
Papillomavirus Infectious Pathways: A Comparison of Systems

Abstract
:1. Introduction

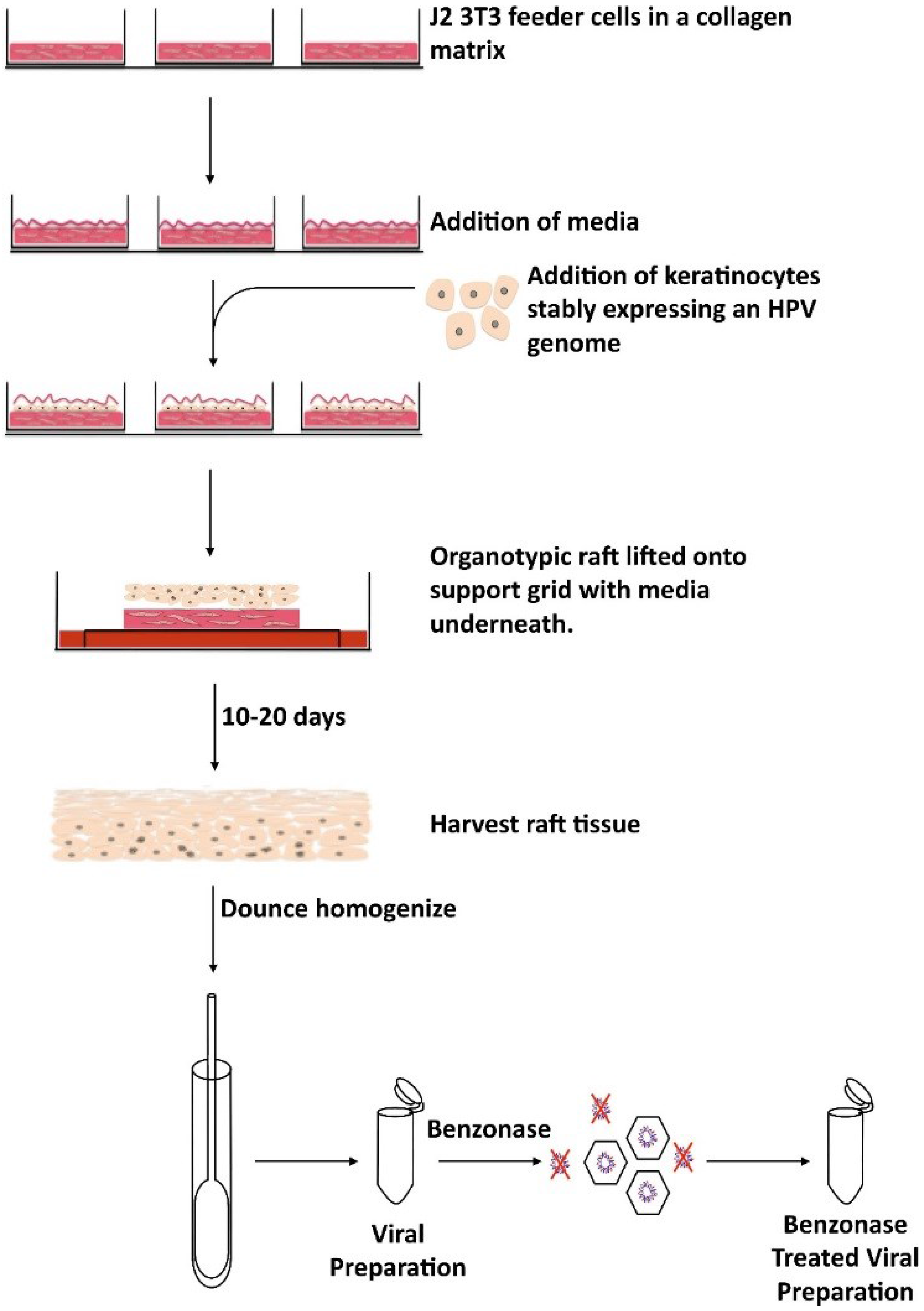{kind=link}
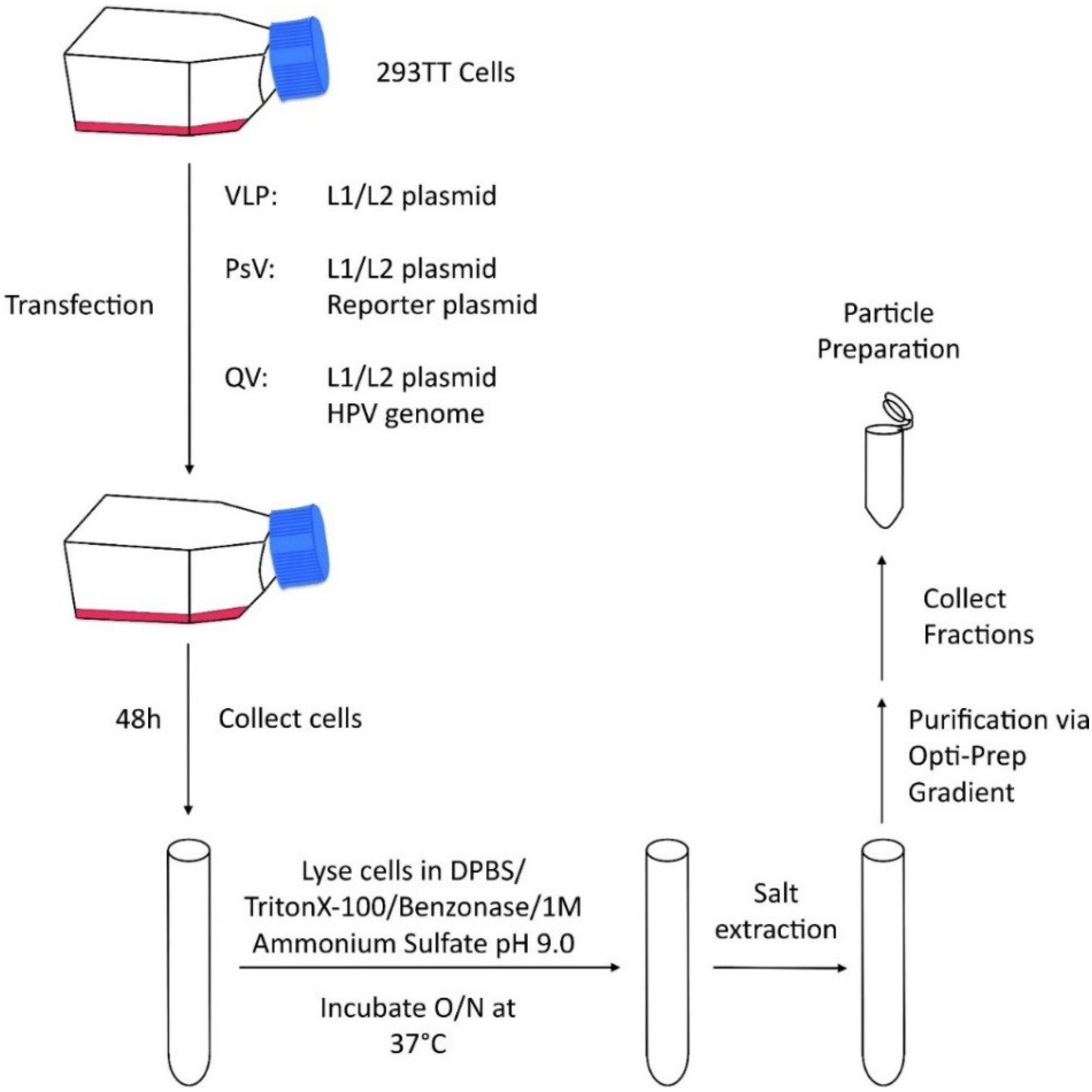{kind=link}
| HPV Particle Type | Method of Production | Particle and System Advantages | Particle and System Disadvantages |
|---|---|---|---|
| Native Virions | Organotypic raft culture system | Contains the full HPV capsid as well as full HPV genome Assembles and matures over a period of 10–20 days in tissue | Expensive Slow production time |
| Virus Like Particles (VLPs) | Transfection based system | Quick and inexpensive particle production | Codon modification of L1 and L2 is necessary for production Over-expression system Particles produced in non-relevant cell lines Particle assembly within 48 hours Maturation occurs overnight Deletion of in frame methionines upstream of the consensus methionine in L1 VLPs contain no genomes, which may alter particle structure |
| Pseudovirions (PsVs) | Transfection based system | Quick and inexpensive particle production Ability to track cellular infectivity | |
| Quasivirions (QVs) | Transfection based system | Quick and inexpensive particle production Contains L1, L2, and a full HPV genome Closest to NVs—retains majority of cell surface exposed conformational epitopes |
2. Papillomavirus Particle Production


3. Maturation and Assembly of Viral Particles
4. Attachment and Internalization
5. Subsequent Steps in HPV Infection
6. Translational Research
7. Concluding Remarks
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Zur Hausen, H. Papillomaviruses in the causation of human cancers—A brief historical account. Virology 2009, 384, 260–265. [Google Scholar]
- Syrjanen, S.; Lodi, G.; von Bültzingslöwen, I.; Aliko, A.; Arduino, P.; Campisi, G.; Challacombe, S.; Ficarra, G.; Flaitz, C.; Zhou, H.M.; et al. Human papillomaviruses in oral carcinoma and oral potentially malignant disorders: A systematic review. Oral Dis. 2011, 17, S58–S72. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, G.; Kreimer, A.R.; Viscidi, R.; Pawlita, M.; Fakhry, C.; Koch, W.M.; Westra, W.H.; Gillison, M.L. Case-control study of human papillomavirus and oropharyngeal cancer. N. Engl. J. Med. 2007, 356, 1944–1956. [Google Scholar] [CrossRef] [PubMed]
- Schiffman, M.; Kjaer, S.K. Chapter 2: Natural history of anogenital human papillomavirus infection and neoplasia. J. Natl. Cancer Inst. Monogr. 2003, 31, 14–19. [Google Scholar] [CrossRef] [PubMed]
- Arbyn, M.; Castellsagué, X.; de Sanjosé, S.; Bruni, L.; Saraiya, M.; Bray, F.; Ferlay, J. Worldwide burden of cervical cancer in 2008. Ann. Oncol. 2011, 22, 2675–2686. [Google Scholar] [CrossRef] [PubMed]
- De Villiers, E.M.; Fauquet, C.; Broker, T.R.; Bernard, H.U.; zur Hausen, H. Classification of papillomaviruses. Virology 2004, 324, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Longworth, M.S.; Laimins, L.A. Pathogenesis of human papillomaviruses in differentiating epithelia. Microbiol. Mol. Biol. Rev. 2004, 68, 362–372. [Google Scholar] [CrossRef] [PubMed]
- Madsen, B.S.; Jensen, H.L.; van den Brule, A.J.; Wohlfahrt, J.; Frisch, M. Risk factors for invasive squamous cell carcinoma of the vulva and vagina—Population-based case-control study in Denmark. Int. J. Cancer 2008, 122, 2827–2834. [Google Scholar] [CrossRef] [PubMed]
- Meyers, C.; Frattini, M.G.; Hudson, J.B.; Laimins, L.A. Biosynthesis of human papillomavirus from a continuous cell line upon epithelial differentiation. Science 1992, 257, 971–973. [Google Scholar] [CrossRef] [PubMed]
- Hummel, M.; Hudson, J.B.; Laimins, L.A. Differentiation-induced and constitutive transcription of human papillomavirus type 31b in cell lines containing viral episomes. J. Virol. 1992, 66, 6070–6080. [Google Scholar] [PubMed]
