. New chimaeric hepatitis B virus core particles carrying hantavirus (serotype Puumala) epitopes: Immunogenicity and protection against virus challenge. J. Biotechnol. 1999, 73, 141–153. [Google Scholar] [CrossRef]
- Arora, U.; Tyagi, P.; Swaminathan, S.; Khanna, N. Chimeric Hepatitis B core antigen virus-like particles displaying the envelope domain III of dengue virus type 2. J. Nanobiotechnol. 2012, 10, e30. [Google Scholar] [CrossRef] [PubMed]
- Gedvilaite, A.; Frömmel, C.; Sasnauskas, K.; Micheel, B.; Ozel, M.; Behrsing, O.; Staniulis, J.; Jandrig, B.; Scherneck, S.; Ulrich, R. Formation of immunogenic virus-like particles by inserting epitopes into surface-exposed regions of hamster polyomavirus major capsid protein. Virology 2000, 273, 21–35. [Google Scholar] [CrossRef] [PubMed]
- Gedvilaite, A.; Zvirbliene, A.; Staniulis, J.; Sasnauskas, K.; Krüger, D.H.; Ulrich, R. Segments of puumala hantavirus nucleocapsid protein inserted into chimeric polyomavirus-derived virus-like particles induce a strong immune response in mice. Viral Immunol. 2004, 17, 51–68. [Google Scholar] [CrossRef] [PubMed]
- Zvirbliene, A.; Samonskyte, L.; Gedvilaite, A.; Voronkova, T.; Ulrich, R.; Sasnauskas, K. Generation of monoclonal antibodies of desired specificity using chimeric polyomavirus-derived virus-like particles. J. Immunol. Methods 2006, 311, 57–70. [Google Scholar] [CrossRef] [PubMed]
- Dorn, D.C.; Lawatscheck, R.; Zvirbliene, A.; Aleksaite, E.; Pecher, G.; Sasnauskas, K.; Ozel, M.; Raftery, M.; Schönrich, G.; Ulrich, R.G.; et al. Cellular and humoral immunogenicity of hamster polyomavirus-derived virus-like particles harboring a mucin 1 cytotoxic T-cell epitope. Viral Immunol. 2008, 21, 12–27. [Google Scholar] [CrossRef] [PubMed]
- Zvirbliene, A.; Kucinskaite-Kodze, I.; Razanskiene, A.; Petraityte-Burneikiene, R.; Klempa, B.; Ulrich, R.G.; Gedvilaite, A. The use of chimeric virus-like particles harbouring a segment of hantavirus Gc glycoprotein to generate a broadly-reactive hantavirus-specific monoclonal antibody. Viruses 2014, 6, 640–660. [Google Scholar] [CrossRef] [PubMed]
- Geldmacher, A.; Skrastina, D.; Borisova, G.; Petrovskis, I.; Krüger, D.H.; Pumpens, P.; Ulrich, R. A hantavirus nucleocapsid protein segment exposed on hepatitis B virus core particles is highly immunogenic in mice when applied without adjuvants or in the presence of pre-existing anti-core antibodies. Vaccine 2005, 23, 3973–3983. [Google Scholar] [CrossRef] [PubMed]
- Sasnauskas, K.; Buzaite, O.; Vogel, F.; Jandrig, B.; Razanskas, R.; Staniulis, J.; Scherneck, S.; Krüger, D.H.; Ulrich, R. Yeast cells allow high-level expression and formation of polyomavirus-like particles. Biol. Chem. 1999, 380, 381–386. [Google Scholar] [CrossRef] [PubMed]
- Sasnauskas, K.; Bulavaite, A.; Hale, A.; Jin, L.; Knowles, W.A.; Gedvilaite, A.; Dargeviciute, A.; Bartkeviciute, D.; Zvirbliene, A.; Staniulis, J.; et al. Generation of recombinant virus-like particles of human and non-human polyomaviruses in yeast Saccharomyces cerevisiae. Intervirology 2002, 45, 308–317. [Google Scholar] [CrossRef] [PubMed]
- Norkiene, M.; Stonyte, J.; Ziogiene, D.; Mazeike, E.; Sasnauskas, K.; Gedvilaite, A. Production of recombinant VP1-derived virus-like particles from novel human polyomaviruses in yeast. BMC Biotechnol 2015, in press. [Google Scholar]
