
Single Amino Acid Substitution N659D in HIV-2 Envelope Glycoprotein (Env) Impairs Viral Release and Hampers BST-2 Antagonism


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Plasmids, Cloning and Site-Directed Mutagenesis
2.3. Cell Infections and Viral Release Quantification
2.4. Subtilisin Treatment
2.5. CRISPR/Cas9 Knockout
2.6. Statistics and Sofware
3. Results
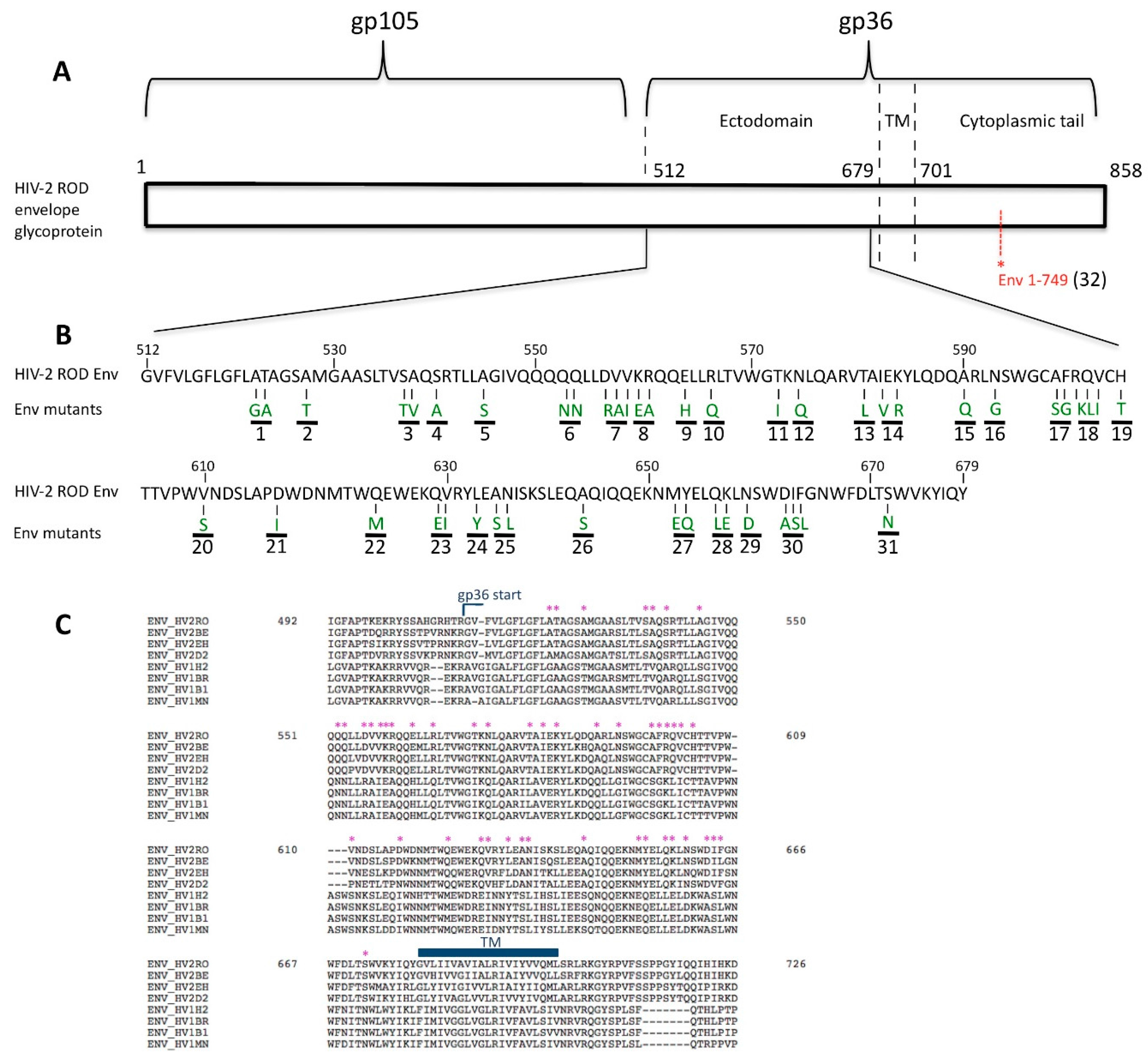
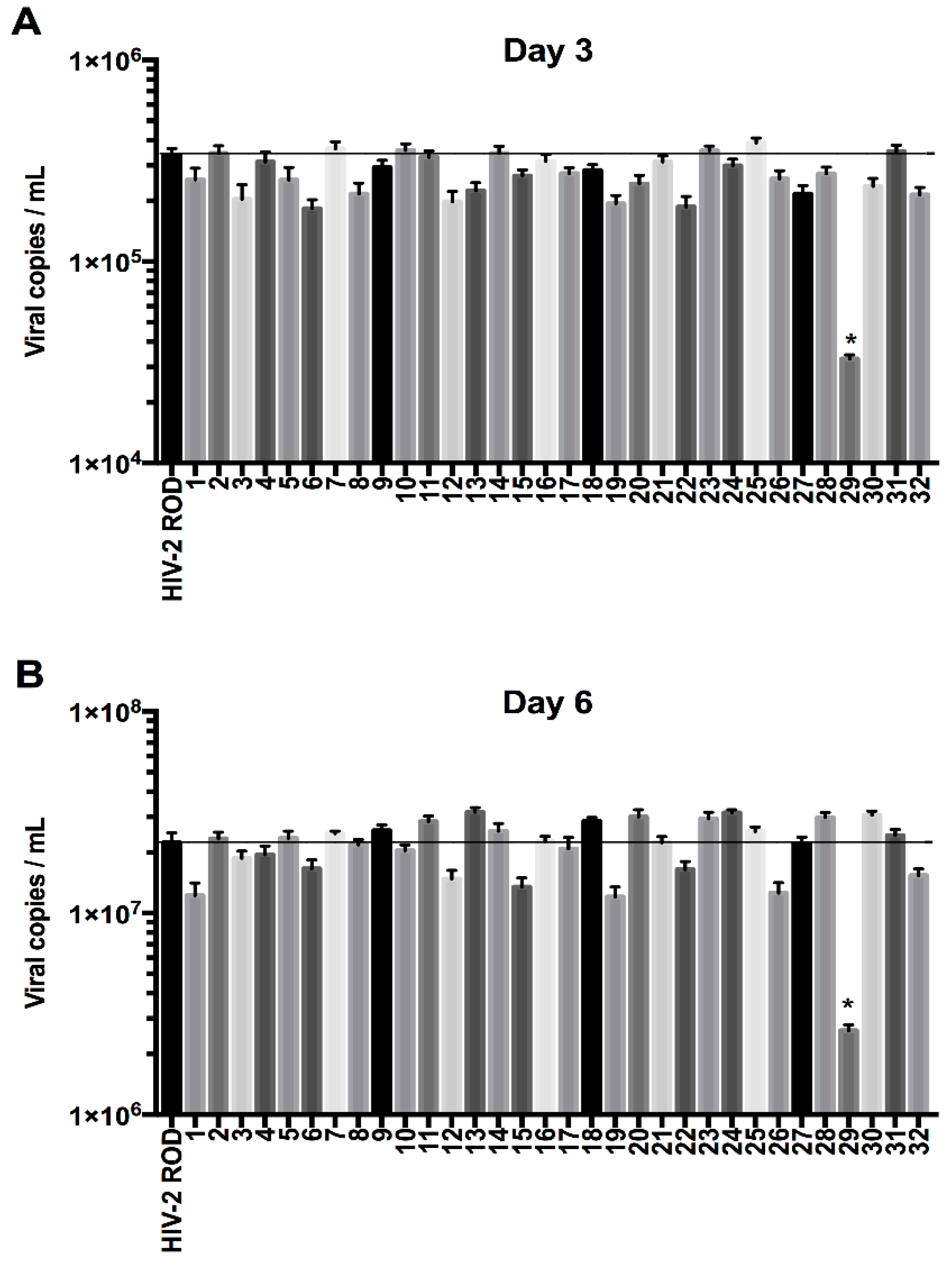
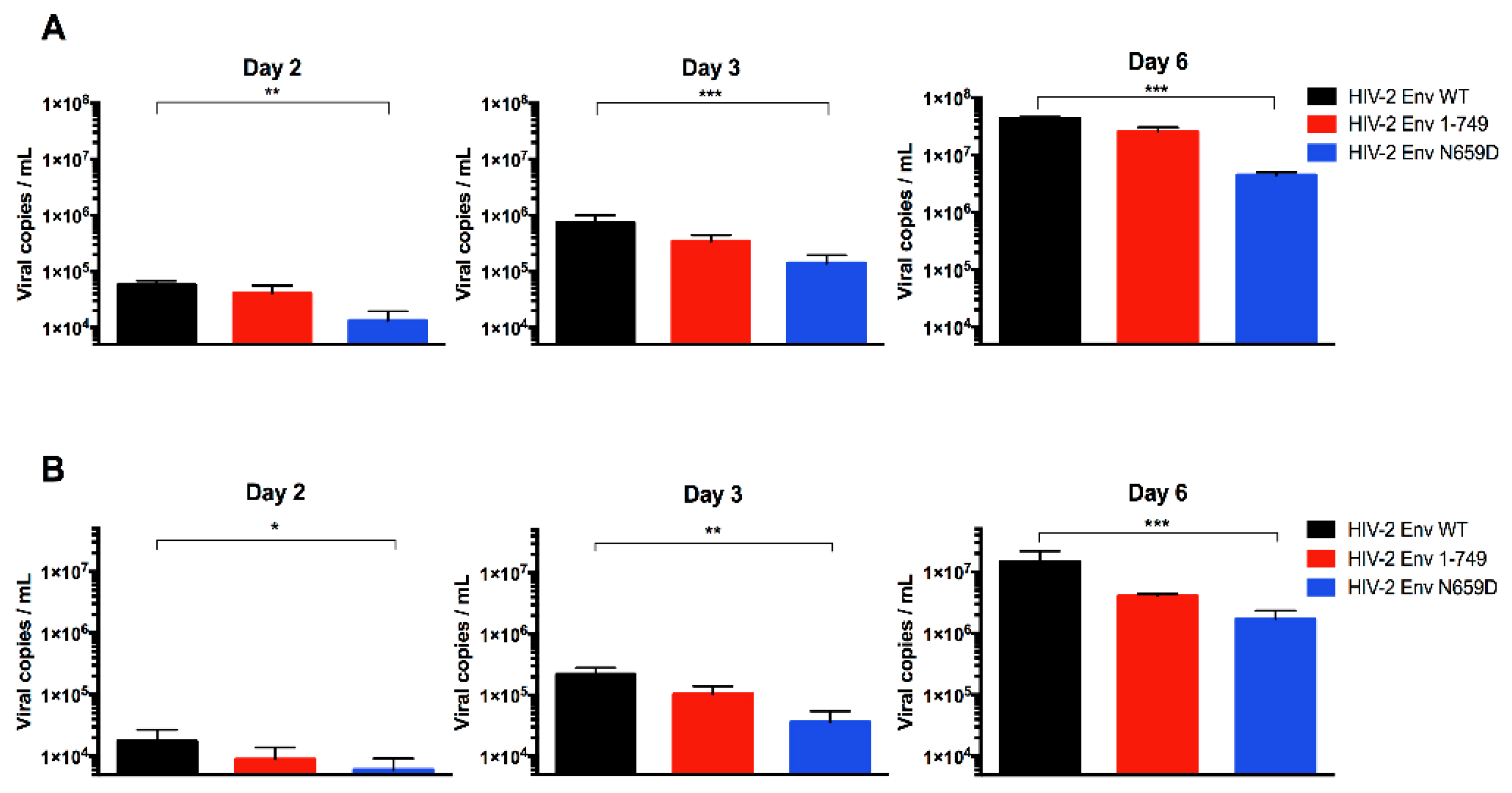
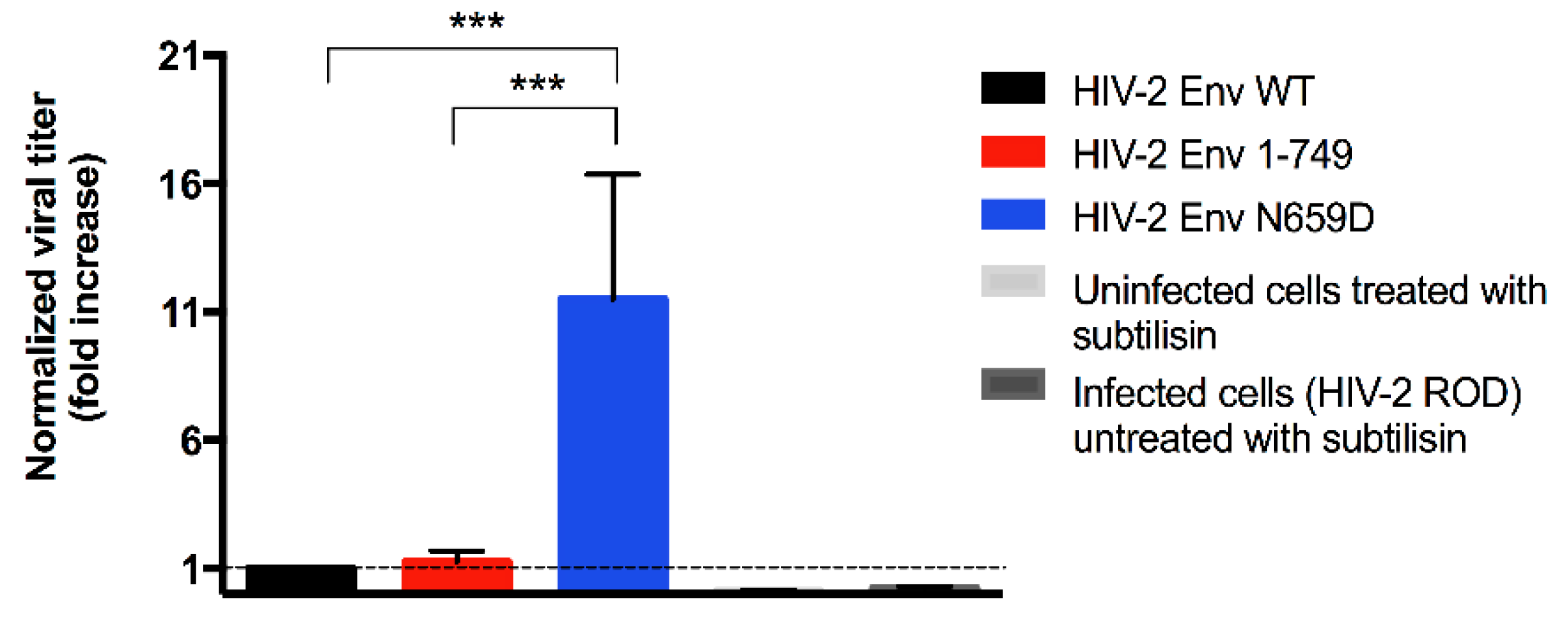
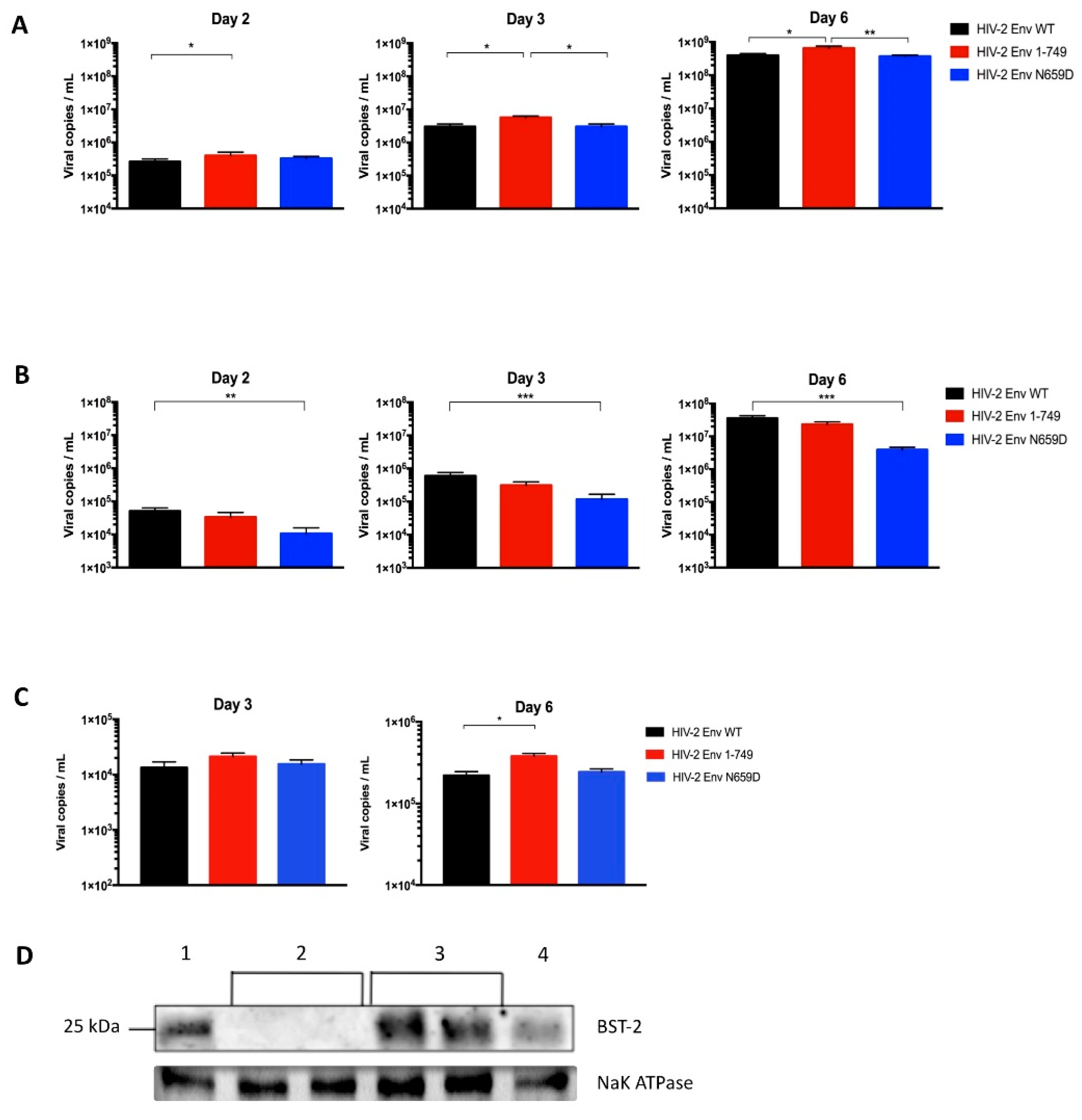
3.1. Residue N659 in the HIV-2 ROD Env Protein Is Involved in the Efficiency of Viral Release
3.2. Viral Release of the Env N659D Mutant Is Restored in BST-2-Depleted H9 Cells
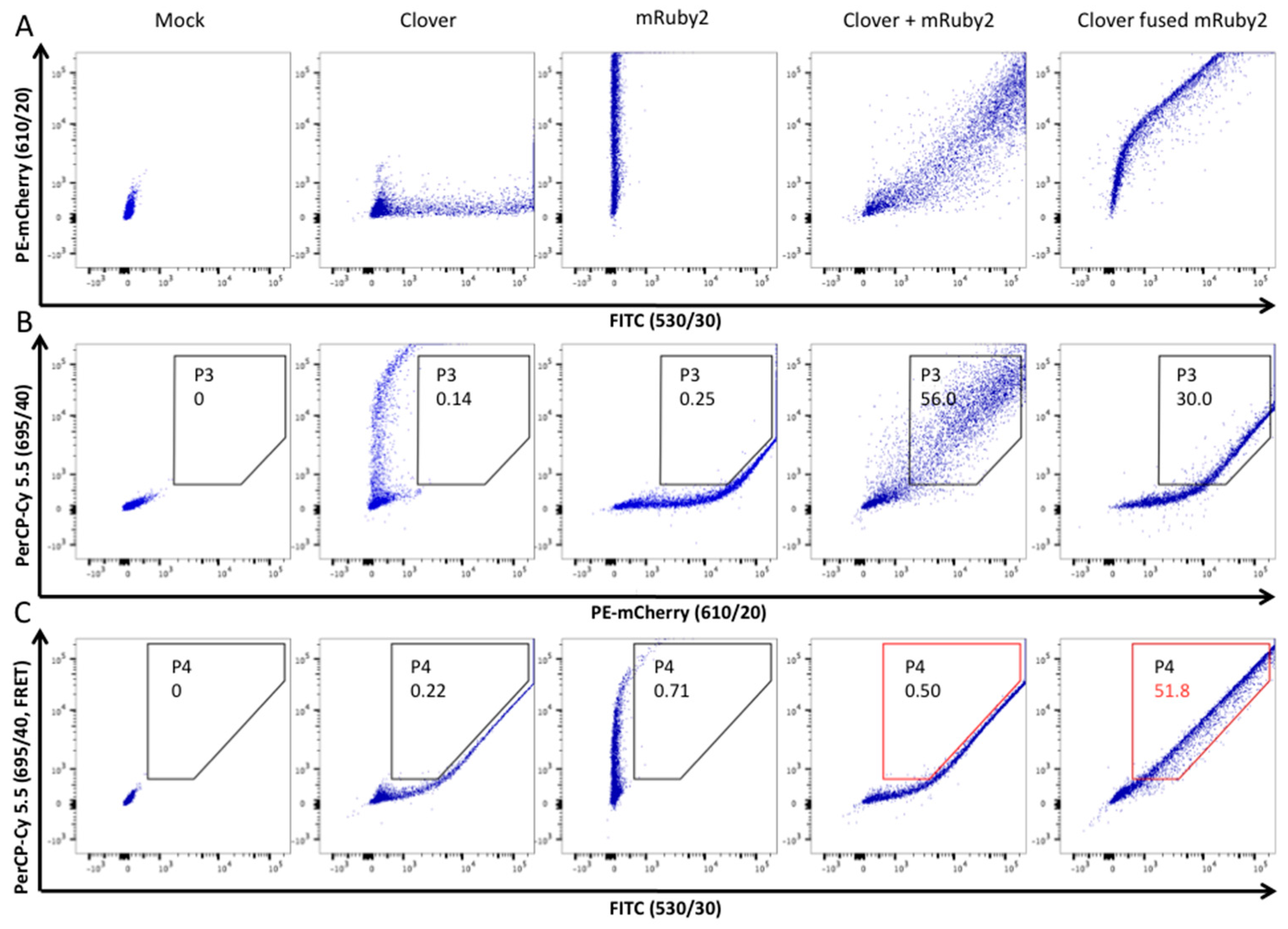
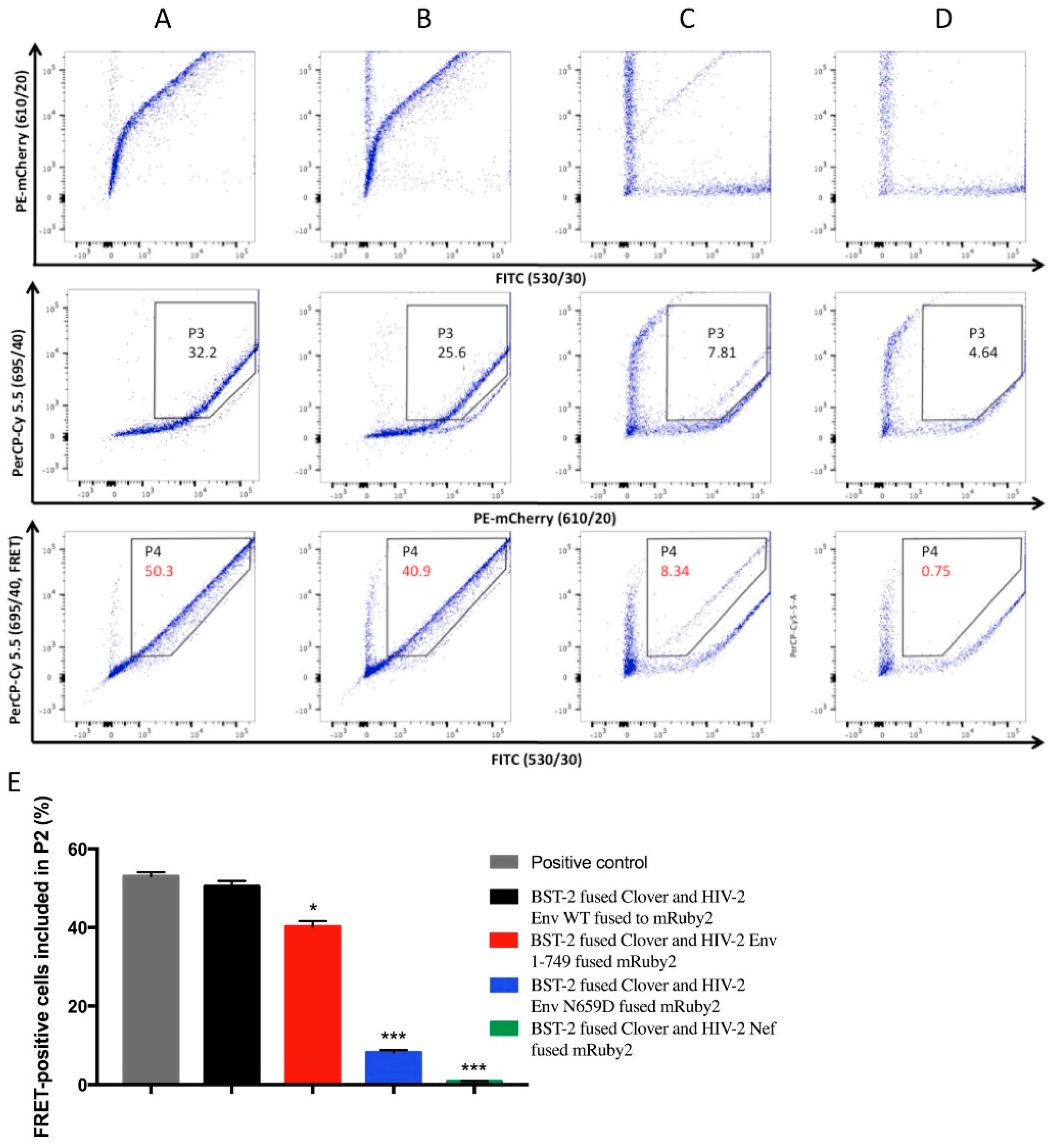
3.3. HIV-2 Env N659D Mutant Is Unable to Bind BST-2
4. Discussion
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Clavel, F.; Guétard, D.; Brun-Vézinet, F.; Chamaret, S.; Rey, M.A.; Santos-Ferreira, M.O.; Laurent, G.; Dauguet, C.; Katlama, C.; Rouzioux, C.; et al. Isolation of a new human retrovirus from West African patients with AIDS. Science 1986, 233, 343–346. [Google Scholar] [CrossRef] [PubMed]
- Sharp, P.M.; Hahn, B.H. Origins of HIV and the AIDS pandemic. Cold Spring Harb. Perspect. Med. 2011, 1, a006841. [Google Scholar] [CrossRef] [PubMed]