- Bedell, M.A.; Hudson, J.B.; Golub, T.R.; Turyk, M.E.; Hosken, M.; Wilbanks, G.D.; Laimins, L.A. Amplification of human papillomavirus genomes in vitro is dependent on epithelial differentiation. J. Virol. 1991, 65, 2254–2260. [Google Scholar] [PubMed]
- Grassmann, K.; Rapp, B.; Maschek, H.; Petry, K.U.; Iftner, T. Identification of a differentiation-inducible promoter in the E7 open reading frame of human papillomavirus type 16 (HPV-16) in raft cultures of a new cell line containing high copy numbers of episomal HPV-16 DNA. J. Virol. 1996, 70, 2339–2349. [Google Scholar] [PubMed]
- Ozbun, M.A.; Meyers, C. Characterization of late gene transcripts expressed during vegetative replication of human papillomavirus type 31b. J. Virol. 1997, 71, 5161–5172. [Google Scholar] [PubMed]
- Richards, K.F.; Mukherjee, S.; Bienkowska-Haba, M.; Pang, J.; Sapp, M. Human papillomavirus species-specific interaction with the basement membrane-resident non-heparan sulfate receptor. Viruses 2014, 6, 4856–4879. [Google Scholar] [CrossRef] [PubMed]
- Cruz, L.; Meyers, C. Differential dependence on host cell glycosaminoglycans for infection of epithelial cells by high-risk HPV types. PLoS ONE 2013, 8, e68379. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin-Drubin, M.E.; Christensen, N.D.; Meyers, C. Propagation, infection, and neutralization of authentic HPV16 virus. Virology 2004, 322, 213–219. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin-Drubin, M.E.; Wilson, S.; Mullikin, B.; Suzich, J.; Meyers, C. Human papillomavirus type 45 propagation, infection, and neutralization. Virology 2003, 312, 1–7. [Google Scholar] [CrossRef]
- Meyers, C.; Mayer, T.J.; Ozbun, M.A. Synthesis of infectious human papillomavirus type 18 in differentiating epithelium transfected with viral DNA. J. Virol. 1997, 71, 7381–7386. [Google Scholar] [PubMed]
- Kirnbauer, R.; Booy, F.; Cheng, N.; Lowy, D.R.; Schiller, J.T. Papillomavirus L1 major capsid protein self-assembles into virus-like particles that are highly immunogenic. Proc. Natl. Acad. Sci. USA 1992, 89, 12180–12184. [Google Scholar] [CrossRef] [PubMed]
- Kirnbauer, R.; Taub, J.; Greenstone, H.; Roden, R.; Dürst, M.; Gissmann, L.; Lowy, D.R.; Schiller, J.T. Efficient self-assembly of human papillomavirus type 16 L1 and L1-L2 into virus-like particles. J. Virol. 1993, 67, 6929–6936. [Google Scholar] [PubMed]
- Rose, R.C.; Bonnez, W.; Reichman, R.C.; Garcea, R.L. Expression of human papillomavirus type 11 L1 protein in insect cells: In vivo and in vitro assembly of viruslike particles. J. Virol. 1993, 67, 1936–1944. [Google Scholar] [PubMed]
- Hagensee, M.E.; Yaegashi, N.; Galloway, D.A. Self-assembly of human papillomavirus type 1 capsids by expression of the L1 protein alone or by coexpression of the L1 and L2 capsid proteins. J. Virol. 1993, 67, 315–322. [Google Scholar] [PubMed]
- Hagensee, M.E.; Olson, N.H.; Baker, T.S.; Galloway, D.A. Three-dimensional structure of vaccinia virus-produced human papillomavirus type 1 capsids. J. Virol. 1994, 68, 4503–4505. [Google Scholar] [PubMed]
- Chen, X.S.; Garcea, R.L.; Goldberg, I.; Casini, G.; Harrison, S.C. Structure of small virus-like particles assembled from the L1 protein of human papillomavirus 16. Mol. Cell 2000, 5, 557–567. [Google Scholar] [CrossRef]
- Zhang, W.; Carmichael, J.; Ferguson, J.; Inglis, S.; Ashrafian, H.; Stanley, M. Expression of human papillomavirus type 16 L1 protein in Escherichia coli: Denaturation, renaturation, and self-assembly of virus-like particles in vitro. Virology 1998, 243, 423–431. [Google Scholar] [CrossRef] [PubMed]
- Culp, T.D.; Christensen, N.D. Kinetics of in vitro adsorption and entry of papillomavirus virions. Virology 2004, 319, 152–161. [Google Scholar] [CrossRef] [PubMed]
