
Hepatitis B Virus Protein X Induces Degradation of Talin-1

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Transfection
2.2. Expression Vectors and Lentiviral Transduction
2.3. Luciferase Assay
2.4. Quantitative PCR and Quantification of HBV Replication in HepG2 Cells
2.5. Western Blotting
2.6. Immunoprecipitation and Mass Spectrometry
2.7. Mass Spectrometry Data Acquisition
2.8. Mass Spectrometry Data Analysis
2.9. Software and Statistics
3. Results
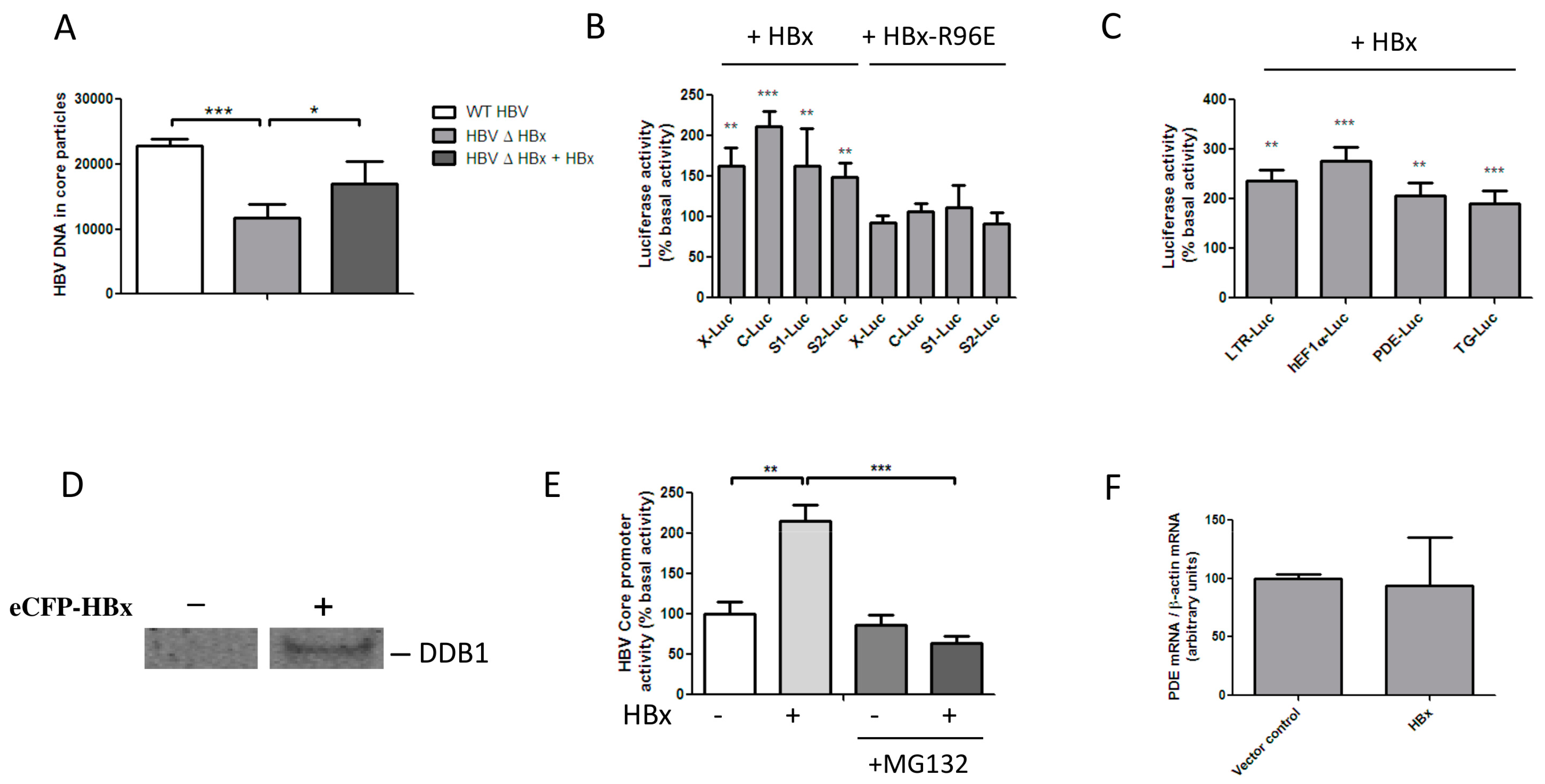
3.1. HBx Transactivates Transcription by Inducing the Degradation of a Host Protein
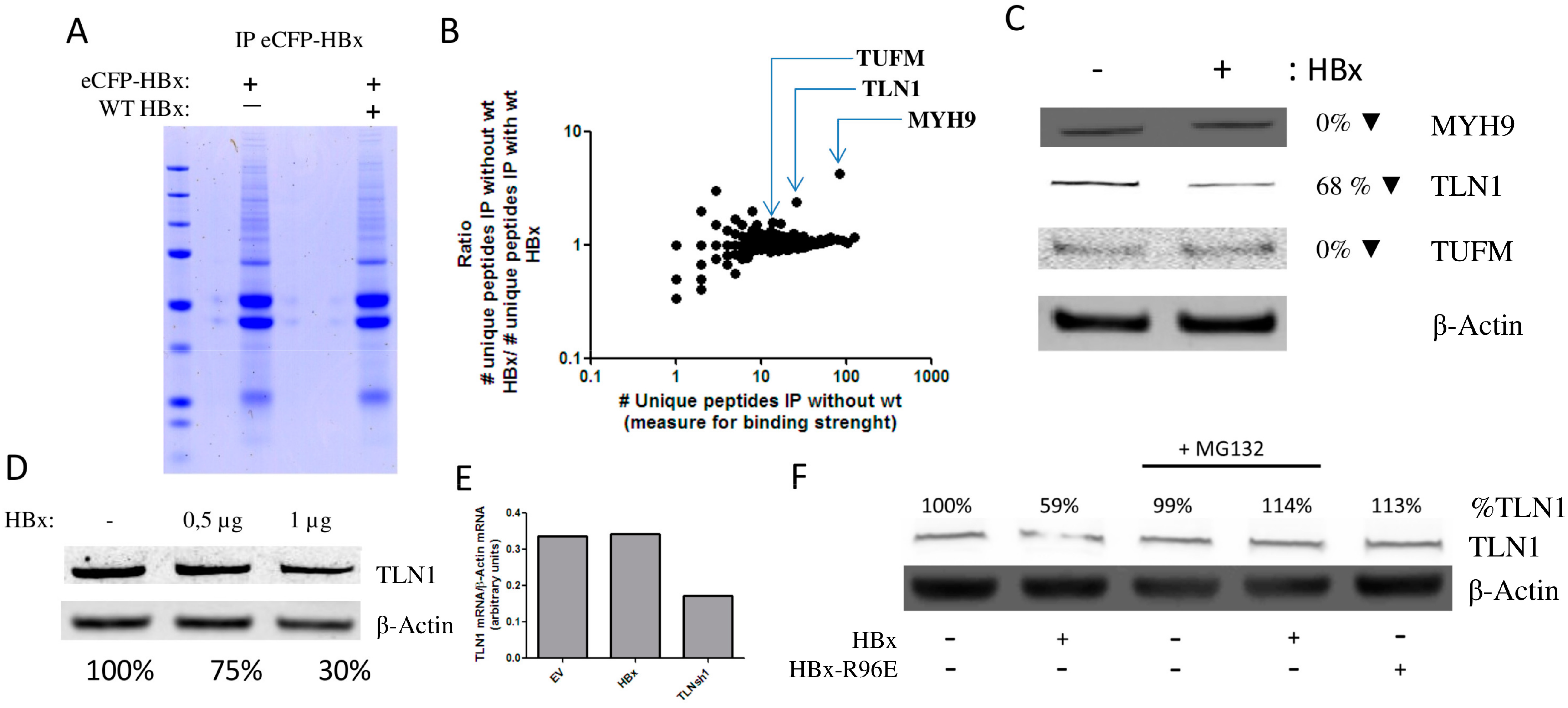
3.2. HBx Induces the Degradation of Talin-1
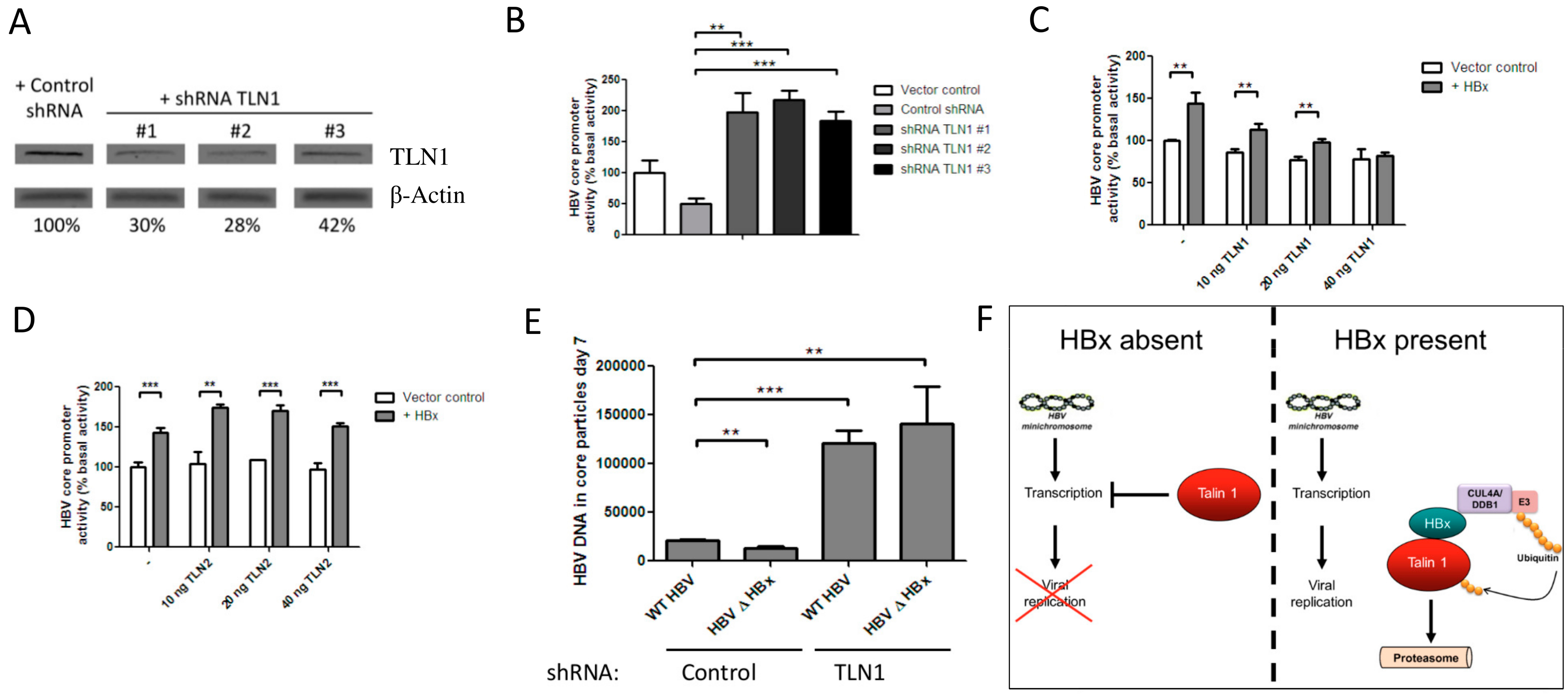
3.3. Talin-1 Suppresses HBV Replication by Interfering with RNA Transcription
4. Discussion
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Chen, H.S.; Kaneko, S.; Girones, R.; Anderson, R.W.; Hornbuckle, W.E.; Tennant, B.C.; Cote, P.J.; Gerin, J.L.; Purcell, R.H.; Miller, R.H. The woodchuck hepatitis virus X gene is important for establishment of virus infection in woodchucks. J. Virol. 1993, 67, 1218–1226. [Google Scholar] [PubMed]
- Lucifora, J.; Arzberger, S.; Durantel, D.; Belloni, L.; Strubin, M.; Levrero, M.; Zoulim, F.; Hantz, O.; Protzer, U. Hepatitis B virus X protein is essential to initiate and maintain virus replication after infection. J. Hepatol. 2011, 55, 996–1003. [Google Scholar] [CrossRef] [PubMed]
- Tsuge, M.; Hiraga, N.; Akiyama, R.; Tanaka, S.; Matsushita, M.; Mitsui, F.; Abe, H.; Kitamura, S.; Hatakeyama, T.; Kimura, T.; et al. HBx protein is indispensable for development of viraemia in human hepatocyte chimeric mice. J. Gen. Virol. 2010, 91, 1854–1864. [Google Scholar] [CrossRef] [PubMed]
- Zoulim, F.; Saputelli, J.; Seeger, C. Woodchuck hepatitis virus X protein is required for viral infection in vivo. J. Virol. 1994, 68, 2026–2030. [Google Scholar] [PubMed]
- Arbuthnot, P.; Capovilla, A.; Kew, M. Putative role of hepatitis B virus X protein in hepatocarcinogenesis: Effects on apoptosis, DNA repair, mitogen-activated protein kinase and JAK/STAT pathways. J. Gastroenterol. Hepatol. 2000, 15, 357–368. [Google Scholar] [CrossRef] [PubMed]
- Lin-Marq, N.; Bontron, S.; Leupin, O.; Strubin, M. Hepatitis B virus X protein interferes with cell viability through interaction with the p127-kDa UV-damaged DNA-binding protein. Virology 2001, 287, 266–274. [Google Scholar] [CrossRef] [PubMed]