- Lemey, P.; Pybus, O.G.; Wang, B.; Saksena, N.K.; Salemi, M.; Vandamme, A.M. Tracing the origin and history of the HIV-2 epidemic. Proc. Natl. Acad. Sci. USA 2003, 100, 6588–6592. [Google Scholar] [CrossRef] [PubMed]
- De Silva, T.I.; Cotten, M.; Rowland-Jones, S.L. HIV-2: The forgotten AIDS virus. Trends Microbiol. 2008, 16, 588–595. [Google Scholar] [CrossRef] [PubMed]
- Peeters, M.; D’Arc, M.; Delaporte, E. The origin and diversity of human retroviruses. AIDS Rev. 2014, 16, 23–34. [Google Scholar] [PubMed]
- Popper, S.; Dieng Sarr, A.; Gueye-Ndiaye, A.; Mboup, S.; Essex, M.E.; Kanki, P.J. Low plasma human immunodeficiency virus type 2 viral load is independent of proviral load: Low virus production in vivo. J. Virol. 2000, 74, 1554–1557. [Google Scholar] [CrossRef] [PubMed]
- Reeves, J.D.; Doms, R.W. Human immunodeficiency virus type 2. J. Gen. Virol. 2002, 83, 1253–1265. [Google Scholar] [CrossRef] [PubMed]
- Damond, F.; Gueudin, M.; Pueyo, S.; Farfara, I.; Robertson, D.L.; Descamps, D.; Chene, G.; Matheron, S.; Campa, P.; Brun-Vezinet, F.; et al. Plasma RNA viral load in human immunodeficiency virus type 2 subtype A and subtype B infections. J. Clin. Microbiol. 2002, 40, 3654–3659. [Google Scholar] [CrossRef] [PubMed]
- Rowland-Jones, S.L.; Whittle, H.C. Out of Africa: What can we learn from HIV-2 about protective immunity to HIV-1? Nat. Immunol. 2007, 8, 329–331. [Google Scholar] [CrossRef] [PubMed]