- Pyeon, D.; Lambert, P.F.; Ahlquist, P. Production of infectious human papillomavirus independently of viral replication and epithelial cell differentiation. Proc. Natl. Acad. Sci. USA 2005, 102, 9311–9316. [Google Scholar] [CrossRef] [PubMed]
- Day, P.M.; Lowy, D.R.; Schiller, J.T. Papillomaviruses infect cells via a clathrin-dependent pathway. Virology 2003, 307, 1–11. [Google Scholar] [CrossRef]
- Embers, M.E.; Budgeon, L.R.; Culp, T.D.; Reed, C.A.; Pickel, M.D.; Christensen, N.D. Differential antibody responses to a distinct region of human papillomavirus minor capsid proteins. Vaccine 2004, 22, 670–680. [Google Scholar] [CrossRef] [PubMed]
- Gambhira, R.; Karanam, B.; Jagu, S.; Roberts, J.N.; Buck, C.B.; Bossis, I.; Alphs, H.; Culp, T.; Christensen, N.D.; Roden, R.B. A protective and broadly cross-neutralizing epitope of human papillomavirus L2. J. Virol. 2007, 81, 13927–13931. [Google Scholar] [CrossRef] [PubMed]
- Buck, C.B.; Cheng, N.; Thompson, C.D.; Lowy, D.R.; Steven, A.C.; Schiller, J.T.; Trus, B.L. Arrangement of L2 within the papillomavirus capsid. J. Virol. 2008, 82, 5190–5197. [Google Scholar] [CrossRef] [PubMed]
- White, W.I.; Wilson, S.D.; Parmer-Hill, F.J.; Woods, R.M.; Ghim, S.; Hewitt, L.A.; Goldman, D.M.; Burke, S.J.; Jerson, A.B.; Koening, S.; et al. Characterization of a major neutralizing epitope on human papillomavirus type 16 L1. J. Virol. 1999, 73, 4882–4889. [Google Scholar] [PubMed]
- Zhou, J.; Liu, W.J.; Peng, S.W.; Sun, X.Y.; Frazer, I. Papillomavirus capsid protein expression level depends on the match between codon usage and tRNA availability. J. Virol. 1999, 73, 4972–4982. [Google Scholar] [PubMed]
- Leder, C.; Kleinschmidt, J.A.; Wiethe, C.; Müller, M. Enhancement of capsid gene expression: Preparing the human papillomavirus type 16 major structural gene L1 for DNA vaccination purposes. J. Virol. 2001, 75, 9201–9209. [Google Scholar] [CrossRef] [PubMed]
- Spencer, P.S.; Barral, J.M. Genetic code redundancy and its influence on the encoded polypeptides. Comput. Struct. Biotechnol. J. 2012, 1, e201204006. [Google Scholar] [CrossRef] [PubMed]
- Fang, N.X.; Frazer, I.H.; Fernando, G.J. Differences in the post-translational modifications of human papillomavirus type 6b major capsid protein expressed from a baculovirus system compared with a vaccinia virus system. Biotechnol. Appl. Biochem. 2000, 32, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Sun, X.Y.; Stenzel, D.J.; Frazer, I.H. Expression of vaccinia recombinant HPV 16 L1 and L2 ORF proteins in epithelial cells is sufficient for assembly of HPV virion-like particles. Virology 1991, 185, 251–257. [Google Scholar] [CrossRef]
- Conway, M.J.; Alam, S.; Ryndock, E.J.; Cruz, L.; Christensen, N.D.; Roden, R.B.; Meyers, C. Tissue-spanning redox gradient-dependent assembly of native human papillomavirus type 16 virions. J. Virol. 2009, 83, 10515–10526. [Google Scholar] [CrossRef] [PubMed]
- Holmgren, S.C.; Patterson, N.A.; Ozbun, M.A.; Lambert, P.F. The minor capsid protein L2 contributes to two steps in the human papillomavirus type 31 life cycle. J. Virol. 2005, 79, 3938–3948. [Google Scholar] [CrossRef] [PubMed]
- Fey, S.J.; Nielsen, T.; Vetner, M.; Storgaard, L.; Smed, V.; Larsen, P.M. Demonstration of in vitro synthesis of human papilloma viral proteins from hand and foot warts. J. Investig. Dermatol. 1989, 92, 817–824. [Google Scholar] [CrossRef] [PubMed]
- Larsen, P.M.; Storgaard, L.; Fey, S.J. Proteins present in bovine papillomavirus particles. J. Virol. 1987, 61, 3596–3601. [Google Scholar] [PubMed]
- Joshi, A.; Nagashima, K.; Freed, E.O. Mutation of dileucine-like motifs in the human immunodeficiency virus type 1 capsid disrupts virus assembly, gag-gag interactions, gag-membrane binding, and virion maturation. J. Virol. 2006, 80, 7939–7951. [Google Scholar] [CrossRef] [PubMed]