- Sominskaya, I.; Pushko, P.; Dreilina, D.; Kozlovskaya, T.; Pumpens, P. Determination of the minimal length of preS1 epitope recognized by a monoclonal antibody which inhibits attachment of hepatitis B virus to hepatocytes. Med. Microbiol. Immunol. 1992, 181, 215–226. [Google Scholar] [CrossRef] [PubMed]
- Nair, D.T.; Singh, K.; Sahu, N.; Rao K, V.; Salunke, D.M. Crystal structure of an antibody bound to an immunodominant peptide epitope: Novel features in peptide-antibody recognition. J. Immunol. 2000, 165, 6949–6955. [Google Scholar] [CrossRef] [PubMed]
- Pontisso, P.; Ruvoletto, M.G.; Gerlich, W.H.; Heermann, K.H.; Bardini, R.; Alberti, A. Identification of an attachment site for human liver plasma membranes on hepatitis B virus particles. Virology 1989, 173, 522–530. [Google Scholar] [CrossRef]
- Pontisso, P.; Petit, M.A.; Bankowski, M.J.; Peeples, M.E. Human liver plasma membranes contain receptors for the hepatitis B virus pre-S1 region and, via polymerized human serum albumin, for the pre-S2 region. J. Virol. 1989, 63, 1981–1988. [Google Scholar] [PubMed]
- Glebe, D.; Urban, S.; Knoop, E.V.; Cag, N.; Krass, P.; Grün, S.; Bulavaite, A.; Sasnauskas, K.; Gerlich, W.H. Mapping of the Hepatitis B Virus Attachment Site by Use of Infection-Inhibiting preS1 Lipopeptides and Tupaia Hepatocytes. Gastroenterology 2005, 129, 234–245. [Google Scholar] [CrossRef] [PubMed]
- Neurath, A.R.; Seto, B.; Strick, N. Antibodies to synthetic peptides from the preS1 region of the hepatitis B virus (HBV) envelope (env) protein are virus-neutralizing and protective. Vaccine 1989, 7, 234–236. [Google Scholar] [CrossRef]
- Heermann, K.H. Large Surface Proteins of Hepatitis B Virus Containing the Pre-s Sequence. J. Virol. 1984, 52, 396–402. [Google Scholar] [PubMed]
- Alexander, J.; Sidney, J.; Southwood, S.; Ruppert, J.; Oseroff, C.; Maewal, A.; Snoke, K.; Serra, H.M.; Kubo, R.T.; Sette, A.; et al. Development of high potency universal DR-restricted helper epitopes by modification of high affinity DR-blocking peptides. Immunity 1994, 1, 751–761. [Google Scholar] [CrossRef]
- Panina-Bordignon, P.; Tan, A.; Termijtelen, A.; Demotz, S.; Corradin, G.; Lanzavecchia, A. Universally immunogenic T cell epitopes: Promiscuous binding to human MHC class II and promiscuous recognition by T cells. Eur. J. Immunol. 1989, 19, 2237–2242. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Su, X.; Zhang, P.; Liang, J.; Wei, H.; Wan, M.; Wu, X.; Yu, Y.; Wang, L. Recombinant heat shock protein 65 carrying PADRE and HBV epitopes activates dendritic cells and elicits HBV-specific CTL responses. Vaccine 2011, 29, 2328–2335. [Google Scholar] [CrossRef] [PubMed]
- Söding, J. Protein homology detection by HMM-HMM comparison. Bioinformatics 2005, 21, 951–960. [Google Scholar] [CrossRef] [PubMed]
- Sali, A.; Blundell, T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Sambrook, J.; Russell, D.W. Molecular Cloning, a Laboratory Manual, 3rd ed.; Cold Spring Harbour Laboratory Press: New York, NY, USA, 2001. [Google Scholar]