- Thiébaut, R.; Matheron, S.; Taied, A.; Brun-Vezinet, F.; Chêne, G.; Autran, B. Long-term nonprogressors and elite controllers in the ANRS CO5 HIV-2 cohort. AIDS 2011, 25, 865–867. [Google Scholar] [CrossRef] [PubMed]
- Gottlieb, G.S.; Hawes, S.E.; Kiviat, N.B.; Sow, P.S. Differences in proviral DNA load between HIV-1-infected and HIV-2-infected patients. AIDS 2008, 22, 1379–1380. [Google Scholar] [CrossRef] [PubMed]
- De Silva, T.I.; van Tienen, C.; Onyango, C.; Jabang, A.; Vincent, T.; Loeff, M.F.; Coutinho, R.A.; Jaye, A.; Rowland-Jones, S.L.; Whittle, H.C.; et al. Population dynamics of HIV-2 in rural West Africa: Comparison with HIV-1 and ongoing transmission at the heart of the epidemic. AIDS 2013, 27, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Sheehy, A.M.; Gaddis, N.C.; Choi, J.D.; Malim, M.H. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature 2002, 418, 646–650. [Google Scholar] [CrossRef] [PubMed]
- Stremlau, M.; Owens, C.M.; Perron, M.J.; Kiessling, M.; Autissier, P.; Sodroski, J. The cytoplasmic body component TRIM5alpha restricts HIV-1 infection in Old World monkeys. Nature 2004, 427, 848–853. [Google Scholar] [CrossRef] [PubMed]
- Neil, S.J.; Zang, T.; Bieniasz, P.D. Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature 2008, 451, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Van Damme, N.; Goff, D.; Katsura, C.; Jorgenson, R.L.; Mitchell, R.; Johnson, M.C.; Stephens, E.B.; Guatelli, J. The interferon-induced protein BST-2 restricts HIV-1 release and is downregulated