- Perez-Berna, A.J.; Ortega-Esteban, A.; Menéndez-Conejero, R.; Winkler, D.C.; Menéndez, M.; Steven, A.C.; Flint, S.J.; de Pablo, P.J.; San Martín, C. The role of capsid maturation on adenovirus priming for sequential uncoating. J. Biol. Chem. 2012, 287, 31582–31595. [Google Scholar] [CrossRef] [PubMed]
- Buck, C.B.; Thompson, C.D.; Pang, Y.Y.; Lowy, D.R.; Schiller, J.T. Maturation of papillomavirus capsids. J. Virol. 2005, 79, 2839–2846. [Google Scholar] [CrossRef] [PubMed]
- Conway, M.J.; Cruz, L.; Alam, S.; Christensen, N.D.; Meyers, C. Differentiation-dependent interpentameric disulfide bond stabilizes native human papillomavirus type 16. PLoS ONE 2011, 6, e22427. [Google Scholar] [CrossRef] [PubMed]
- Cardone, G.; Moyer, A.L.; Cheng, N.; Thompson, C.D.; Dvoretzky, I.; Lowy, D.R.; Schiller, J.T.; Steven, A.C.; Buck, C.B.; Trus, B.L. Maturation of the human papillomavirus 16 capsid. MBio 2014, 5, e01104-14. [Google Scholar] [CrossRef] [PubMed]
- Fligge, C.; Schäfer, F.; Selinka, H.C.; Sapp, C.; Sapp, M. DNA-induced structural changes in the papillomavirus capsid. J. Virol. 2001, 75, 7727–7731. [Google Scholar] [CrossRef] [PubMed]
- Ishii, Y.; Tanaka, K.; Kanda, T. Mutational analysis of human papillomavirus type 16 major capsid protein L1: The cysteines affecting the intermolecular bonding and structure of L1-capsids. Virology 2003, 308, 128–136. [Google Scholar] [CrossRef]
- Wolf, M.; Garcea, R.L.; Grigorieff, N.; Harrison, S.C. Subunit interactions in bovine papillomavirus. Proc. Natl. Acad. Sci. USA 2010, 107, 6298–6303. [Google Scholar] [CrossRef] [PubMed]
- Modis, Y.; Trus, B.L.; Harrison, S.C. Atomic model of the papillomavirus capsid. EMBO J. 2002, 21, 4754–4762. [Google Scholar] [CrossRef] [PubMed]
- Ryndock, E.J.; Conway, M.J.; Alam, S.; Gul, S.; Murad, S.; Christensen, N.D.; Meyers, C. Roles for human papillomavirus type 16 l1 cysteine residues 161, 229, and 379 in genome encapsidation and capsid stability. PLoS ONE 2014, 9, e99488. [Google Scholar] [CrossRef] [PubMed]
- Ishii, Y.; Ozaki, S.; Tanaka, K.; Kanda, T. Human papillomavirus 16 minor capsid protein L2 helps capsomeres assemble independently of intercapsomeric disulfide bonding. Virus Genes 2005, 31, 321–328. [Google Scholar] [CrossRef] [PubMed]
- Campos, S.K.; Ozbun, M.A. Two highly conserved cysteine residues in HPV16 L2 form an intramolecular disulfide bond and are critical for infectivity in human keratinocytes. PLoS ONE 2009, 4, e4463. [Google Scholar] [CrossRef] [PubMed]
- Gambhira, R.; Jagu, S.; Karanam, B.; Day, P.M.; Roden, R. Role of L2 cysteines in papillomavirus infection and neutralization. Virol. J. 2009, 6, e176. [Google Scholar] [CrossRef] [PubMed]
- Conway, M.J.; Alam, S.; Christensen, N.D.; Meyers, C. Overlapping and independent structural roles for human papillomavirus type 16 L2 conserved cysteines. Virology 2009, 393, 295–303. [Google Scholar] [CrossRef] [PubMed]
- Kondo, K.; Ishii, Y.; Ochi, H.; Matsumoto, T.; Yoshikawa, H.; Kanda, T. Neutralization of HPV16, 18, 31, and 58 pseudovirions with antisera induced by immunizing rabbits with synthetic peptides representing segments of the HPV16 minor capsid protein L2 surface region. Virology 2007, 358, 266–272. [Google Scholar] [CrossRef] [PubMed]
- Roden, R.B.; Greenstone, H.L.; Kirnbauer, R.; Booy, F.P.; Jessie, J.; Lowy, D.R.; Schiller, J.T. In vitro generation and type-specific neutralization of a human papillomavirus type 16 virion pseudotype. J. Virol. 1996, 70, 5875–5883. [Google Scholar] [PubMed]
- Zhao, K.N.; Sun, X.Y.; Frazer, I.H.; Zhou, J. DNA packaging by L1 and L2 capsid proteins of bovine papillomavirus type 1. Virology 1998, 243, 482–491. [Google Scholar] [CrossRef] [PubMed]