- Martin-Lluesma, S.; Schaeffer, C.; Robert, E.I.; van Breugel, P.C.; Leupin, O.; Hantz, O.; Strubin, M. Hepatitis B virus X protein affects S phase progression leading to chromosome segregation defects by binding to damaged DNA binding protein 1. Hepatology 2008, 48, 1467–1476. [Google Scholar] [CrossRef] [PubMed]
- Murakami, S. Hepatitis B virus X protein: A multifunctional viral regulator. J. Gastroenterol. 2001, 36, 651–660. [Google Scholar] [CrossRef] [PubMed]
- Natoli, G.; Avantaggiati, M.L.; Chirillo, P.; De, M.E.; Collepardo, D.; Falco, M.; Balsano, C.; Levrero, M. Modulation of intracellular signal transduction pathways by the hepatitis B virus transactivator pX. J. Hepatol. 1995, 22, 14–20. [Google Scholar] [PubMed]
- Levrero, M.; Pollicino, T.; Petersen, J.; Belloni, L.; Raimondo, G.; Dandri, M. Control of cccDNA function in hepatitis B virus infection. J. Hepatol. 2009, 51, 581–592. [Google Scholar] [CrossRef] [PubMed]
- Luo, L.; Chen, S.; Gong, Q.; Luo, N.; Lei, Y.; Guo, J.; He, S. Hepatitis B virus X protein modulates remodelling of minichromosomes related to hepatitis B virus replication in HepG2 cells. Int. J. Mol. Med. 2013, 31, 197–204. [Google Scholar] [PubMed]
- Quasdorff, M.; Protzer, U. Control of hepatitis B virus at the level of transcription. J. Viral Hepat. 2010, 17, 527–536. [Google Scholar] [CrossRef] [PubMed]
- Glebe, D.; Berting, A.; Broehl, S.; Naumann, H.; Schuster, R.; Fiedler, N.; Tolle, T.K.; Nitsche, S.; Seifer, M.; Gerlich, W.H.; Schaefer, S. Optimised conditions for the production of hepatitis B virus from cell culture. Intervirology 2001, 44, 370–378. [Google Scholar] [CrossRef] [PubMed]
- Keasler, V.V.; Hodgson, A.J.; Madden, C.R.; Slagle, B.L. Enhancement of hepatitis B virus replication by the regulatory X protein in vitro and in vivo. J. Virol. 2007, 81, 2656–2662. [Google Scholar] [CrossRef] [PubMed]
- Slagle, B.L.; Andrisani, O.M.; Bouchard, M.J.; Lee, C.G.; Ou, J.H.; Siddiqui, A. Technical standards for hepatitis B virus X protein (HBx) research. Hepatology 2015, 61, 1416–1424. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Oishi, N.; Kaneko, S.; Murakami, S. Molecular functions and biological roles of hepatitis B virus X protein. Cancer Sci. 2006, 97, 977–983. [Google Scholar] [CrossRef] [PubMed]
- Van Breugel, P.C.; Robert, E.I.; Mueller, H.; Decorsiere, A.; Zoulim, F.; Hantz, O.; Strubin, M. Hepatitis B virus X protein stimulates gene expression selectively from extrachromosomal DNA templates. Hepatology 2012, 56, 2116–2124. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Neuveut, C.; Tiollais, P.; Buendia, M.A. Molecular biology of the hepatitis B virus and role of the X gene. Pathol. Biol. 2010, 58, 267–272. [Google Scholar] [CrossRef] [PubMed]