- Pleckaityte, M.; Zvirbliene, A.; Sezaite, I.; Gedvilaite, A. Production in yeast of pseudotype virus-like particles harboring functionally active antibody fragments neutralizing the cytolytic activity of vaginolysin. Microb. Cell. Fact. 2011, 10, e109. [Google Scholar] [CrossRef] [PubMed]
- Köhler, G.; Milstein, C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 1975, 256, 495–497. [Google Scholar] [CrossRef] [PubMed]
- Teunissen, E.A.; de Raad, M.; Mastrobattista, E. Production and biomedical applications of virus-like particles derived from polyomaviruses. J. Control. Release 2013, 172, 305–321. [Google Scholar] [CrossRef] [PubMed]
- Pushko, P.; Pearce, M.B.; Ahmad, A.; Tretyakova, I.; Smith, G.; Belser, J.A.; Tumpey, T.M. Influenza virus-like particle can accommodate multiple subtypes of hemagglutinin and protect from multiple influenza types and subtypes. Vaccine 2011, 29, 5911–5918. [Google Scholar] [CrossRef] [PubMed]
- Lua, L.H.L.; Connors, N.K.; Sainsbury, F.; Chuan, Y.P.; Wibowo, N.; Middelberg, A.P.J. Bioengineering virus-like particles as vaccines. Biotechnol. Bioeng. 2014, 111, 425–440. [Google Scholar] [CrossRef] [PubMed]
- Leavitt, A.D.; Roberts, T.M.; Garcea, R.L. Polyoma virus major capsid protein, VP1. Purification after high level expression in Escherichia coli. J. Biol. Chem. 1985, 260, 12803–12809. [Google Scholar] [PubMed]
- Salunke, D.M.; Caspar, D.L.; Garcea, R.L. Self-Assembly of purified polyomavirus capsid protein VP1. Cell 1986, 46, 895–904. [Google Scholar] [CrossRef]
- Griffith, J.P.; Griffith, D.L.; Rayment, I.; Murakami, W.T.; Caspar, D.L. Inside polyomavirus at 25-A resolution. Nature 1992, 355, 652–654. [Google Scholar] [CrossRef] [PubMed]
- Caparrós-Wanderley, W.; Clark, B.; Griffin, B.E. Effect of dose and long-term storage on the immunogenicity of murine polyomavirus VP1 virus-like particles. Vaccine 2004, 22, 352–361. [Google Scholar] [CrossRef] [PubMed]
- Heidari, S.; Vlastos, A.; Ramqvist, T.; Clark, B.; Griffin, B.E.; Garcia, M.I.; Perez, M.; Amati, P.; Dalianis, T. Immunization of T-cell deficient mice against polyomavirus infection using viral pseudocapsids or temperature sensitive mutants. Vaccine 2002, 20, 1571–1578. [Google Scholar] [CrossRef]
- Shin, Y.C.; Folk, W.R. Formation of polyomavirus-like particles with different VP1 molecules that bind the urokinase plasminogen activator receptor. J. Virol. 2003, 77, 11491–11498. [Google Scholar] [CrossRef] [PubMed]
- Langner, J.; Neumann, B.; Goodman, S.L.; Pawlita, M. RGD-mutants of B-lymphotropic polyomavirus capsids specifically bind to alpha(v)beta3 integrin. Arch. Virol. 2004, 149, 1877–1896. [Google Scholar] [PubMed]
- Kawano, M.; Morikawa, K.; Suda, T.; Ohno, N.; Matsushita, S.; Akatsuka, T.; Handa, H.; Matsui, M. Chimeric SV40 virus-like particles induce specific cytotoxicity and protective immunity against influenza A virus without the need of adjuvants. Virology 2014, 448, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Pawlita, M.; Müller, M.; Oppenländer, M.; Zentgraf, H.; Herrmann, M. DNA encapsidation by viruslike particles assembled in insect cells from the major capsid protein VP1 of B-lymphotropic papovavirus. J. Virol. 1996, 70, 7517–7526. [Google Scholar] [PubMed]