- Trus, B.L.; Roden, R.B.; Greenstone, H.L.; Vrhel, M.; Schiller, J.T.; Booy, F.P. Novel structural features of bovine papillomavirus capsid revealed by a three-dimensional reconstruction to 9 Å resolution. Nat. Struct. Biol. 1997, 4, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Doorbar, J.; Gallimore, P.H. Identification of proteins encoded by the L1 and L2 open reading frames of human papillomavirus 1a. J. Virol. 1987, 61, 2793–2799. [Google Scholar] [PubMed]
- Meyers, C.; Bromberg-White, J.L.; Zhang, J.; Kaupas, M.E.; Bryan, J.T.; Lowe, R.S.; Jansen, K.U. Infectious virions produced from a human papillomavirus type 18/16 genomic DNA chimera. J. Virol. 2002, 76, 4723–4733. [Google Scholar] [CrossRef] [PubMed]
- Bowser, B.S.; Chen, H.S.; Conway, M.J.; Christensen, N.D.; Meyers, C. Human papillomavirus type 18 chimeras containing the L2/L1 capsid genes from evolutionarily diverse papillomavirus types generate infectious virus. Virus Res. 2011, 160, 246–255. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.S.; Conway, M.J.; Christensen, N.D.; Alam, S.; Meyers, C. Papillomavirus capsid proteins mutually impact structure. Virology 2011, 412, 378–383. [Google Scholar] [CrossRef] [PubMed]
- Kines, R.C.; Thompson, C.D.; Lowy, D.R.; Schiller, J.T.; Day, P.M. The initial steps leading to papillomavirus infection occur on the basement membrane prior to cell surface binding. Proc. Natl. Acad. Sci. USA 2009, 106, 20458–20463. [Google Scholar] [CrossRef] [PubMed]
- Sapp, M.; Bienkowska-Haba, M. Viral entry mechanisms: Human papillomavirus and a long journey from extracellular matrix to the nucleus. FEBS J. 2009, 276, 7206–7216. [Google Scholar] [CrossRef] [PubMed]
- Giroglou, T.; Florin, L.; Schäfer, F.; Streeck, R.E.; Sapp, M. Human papillomavirus infection requires cell surface heparan sulfate. J. Virol. 2001, 75, 1565–1570. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.M.; Kines, R.C.; Roberts, J.N.; Lowy, D.R.; Schiller, J.T.; Day, P.M. Role of heparan sulfate in attachment to and infection of the murine female genital tract by human papillomavirus. J. Virol. 2009, 83, 2067–2074. [Google Scholar] [CrossRef] [PubMed]
- Richards, R.M.; Lowy, D.R.; Schiller, J.T.; Day, P.M. Cleavage of the papillomavirus minor capsid protein, L2, at a furin consensus site is necessary for infection. Proc. Natl. Acad. Sci. USA 2006, 103, 1522–1527. [Google Scholar] [CrossRef] [PubMed]
- Day, P.M.; Gambhira, R.; Roden, R.B.; Lowy, D.R.; Schiller, J.T. Mechanisms of human papillomavirus type 16 neutralization by l2 cross-neutralizing and l1 type-specific antibodies. J. Virol. 2008, 82, 4638–4646. [Google Scholar] [CrossRef] [PubMed]
- Knappe, M.; Bodevin, S.; Selinka, H.C.; Spillmann, D.; Streeck, R.E.; Chen, X.S.; Lindahl, U.; Sapp, M. Surface-exposed amino acid residues of HPV16 L1 protein mediating interaction with cell surface heparan sulfate. J. Biol. Chem. 2007, 282, 27913–27922. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Jacob, T.; Abban, C.Y.; Meneses, P.I. Intermediate heparan sulfate binding during HPV-16 infection in HaCaTs. Am. J. Ther. 2014, 21, 331–342. [Google Scholar] [CrossRef] [PubMed]
- Joyce, J.G.; Tung, J.S.; Przysiecki, C.T.; Cook, J.C.; Lehman, E.D.; Sands, J.A.; Jansen, K.U.; Keller, P.M. The L1 major capsid protein of human papillomavirus type 11 recombinant virus-like particles interacts with heparin and cell-surface glycosaminoglycans on human keratinocytes. J. Biol. Chem. 1999, 274, 5810–5822. [Google Scholar] [CrossRef] [PubMed]
- Patterson, N.A.; Smith, J.L.; Ozbun, M.A. Human papillomavirus type 31b infection of human keratinocytes does not require heparan sulfate. J. Virol. 2005, 79, 6838–6847. [Google Scholar] [CrossRef] [PubMed]
- Bishop, B.; Dasgupta, J.; Klein, M.; Garcea, R.L.; Christensen, N.D.; Zhao, R.; Chen, X.S. Crystal structures of four types of human papillomavirus L1 capsid proteins: Understanding the specificity of neutralizing monoclonal antibodies. J. Biol. Chem. 2007, 282, 31803–31811. [Google Scholar] [CrossRef] [PubMed]