- Wollersheim, M.; Debelka, U.; Hofschneider, P.H. A transactivating function encoded in the hepatitis B virus X gene is conserved in the integrated state. Oncogene 1988, 3, 545–552. [Google Scholar] [PubMed]
- Tang, H.; Delgermaa, L.; Huang, F.; Oishi, N.; Liu, L.; He, F.; Zhao, L.; Murakami, S. The transcriptional transactivation function of HBx protein is important for its augmentation role in hepatitis B virus replication. J. Virol. 2005, 79, 5548–5556. [Google Scholar] [CrossRef] [PubMed]
- Bergametti, F.; Sitterlin, D.; Transy, C. Turnover of hepatitis B virus X protein is regulated by damaged DNA-binding complex. J. Virol. 2002, 76, 6495–6501. [Google Scholar] [CrossRef] [PubMed]
- Hodgson, A.J.; Hyser, J.M.; Keasler, V.V.; Cang, Y.; Slagle, B.L. Hepatitis B virus regulatory HBx protein binding to DDB1 is required but is not sufficient for maximal HBV replication. Virology 2012, 426, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.H.; Elledge, S.J.; Butel, J.S. Hepatitis B virus X protein interacts with a probable cellular DNA repair protein. J. Virol. 1995, 69, 1107–1114. [Google Scholar] [PubMed]
- Leupin, O.; Bontron, S.; Strubin, M. Hepatitis B virus X protein and simian virus 5 V protein exhibit similar UV-DDB1 binding properties to mediate distinct activities. J. Virol. 2003, 77, 6274–6283. [Google Scholar] [CrossRef] [PubMed]
- Leupin, O.; Bontron, S.; Schaeffer, C.; Strubin, M. Hepatitis B virus X protein stimulates viral genome replication via a DDB1-dependent pathway distinct from that leading to cell death. J. Virol. 2005, 79, 4238–4245. [Google Scholar] [CrossRef] [PubMed]
- Sitterlin, D.; Lee, T.H.; Prigent, S.; Tiollais, P.; Butel, J.S.; Transy, C. Interaction of the UV-damaged DNA-binding protein with hepatitis B virus X protein is conserved among mammalian hepadnaviruses and restricted to transactivation-proficient X-insertion mutants. J. Virol. 1997, 71, 6194–6199. [Google Scholar] [PubMed]
- Sitterlin, D.; Bergametti, F.; Tiollais, P.; Tennant, B.C.; Transy, C. Correct binding of viral X protein to UVDDB-p127 cellular protein is critical for efficient infection by hepatitis B viruses. Oncogene 2000, 19, 4427–4431. [Google Scholar] [CrossRef] [PubMed]
- Laguette, N.; Sobhian, B.; Casartelli, N.; Ringeard, M.; Chable-Bessia, C.; Segeral, E.; Yatim, A.; Emiliani, S.; Schwartz, O.; Benkirane, M. SAMHD1 is the dendritic- and myeloid-cell-specific HIV-1 restriction factor counteracted by Vpx. Nature 2011, 474, 654–657. [Google Scholar] [CrossRef] [PubMed]