from the cell surface by the viral Vpu protein. Cell Host Microbe 2008, 3, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Perez-Caballero, D.; Zang, T.; Ebrahimi, A.; McNatt, M.W.; Gregory, D.A.; Johnson, M.C.; Bieniasz, P.D. Tetherin inhibits HIV-1 release by directly tethering virions to cells. Cell 2009, 139, 499–511. [Google Scholar] [CrossRef] [PubMed]
- Jouvenet, N.; Neil, S.J.; Zhadina, M.; Zang, T.; Kratovac, Z.; Lee, Y.; McNatt, M.; Hatziioannou, T.; Bieniasz, P.D. Broad-spectrum inhibition of retroviral and filoviral particle release by tetherin. J. Virol. 2009, 83, 1837–1844. [Google Scholar] [CrossRef] [PubMed]
- Tokarev, A.; Skasko, M.; Fitzpatrick, K.; Guatelli, J. Antiviral activity of the interferon-induced cellular protein BST-2/tetherin. AIDS Res. Hum. Retrovir. 2009, 25, 1197–1210. [Google Scholar] [CrossRef] [PubMed]
- Evans, D.T.; Serra-Moreno, R.; Singh, R.K.; Guatelli, J.C. BST-2/tetherin: A new component of the innate immune response to enveloped viruses. Trends Microbiol. 2010, 18, 388–396. [Google Scholar] [CrossRef] [PubMed]
- Neil, S.J. The antiviral activities of tetherin. Curr. Top. Microbiol. Immunol. 2013, 371, 67–104. [Google Scholar] [PubMed]
- Hinz, A.; Miguet, N.; Natrajan, G.; Usami, Y.; Yamanaka, H.; Renesto, P.; Hartlieb, B.; McCarthy, A.A.; Simorre, J.P.; Gottlinger, H.; et al. Structural basis of HIV-1 tethering to membranes by the BST-2/tetherin ectodomain. Cell Host Microbe 2010, 7, 314–323. [Google Scholar] [CrossRef] [PubMed]