- Broutian, T.R.; Brendle, S.A.; Christensen, N.D. Differential binding patterns to host cells associated with particles of several human alphapapillomavirus types. J. Gen. Virol. 2010, 91, 531–540. [Google Scholar] [CrossRef] [PubMed]
- Sapp, M.; Day, P.M. Structure, attachment and entry of polyoma- and papillomaviruses. Virology 2009, 384, 400–409. [Google Scholar] [CrossRef] [PubMed]
- Bauer, P.H.; Cui, C.; Liu, W.R.; Stehle, T.; Harrison, S.C.; DeCaprio, J.A.; Benjamin, T.L. Discrimination between sialic acid-containing receptors and pseudoreceptors regulates polyomavirus spread in the mouse. J. Virol. 1999, 73, 5826–5832. [Google Scholar] [CrossRef] [PubMed]
- Neu, U.; Maginnis, M.S.; Palma, A.S.; Ströh, L.J.; Nelson, C.D.; Feizi, T.; Atwood, W.J.; Stehle, T. Structure-function analysis of the human JC polyomavirus establishes the LSTc pentasaccharide as a functional receptor motif. Cell Host Microbe 2010, 8, 309–319. [Google Scholar] [CrossRef] [PubMed]
- Horvath, C.A.; Boulet, G.A.; Renoux, V.M.; Delvenne, P.O.; Bogers, J.P. Mechanisms of cell entry by human papillomaviruses: An overview. Virol. J. 2010, 7, e11. [Google Scholar] [CrossRef] [PubMed]
- Spoden, G.; Freitag, K.; Husmann, M.; Boller, K.; Sapp, M.; Lambert, C.; Florin, L. Clathrin- and caveolin-independent entry of human papillomavirus type 16—Involvement of tetraspanin-enriched microdomains (TEMs). PLoS ONE 2008, 3, e3313. [Google Scholar] [CrossRef] [PubMed]
- Selinka, H.C.; Florin, L.; Patel, H.D.; Freitag, K.; Schmidtke, M.; Makarov, V.A.; Sapp, M. Inhibition of transfer to secondary receptors by heparan sulfate-binding drug or antibody induces noninfectious uptake of human papillomavirus. J. Virol. 2007, 81, 10970–10980. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.L.; Campos, S.K.; Ozbun, M.A. Human papillomavirus type 31 uses a caveolin 1- and dynamin 2-mediated entry pathway for infection of human keratinocytes. J. Virol. 2007, 81, 9922–9931. [Google Scholar] [CrossRef] [PubMed]
- Day, P.M.; Lowy, D.R.; Schiller, J.T. Heparan sulfate-independent cell binding and infection with furin-precleaved papillomavirus capsids. J. Virol. 2008, 82, 12565–12568. [Google Scholar] [CrossRef] [PubMed]
- Bousarghin, L.; Touzé, A.; Sizaret, P.Y.; Coursaget, P. Human papillomavirus types 16, 31, and 58 use different endocytosis pathways to enter cells. J. Virol. 2003, 77, 3846–3850. [Google Scholar] [CrossRef] [PubMed]
- Hindmarsh, P.L.; Laimins, L.A. Mechanisms regulating expression of the HPV 31 L1 and L2 capsid proteins and pseudovirion entry. Virol. J. 2007, 4, e19. [Google Scholar] [CrossRef] [PubMed]
- Laniosz, V.; Dabydeen, S.A.; Havens, M.A.; Meneses, P.I. Human papillomavirus type 16 infection of human keratinocytes requires clathrin and caveolin-1 and is brefeldin a sensitive. J. Virol. 2009, 83, 8221–8232. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Kazakov, T.; Popa, A.; DiMaio, D. Vesicular trafficking of incoming human papillomavirus 16 to the Golgi apparatus and endoplasmic reticulum requires gamma-secretase activity. MBio 2014, 5, e01777-14. [Google Scholar] [CrossRef] [PubMed]
- Schelhaas, M.; Shah, B.; Holzer, M.; Blattmann, P.; Kühling, L.; Day, P.M.; Schiller, J.T.; Helenius, A. Entry of human papillomavirus type 16 by actin-dependent, clathrin- and lipid raft-independent endocytosis. PLoS Pathog. 2012, 8, e1002657. [Google Scholar] [CrossRef] [PubMed]
- Spoden, G.; Kühling, L.; Cordes, N.; Frenzel, B.; Sapp, M.; Boller, K.; Florin, L.; Schelhaas, M. Human papillomavirus types 16, 18, and 31 share similar endocytic requirements for entry. J. Virol. 2013, 87, 7765–7773. [Google Scholar] [CrossRef] [PubMed]
- Evander, M.; Frazer, I.H.; Payne, E.; Qi, Y.M.; Hengst, K.; McMillan, N.A. Identification of the alpha6 integrin as a candidate receptor for papillomaviruses. J. Virol. 1997, 71, 2449–2456. [Google Scholar] [PubMed]