- Ahn, J.; Vu, T.; Novince, Z.; Guerrero-Santoro, J.; Rapic-Otrin, V.; Gronenborn, A.M. HIV-1 Vpr loads uracil DNA glycosylase-2 onto DCAF1, a substrate recognition subunit of a cullin 4A-ring E3 ubiquitin ligase for proteasome-dependent degradation. J. Biol. Chem. 2010, 285, 37333–37341. [Google Scholar] [CrossRef] [PubMed]
- Precious, B.; Childs, K.; Fitzpatrick-Swallow, V.; Goodbourn, S.; Randall, R.E. Simian virus 5 V protein acts as an adaptor, linking DDB1 to STAT2, to facilitate the ubiquitination of STAT1. J. Virol. 2005, 79, 13434–13441. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Robert, E.I.; van Breugel, P.C.; Strubin, M.; Zheng, N. A promiscuous alpha-helical motif anchors viral hijackers and substrate receptors to the CUL4-DDB1 ubiquitin ligase machinery. Nat. Struct. Mol. Biol. 2010, 17, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Protzer, U.; Hu, Z.; Jacob, J.; Liang, T.J. Inhibition of cellular proteasome activities enhances hepadnavirus replication in an HBX-dependent manner. J. Virol. 2004, 78, 4566–4572. [Google Scholar] [CrossRef] [PubMed]
- Decorsière, A.; Mueller, H.; van Breugel, P.C.; van Abdul, F.; Gerossier, L.; Beran, R.K.; Livingston, C.M.; Niu, C.; Fletcher, S.P.; Hantz, O.; et al. Hepatitis B virus X protein identifies the Smc5/6 complex as a host restriction factor. Nature 2016, 531, 386–389. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Sun, E.; Ou, J.H.; Liang, T.J. Inhibition of cellular proteasome activities mediates HBX-independent hepatitis B virus replication in vivo. J. Virol. 2010, 84, 9326–9331. [Google Scholar] [CrossRef] [PubMed]
- Van de Klundert, M.A.; van Hemert, F.J.; Zaaijer, H.L.; Kootstra, N.A. The hepatitis B virus X protein inhibits thymine DNA glycosylase initiated base excision repair. PLoS ONE 2012, 7, e48940. [Google Scholar] [CrossRef] [PubMed]
- Jeeninga, R.E.; Hoogenkamp, M.; Armand-Ugon, M.; de, B.M.; Verhoef, K.; Berkhout, B. Functional differences between the long terminal repeat transcriptional promoters of human immunodeficiency virus type 1 subtypes A through G. J. Virol. 2000, 74, 3740–3751. [Google Scholar] [CrossRef] [PubMed]
- Klaver, B.; Berkhout, B. Comparison of 5′ and 3′ long terminal repeat promoter function in human immunodeficiency virus. J. Virol. 1994, 68, 3830–3840. [Google Scholar] [PubMed]