- Voronkova, T.; Kazaks, A.; Ose, V.; Ozel, M.; Scherneck, S.; Pumpens, P.; Ulrich, R. Hamster polyomavirus-derived virus-like particles are able to transfer in vitro encapsidated plasmid DNA to mammalian cells. Virus Genes 2007, 34, 303–314. [Google Scholar] [CrossRef] [PubMed]
- Dalianis, T.; Hirsch, H.H. Human polyomaviruses in disease and cancer. Virology 2013, 437, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Buck, C.B.; Phan, G.Q.; Raiji, M.T.; Murphy, P.M.; McDermott, D.H.; McBride, A.A. Complete genome sequence of a tenth human polyomavirus. J. Virol. 2012, 86, 10887. [Google Scholar] [CrossRef] [PubMed]
- Lim, E.S.; Reyes, A.; Antonio, M.; Saha, D.; Ikumapayi, U.N.; Adeyemi, M.; Stine, O.C.; Skelton, R.; Brennan, D.C.; Mkakosya, R.S.; et al. Discovery of STL polyomavirus, a polyomavirus of ancestral recombinant origin that encodes a unique T antigen by alternative splicing. Virology 2013, 436, 295–303. [Google Scholar] [CrossRef] [PubMed]
- Korup, S.; Rietscher, J.; Calvignac-Spencer, S.; Trusch, F.; Hofmann, J.; Moens, U.; Sauer, I.; Voigt, S.; Schmuck, R.; Ehlers, B. Identification of a novel human polyomavirus in organs of the gastrointestinal tract. PLoS ONE 2013, 8, e58021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Meijden, E.; Janssens, R.W.A.; Lauber, C.; Bouwes Bavinck, J.N.; Gorbalenya, A.E.; Feltkamp, M.C.W. Discovery of a new human polyomavirus associated with trichodysplasia spinulosa in an immunocompromized patient. PLoS Pathog. 2010, 6, e1001024. [Google Scholar] [CrossRef] [PubMed]
- Pumpens, P.; Grens, E. HBV core particles as a carrier for B cell/T cell epitopes. Intervirology 2001, 44, 98–114. [Google Scholar] [CrossRef] [PubMed]
- Lawatscheck, R.; Aleksaite, E.; Schenk, J.A.; Micheel, B.; Jandrig, B.; Holland, G.; Sasnauskas, K.; Gedvilaite, A.; Ulrich, R.G. Chimeric polyomavirus-derived virus-like particles: The immunogenicity of an inserted peptide applied without adjuvant to mice depends on its insertion site and its flanking linker sequence. Viral Immunol. 2007, 20, 453–460. [Google Scholar]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gedvilaite, A.; Kucinskaite-Kodze, I.; Lasickiene, R.; Timinskas, A.; Vaitiekaite, A.; Ziogiene, D.; Zvirbliene, A. Evaluation of Trichodysplasia Spinulosa-Associated Polyomavirus Capsid Protein as a New Carrier for Construction of Chimeric Virus-Like Particles Harboring Foreign Epitopes. Viruses 2015, 7, 4204-4229. https://0-doi-org.brum.beds.ac.uk/10.3390/v7082818
Gedvilaite A, Kucinskaite-Kodze I, Lasickiene R, Timinskas A, Vaitiekaite A, Ziogiene D, Zvirbliene A. Evaluation of Trichodysplasia Spinulosa-Associated Polyomavirus Capsid Protein as a New Carrier for Construction of Chimeric Virus-Like Particles Harboring Foreign Epitopes. Viruses. 2015; 7(8):4204-4229. https://0-doi-org.brum.beds.ac.uk/10.3390/v7082818
Chicago/Turabian StyleGedvilaite, Alma, Indre Kucinskaite-Kodze, Rita Lasickiene, Albertas Timinskas, Ausra Vaitiekaite, Danguole Ziogiene, and Aurelija Zvirbliene. 2015. "Evaluation of Trichodysplasia Spinulosa-Associated Polyomavirus Capsid Protein as a New Carrier for Construction of Chimeric Virus-Like Particles Harboring Foreign Epitopes" Viruses 7, no. 8: 4204-4229. https://0-doi-org.brum.beds.ac.uk/10.3390/v7082818