- Rollason, R.; Korolchuk, V.; Hamilton, C.; Schu, P.; Banting, G. Clathrin-mediated endocytosis of a lipid-raft-associated protein is mediated through a dual tyrosine motif. J. Cell Sci. 2007, 120, 3850–3858. [Google Scholar] [CrossRef] [PubMed]
- Sauter, D. Counteraction of the multifunctional restriction factor tetherin. Front. Microbiol. 2014, 5, 163. [Google Scholar] [CrossRef] [PubMed]
- Hotter, D.; Sauter, D.; Kirchhoff, F. Emerging role of the host restriction factor tetherin in viral immune sensing. J. Mol. Biol. 2013, 425, 4956–4964. [Google Scholar] [CrossRef] [PubMed]
- Kupzig, S.; Korolchuk, V.; Rollason, R.; Sugden, A.; Wilde, A.; Banting, G. Bst-2/HM1.24 is a raft-associated apical membrane protein with an unusual topology. Traffic 2003, 4, 694–709. [Google Scholar] [CrossRef] [PubMed]
- Le Tortorec, A.; Willey, S.; Neil, S.J. Antiviral inhibition of enveloped virus release by tetherin/BST-2: Action and counteraction. Viruses 2011, 3, 520–540. [Google Scholar] [CrossRef] [PubMed]
- Billcliff, P.G.; Gorleku, O.A.; Chamberlain, L.H.; Banting, G. The cytosolic N-terminus of CD317/tetherin is a membrane microdomain exclusion motif. Biol. Open 2013, 2, 1253–1263. [Google Scholar] [CrossRef] [PubMed]
- Venkatesh, S.; Bieniasz, P.D. Mechanism of HIV-1 virion entrapment by tetherin. PLoS Pathog. 2013, 9, e1003483. [Google Scholar] [CrossRef] [PubMed]