- Bol, S.M.; Booiman, T.; Bunnik, E.M.; Moerland, P.D.; van, D.K.; Strauss, J.F., III; Sieberer, M.; Schuitemaker, H.; Kootstra, N.A.; van ’t Wout, A.B. Polymorphism in HIV-1 dependency factor PDE8A affects mRNA level and HIV-1 replication in primary macrophages. Virology 2011, 420, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Zufferey, R.; Dull, T.; Mandel, R.J.; Bukovsky, A.; Quiroz, D.; Naldini, L.; Trono, D. Self-inactivating lentivirus vector for safe and efficient in vivo gene delivery. J. Virol. 1998, 72, 9873–9880. [Google Scholar] [PubMed]
- Van Engelen, K.; Mommersteeg, M.T.; Baars, M.J.; Lam, J.; Ilgun, A.; van Trotsenburg, A.S.; Smets, A.M.; Christoffels, V.M.; Mulder, B.J.; Postma, A.V. The Ambiguous Role of NKX2–5 Mutations in Thyroid Dysgenesis. PLoS ONE 2012, 7, e52685. [Google Scholar] [CrossRef] [PubMed]
- Baumert, T.F.; Rogers, S.A.; Hasegawa, K.; Liang, T.J. Two core promotor mutations identified in a hepatitis B virus strain associated with fulminant hepatitis result in enhanced viral replication. J. Clin. Investig. 1996, 98, 2268–2276. [Google Scholar] [CrossRef] [PubMed]
- Baumert, T.F.; Marrone, A.; Vergalla, J.; Liang, T.J. Naturally occurring mutations define a novel function of the hepatitis B virus core promoter in core protein expression. J. Virol. 1998, 72, 6785–6795. [Google Scholar] [PubMed]
- Franco, S.J.; Rodgers, M.A.; Perrin, B.J.; Han, J.; Bennin, D.A.; Critchley, D.R.; Huttenlocher, A. Calpain-mediated proteolysis of talin regulates adhesion dynamics. Nat. Cell Biol. 2004, 6, 977–983. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Zhou, Y.; Fu, Q.X.; Gong, W.L.; Zhao, F.; Peng, J.C.; Zhan, L.S. Bioluminescence imaging of hepatitis B virus enhancer and promoter activities in mice. FEBS Lett. 2008, 582, 3552–2556. [Google Scholar] [CrossRef] [PubMed]
- Shevchenko, A.; Tomas, H.; Havlis, J.; Olsen, J.V.; Mann, M. In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat. Protoc. 2006, 1, 2856–2860. [Google Scholar] [CrossRef] [PubMed]
- Rappsilber, J.; Ishihama, Y.; Mann, M. Stop and go extraction tips for matrix-assisted laser desorption/ionization, nanoelectrospray, and LC/MS sample pretreatment in proteomics. Anal. Chem. 2003, 75, 663–670. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef] [PubMed]
- UniProt Consortium. The Universal Protein Resource (UniProt) in 2010. Nucleic Acids Res. 2010, 38 (Database issue), D142–D148. [Google Scholar]