- Yoon, C.S.; Kim, K.D.; Park, S.N.; Cheong, S.W. Alpha(6) integrin is the main receptor of human papillomavirus type 16 VLP. Biochem. Biophys. Res. Commun. 2001, 283, 668–673. [Google Scholar] [CrossRef] [PubMed]
- Sibbet, G.; Romero-Graillet, C.; Meneguzzi, G.; Campo, M.S. Alpha6 integrin is not the obligatory cell receptor for bovine papillomavirus type 4. J. Gen. Virol. 2000, 81, 327–334. [Google Scholar] [PubMed]
- Aksoy, P.; Abban, C.Y.; Kiyashka, E.; Qiang, W.; Meneses, P.I. HPV16 infection of HaCaTs is dependent on beta4 integrin, and alpha6 integrin processing. Virology 2014, 449, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Fothergill, T.; McMillan, N.A. Papillomavirus virus-like particles activate the PI3-kinase pathway via alpha-6 beta-4 integrin upon binding. Virology 2006, 352, 319–328. [Google Scholar] [CrossRef] [PubMed]
- Surviladze, Z.; Dziduszko, A.; Ozbun, M.A. Essential roles for soluble virion-associated heparan sulfonated proteoglycans and growth factors in human papillomavirus infections. PLoS Pathog. 2012, 8, e1002519. [Google Scholar] [CrossRef] [PubMed]
- Waisman, D.M. Annexin II tetramer: Structure and function. Mol. Cell. Biochem. 1995, 149–150, 301–322. [Google Scholar] [CrossRef] [PubMed]
- Woodham, A.W.; da Silva, D.M.; Skeate, J.G.; Raff, A.B.; Ambroso, M.R.; Brand, H.E.; Isas, J.M.; Langen, R.; Kast, W.M. The S100A10 subunit of the annexin A2 heterotetramer facilitates L2-mediated human papillomavirus infection. PLoS ONE 2012, 7, e43519. [Google Scholar] [CrossRef] [PubMed]
- Raff, A.B.; Woodham, A.W.; Raff, L.M.; Skeate, J.G.; Yan, L.; da Silva, D.M.; Schelhaas, M.; Kast, W.M. The evolving field of human papillomavirus receptor research: A review of binding and entry. J. Virol. 2013, 87, 6062–6072. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.L.; Lidke, D.S.; Ozbun, M.A. Virus activated filopodia promote human papillomavirus type 31 uptake from the extracellular matrix. Virology 2008, 381, 16–21. [Google Scholar] [CrossRef] [PubMed]
- Selinka, H.C.; Giroglou, T.; Sapp, M. Analysis of the infectious entry pathway of human papillomavirus type 33 pseudovirions. Virology 2002, 299, 279–287. [Google Scholar] [CrossRef] [PubMed]
- Bienkowska-Haba, M.; Williams, C.; Kim, S.M.; Garcea, R.L.; Sapp, M. Cyclophilins facilitate dissociation of the human papillomavirus type 16 capsid protein L1 from the L2/DNA complex following virus entry. J. Virol. 2012, 86, 9875–9887. [Google Scholar] [CrossRef] [PubMed]
- Aydin, I.; Weber, S.; Snijder, B.; Samperio Ventayol, P.; Kühbacher, A.; Becker, M.; Day, P.M.; Schiller, J.T.; Kann, M.; Pelkmans, L.; et al. Large scale RNAi reveals the requirement of nuclear envelope breakdown for nuclear import of human papillomaviruses. PLoS Pathog. 2014, 10, e1004162. [Google Scholar] [CrossRef] [PubMed]
- Lipovsky, A.; Popa, A.; Pimienta, G.; Wyler, M.; Bhan, A.; Kuruvilla, L.; Guie, M.A.; Poffenberger, A.C.; Nelson, C.D.; Atwood, W.J.; et al. Genome-wide siRNA screen identifies the retromer as a cellular entry factor for human papillomavirus. Proc. Natl. Acad. Sci. USA 2013, 110, 7452–7457. [Google Scholar] [CrossRef] [PubMed]
- Day, P.M.; Thompson, C.D.; Schowalter, R.M.; Lowy, D.R.; Schiller, J.T. Identification of a role for the trans-Golgi network in human papillomavirus 16 pseudovirus infection. J. Virol. 2013, 87, 3862–3870. [Google Scholar] [CrossRef] [PubMed]
- DiGiuseppe, S.; Bienkowska-Haba, M.; Hilbig, L.; Sapp, M. The nuclear retention signal of HPV16 L2 protein is essential for incoming viral genome to transverse the trans-Golgi network. Virology 2014, 458–459, 93–105. [Google Scholar] [CrossRef] [PubMed]
- Attar, N.; Cullen, P.J. The retromer complex. Adv. Enzyme Regul. 2010, 50, 216–236. [Google Scholar] [CrossRef] [PubMed]