- Andrew, A.J.; Kao, S.; Strebel, K. C-terminal hydrophobic region in human bone marrow stromal cell antigen 2 (BST-2)/tetherin protein functions as second transmembrane motif. J. Biol. Chem. 2011, 286, 39967–39981. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.K.; Hue, S.; Schaller, T.; Verschoor, E.; Pillay, D.; Towers, G.J. Mutation of a single residue renders human tetherin resistant to HIV-1 Vpu-mediated depletion. PLoS Pathog. 2009, 5, e1000443. [Google Scholar] [CrossRef] [PubMed]
- McNatt, M.W.; Zang, T.; Hatziioannou, T.; Bartlett, M.; Fofana, I.B.; Johnson, W.E.; Neil, S.J.; Bieniasz, P.D. Species-specific activity of HIV-1 Vpu and positive selection of tetherin transmembrane domain variants. PLoS Pathog. 2009, 5, e1000300. [Google Scholar] [CrossRef] [PubMed]
- Sauter, D.; Specht, A.; Kirchhoff, F. Tetherin: Holding on and letting go. Cell 2010, 141, 392–398. [Google Scholar] [CrossRef] [PubMed]
- Heusinger, E.; Kluge, S.F.; Kirchhoff, F.; Sauter, D. Early vertebrate evolution of the host restriction factor tetherin. J. Virol. 2015, 89, 12154–12165. [Google Scholar] [CrossRef] [PubMed]
- Strebel, K. HIV accessory proteins versus host restriction factors. Curr. Opin. Virol. 2013, 3, 692–699. [Google Scholar] [CrossRef] [PubMed]