- Van de Klundert, M.A.; Zaaijer, H.L.; Kootstra, N.A. Identification of FDA-approved drugs that target hepatitis B virus transcription. J. Viral Hepat. 2016, 23, 191–201. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.M.; Sun, D.C.; Lou, S.; Bo, X.C.; Lu, Z.; Qian, X.H.; Wang, S.Q. HBx protein of hepatitis B virus (HBV) can form complex with mitochondrial HSP60 and HSP70. Arch. Virol. 2005, 150, 1579–1590. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, J.; Liu, H.; He, Y.; Yi, R.; Niu, Y.; Chen, T.; Yang, Q.; Zhao, Y. Comparative study of the different activities of hepatitis B virus whole-X protein and HBx in hepatocarcinogenesis by proteomics and bioinformatics analysis. Arch. Virol. 2015, 160, 1645–1656. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; Hu, J. Formation of hepatitis B virus covalently closed circular DNA: Removal of genome-linked protein. J. Virol. 2007, 81, 6164–6174. [Google Scholar] [CrossRef] [PubMed]
- Critchley, D.R.; Gingras, A.R. Talin at a glance. J. Cell Sci. 2008, 121, 1345–1347. [Google Scholar] [CrossRef] [PubMed]
- Nayal, A.; Webb, D.J.; Horwitz, A.F. Talin: An emerging focal point of adhesion dynamics. Curr. Opin. Cell Biol. 2004, 16, 94–98. [Google Scholar] [CrossRef] [PubMed]
- Goksoy, E.; Ma, Y.Q.; Wang, X.; Kong, X.; Perera, D.; Plow, E.F.; Qin, J. Structural basis for the autoinhibition of talin in regulating integrin activation. Mol. Cell. 2008, 31, 124–133. [Google Scholar] [CrossRef] [PubMed]
- Gupta, N.; Li, W.; Willard, B.; Silverstein, R.L.; McIntyre, T.M. Proteasome proteolysis supports stimulated platelet function and thrombosis. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 160–168. [Google Scholar] [CrossRef] [PubMed]
- Wegener, K.L.; Partridge, A.W.; Han, J.; Pickford, A.R.; Liddington, R.C.; Ginsberg, M.H.; Campbell, I.D. Structural basis of integrin activation by talin. Cell 2007, 128, 171–182. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Feng, H.; Chen, W.N.; Chan, V. Hepatitis B virus induced coupling of deadhesion and migration of HepG2 cells on thermo-responsive polymer. Biomaterials 2010, 31, 1894–1903. [Google Scholar] [CrossRef] [PubMed]
- Tan, T.L.; Feng, Z.; Lu, Y.W.; Chan, V.; Chen, W.N. Adhesion contact kinetics of HepG2 cells during Hepatitis B virus replication: Involvement of SH3-binding motif in HBX. Biochim. Biophys. Acta 2006, 1762, 755–766. [Google Scholar] [CrossRef] [PubMed]
- Bouchard, M.J.; Wang, L.; Schneider, R.J. Activation of focal adhesion kinase by hepatitis B virus HBx protein: Multiple functions in viral replication. J. Virol. 2006, 80, 4406–4414. [Google Scholar] [CrossRef] [PubMed]
- Balda, M.S.; Matter, K. Epithelial cell adhesion and the regulation of gene expression. Trends Cell Biol. 2003, 13, 310–318. [Google Scholar] [CrossRef]