- Burd, C.; Cullen, P.J. Retromer: A master conductor of endosome sorting. Cold Spring Harb. Perspect. Biol. 2014, 6, a016774. [Google Scholar] [CrossRef] [PubMed]
- Popa, A.; Zhang, W.; Harrison, M.S.; Goodner, K.; Kazakov, T.; Goodwin, E.C.; Lipovsky, A.; Burd, C.G.; DiMaio, D. Direct binding of retromer to human papillomavirus type 16 minor capsid protein L2 mediates endosome exit during viral infection. PLoS Pathog. 2015, 11, e1004699. [Google Scholar] [CrossRef] [PubMed]
- Bossis, I.; Roden, R.B.; Gambhira, R.; Yang, R.; Tagaya, M.; Howley, P.M.; Meneses, P.I. Interaction of tSNARE syntaxin 18 with the papillomavirus minor capsid protein mediates infection. J. Virol. 2005, 79, 6723–6731. [Google Scholar] [CrossRef] [PubMed]
- Laniosz, V.; Nguyen, K.C.; Meneses, P.I. Bovine papillomavirus type 1 infection is mediated by SNARE syntaxin 18. J. Virol. 2007, 81, 7435–7448. [Google Scholar] [CrossRef] [PubMed]
- Campos, S.K.; Chapman, J.A.; Deymier, M.J.; Bronnimann, M.P.; Ozbun, M.A. Opposing effects of bacitracin on human papillomavirus type 16 infection: Enhancement of binding and entry and inhibition of endosomal penetration. J. Virol. 2012, 86, 4169–4181. [Google Scholar] [CrossRef] [PubMed]
- Pyeon, D.; Pearce, S.M.; Lank, S.M.; Ahlquist, P.; Lambert, P.F. Establishment of human papillomavirus infection requires cell cycle progression. PLoS Pathog. 2009, 5, e1000318. [Google Scholar] [CrossRef] [PubMed]
- Bronnimann, M.P.; Chapman, J.A.; Park, C.K.; Campos, S.K. A transmembrane domain and GxxxG motifs within L2 are essential for papillomavirus infection. J. Virol. 2013, 87, 464–473. [Google Scholar] [CrossRef] [PubMed]
- Christensen, N.D.; Cladel, N.M.; Reed, C.A.; Budgeon, L.R.; Embers, M.E.; Skulsky, D.M.; McClements, W.L.; Ludmerer, S.W.; Jansen, K.U. Hybrid papillomavirus L1 molecules assemble into virus-like particles that reconstitute conformational epitopes and induce neutralizing antibodies to distinct HPV types. Virology 2001, 291, 324–334. [Google Scholar] [CrossRef] [PubMed]
- Day, P.M.; Thompson, C.D.; Buck, C.B.; Pang, Y.Y.; Lowy, D.R.; Schiller, J.T. Neutralization of human papillomavirus with monoclonal antibodies reveals different mechanisms of inhibition. J. Virol. 2007, 81, 8784–8792. [Google Scholar] [CrossRef] [PubMed]
- Christensen, N.D.; Reed, C.A.; Culp, T.D.; Hermonat, P.L.; Howett, M.K.; Anderson, R.A.; Zaneveld, L.J. Papillomavirus microbicidal activities of high-molecular-weight cellulose sulfate, dextran sulfate, and polystyrene sulfonate. Antimicrob. Agents Chemother. 2001, 45, 3427–3432. [Google Scholar] [CrossRef] [PubMed]
- Buck, C.B.; Thompson, C.D.; Roberts, J.N.; Müller, M.; Lowy, D.R.; Schiller, J.T. Carrageenan is a potent inhibitor of papillomavirus infection. PLoS Pathog. 2006, 2, e69. [Google Scholar] [CrossRef] [PubMed]
- CDC Guideline for Disinfection and Sterilization in Healthcare Facilities, 2008. Available online: http://www.cdc.gov/hicpac/Disinfection_Sterilization/3_1deLaparoArthro.html (accessed on 13 May 2015).
- Meyers, J.; Ryndock, E.; Conway, M.J.; Meyers, C.; Robison, R. Susceptibility of high-risk human papillomavirus type 16 to clinical disinfectants. J. Antimicrob. Chemother. 2014, 69, 1546–1550. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Biryukov, J.; Meyers, C. Papillomavirus Infectious Pathways: A Comparison of Systems. Viruses 2015, 7, 4303-4325. https://0-doi-org.brum.beds.ac.uk/10.3390/v7082823
Biryukov J, Meyers C. Papillomavirus Infectious Pathways: A Comparison of Systems. Viruses. 2015; 7(8):4303-4325. https://0-doi-org.brum.beds.ac.uk/10.3390/v7082823
Chicago/Turabian StyleBiryukov, Jennifer, and Craig Meyers. 2015. "Papillomavirus Infectious Pathways: A Comparison of Systems" Viruses 7, no. 8: 4303-4325. https://0-doi-org.brum.beds.ac.uk/10.3390/v7082823