- Dube, M.; Roy, B.B.; Guiot-Guillain, P.; Binette, J.; Mercier, J.; Chiasson, A.; Cohen, E.A. Antagonism of tetherin restriction of HIV-1 release by Vpu involves binding and sequestration of the restriction factor in a perinuclear compartment. PLoS Pathog. 2010, 6, e1000856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goffinet, C.; Allespach, I.; Homann, S.; Tervo, H.M.; Habermann, A.; Rupp, D.; Oberbremer, L.; Kern, C.; Tibroni, N.; Welsch, S.; et al. HIV-1 antagonism of CD317 is species specific and involves Vpu-mediated proteasomal degradation of the restriction factor. Cell Host Microbe 2009, 5, 285–297. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Ode, H.; Yoshida, T.; Sato, K.; Gee, P.; Yamamoto, S.P.; Ebina, H.; Strebel, K.; Sato, H.; Koyanagi, Y. Identification of amino acids in the human tetherin transmembrane domain responsible for HIV-1 Vpu interaction and susceptibility. J. Virol. 2011, 85, 932–945. [Google Scholar] [CrossRef] [PubMed]
- Pang, X.; Hu, S.; Li, J.; Xu, F.; Mei, S.; Zhou, J.; Cen, S.; Jin, Q.; Guo, F. Identification of novel key amino acids at the interface of the transmembrane domains of human BST-2 and HIV-1 Vpu. Retrovirology 2013, 10, 84. [Google Scholar] [CrossRef] [PubMed]
- Vigan, R.; Neil, S.J. Determinants of tetherin antagonism in the transmembrane domain of the human immunodeficiency virus type 1 Vpu protein. J. Virol. 2010, 84, 12958–12970. [Google Scholar] [CrossRef] [PubMed]
- McNatt, M.W.; Zang, T.; Bieniasz, P.D. Vpu binds directly to tetherin and displaces it from nascent virions. PLoS Pathog. 2013, 9, e1003299. [Google Scholar] [CrossRef] [PubMed]