- Mammoto, A.; Mammoto, T.; Ingber, D.E. Mechanosensitive mechanisms in transcriptional regulation. J. Cell Sci. 2012, 125, 3061–3073. [Google Scholar] [CrossRef] [PubMed]
- Becam, I.E.; Tanentzapf, G.; Lepesant, J.A.; Brown, N.H.; Huynh, J.R. Integrin-independent repression of cadherin transcription by talin during axis formation in Drosophila. Nat. Cell Biol. 2005, 7, 510–516. [Google Scholar] [CrossRef] [PubMed]
- Tepass, U.; Godt, D. Talin’s second persona. Nat. Cell Biol. 2005, 7, 443–444. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.K.; Kim, M.H.; Cheong, J.Y.; Cho, S.W.; Yang, S.J.; Kwack, K. Integrin alpha V polymorphisms and haplotypes in a Korean population are associated with susceptibility to chronic hepatitis and hepatocellular carcinoma. Liver Int. 2009, 29, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Belloni, L.; Pollicino, T.; De, N.F.; Guerrieri, F.; Raffa, G.; Fanciulli, M.; Raimondo, G.; Levrero, M. Nuclear HBx binds the HBV minichromosome and modifies the epigenetic regulation of cccDNA function. Proc. Natl. Acad. Sci. USA 2009, 106, 19975–19979. [Google Scholar] [CrossRef] [PubMed]
- Bellissent-Waydelich, A.; Vanier, M.T.; Albiges-Rizo, C.; Simon-Assmann, P. Talin concentrates to the midbody region during mammalian cell cytokinesis. J. Histochem. Cytochem. 1999, 47, 1357–1368. [Google Scholar] [CrossRef] [PubMed]
- Hibi, M.; Nagasaki, A.; Takahashi, M.; Yamagishi, A.; Uyeda, T.Q. Dictyostelium discoideum talin A is crucial for myosin II-independent and adhesion-dependent cytokinesis. J. Muscle Res. Cell Motil. 2004, 25, 127–140. [Google Scholar] [CrossRef] [PubMed]
- Sze, K.M.; Chu, G.K.; Lee, J.M.; Ng, I.O. C-terminal truncated hepatitis B virus x protein is associated with metastasis and enhances invasiveness by C-Jun/matrix metalloproteinase protein 10 activation in hepatocellular carcinoma. Hepatology 2013, 57, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.M.; Koike, K.; Saito, I.; Miyamura, T.; Jay, G. HBx gene of hepatitis B virus induces liver cancer in transgenic mice. Nature 1991, 351, 317–320. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.Y.; Moon, H.B.; Son, J.K.; Jeong, S.; Yu, S.L.; Yoon, H.; Han, Y.M.; Lee, C.S.; Park, J.S.; Lee, C.H.; et al. Incidence of hepatocellular carcinoma in transgenic mice expressing the hepatitis B virus X-protein. J. Hepatol. 1999, 31, 123–132. [Google Scholar] [CrossRef]
- Kanamori, H.; Kawakami, T.; Effendi, K.; Yamazaki, K.; Mori, T.; Ebinuma, H.; Masugi, Y.; Du, W.; Nagasaka, K.; Ogiwara, A.; et al. Identification by differential tissue proteome analysis of talin-1 as a novel molecular marker of progression of hepatocellular carcinoma. Oncology 2011, 80, 406–415. [Google Scholar] [CrossRef] [PubMed]
- Quasdorff, M.; Hosel, M.; Odenthal, M.; Zedler, U.; Bohne, F.; Gripon, P.; Dienes, H.P.; Drebber, U.; Stippel, D.; Goeser, T.; et al. A concerted action of HNF4alpha and HNF1alpha links hepatitis B virus replication to hepatocyte differentiation. Cell Microbiol. 2008, 10, 1478–1490. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.; Morham, S.G.; Walsh, D.; Naghavi, M.H. Focal adhesion proteins talin-1 and vinculin negatively affect paxillin phosphorylation and limit retroviral infection. J. Mol. Biol. 2011, 410, 761–777. [Google Scholar] [CrossRef] [PubMed]
- Stanton, R.J.; Prod’homme, V.; Purbhoo, M.A.; Moore, M.; Aicheler, R.J.; Heinzmann, M.; Bailer, S.M.; Haas, J.; Antrobus, R.; Weekes, M.P.; et al. HCMV pUL135 remodels the actin cytoskeleton to impair immune recognition of infected cells. Cell Host Microbe 2014, 16, 201–214. [Google Scholar] [CrossRef] [PubMed]
- Alon, R. Chemokine arrest signals to leukocyte integrins trigger bi-directional-occupancy of individual heterodimers by extracellular and cytoplasmic ligands. Cell Adhes. Migr. 2010, 4, 211–214. [Google Scholar] [CrossRef]
- Nolz, J.C.; Medeiros, R.B.; Mitchell, J.S.; Zhu, P.; Freedman, B.D.; Shimizu, Y.; Billadeau, D.D. WAVE2 regulates high-affinity integrin binding by recruiting vinculin and talin to the immunological synapse. Mol Cell Biol. 2007, 27, 5986–6000. [Google Scholar] [CrossRef] [PubMed]
- Simonson, W.T.; Franco, S.J.; Huttenlocher, A. Talin1 regulates TCR-mediated LFA-1 function. J. Immunol. 2006, 177, 7707–7714. [Google Scholar] [CrossRef] [PubMed]



© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Van de Klundert, M.A.A.; Van den Biggelaar, M.; Kootstra, N.A.; Zaaijer, H.L. Hepatitis B Virus Protein X Induces Degradation of Talin-1. Viruses 2016, 8, 281. https://0-doi-org.brum.beds.ac.uk/10.3390/v8100281
Van de Klundert MAA, Van den Biggelaar M, Kootstra NA, Zaaijer HL. Hepatitis B Virus Protein X Induces Degradation of Talin-1. Viruses. 2016; 8(10):281. https://0-doi-org.brum.beds.ac.uk/10.3390/v8100281
Chicago/Turabian StyleVan de Klundert, Maarten A. A., Maartje Van den Biggelaar, Neeltje A. Kootstra, and Hans L. Zaaijer. 2016. "Hepatitis B Virus Protein X Induces Degradation of Talin-1" Viruses 8, no. 10: 281. https://0-doi-org.brum.beds.ac.uk/10.3390/v8100281