- Douglas, J.L.; Viswanathan, K.; McCarroll, M.N.; Gustin, J.K.; Fruh, K.; Moses, A.V. Vpu directs the degradation of the human immunodeficiency virus restriction factor BST-2/tetherin via a βTrCP-dependent mechanism. J. Virol. 2009, 83, 7931–7947. [Google Scholar] [CrossRef] [PubMed]
- Arias, J.F.; Iwabu, Y.; Tokunaga, K. Structural basis for the antiviral activity of BST-2/tetherin and its viral antagonism. Front. Microbiol. 2011, 2, 250. [Google Scholar] [CrossRef] [PubMed]
- Hauser, H.; Lopez, L.A.; Yang, S.J.; Oldenburg, J.E.; Exline, C.M.; Guatelli, J.C.; Cannon, P.M. HIV-1 Vpu and HIV-2 env counteract BST-2/tetherin by sequestration in a perinuclear compartment. Retrovirology 2010, 7, 51. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Wilson, S.J.; Landford, W.C.; Virgen, B.; Gregory, D.; Johnson, M.C.; Munch, J.; Kirchhoff, F.; Bieniasz, P.D.; Hatziioannou, T. Nef proteins from simian immunodeficiency viruses are tetherin antagonists. Cell Host Microbe 2009, 6, 54–67. [Google Scholar] [CrossRef] [PubMed]
- Serra-Moreno, R.; Zimmermann, K.; Stern, L.J.; Evans, D.T. Tetherin/BST-2 antagonism by Nef depends on a direct physical interaction between Nef and tetherin, and on clathrin-mediated endocytosis. PLoS Pathog. 2013, 9, e1003487. [Google Scholar] [CrossRef] [PubMed]
- Jia, B.; Serra-Moreno, R.; Neidermyer, W.; Rahmberg, A.; Mackey, J.; Fofana, I.B.; Johnson, W.E.; Westmoreland, S.; Evans, D.T. Species-specific activity of SIV Nef and HIV-1 Vpu in overcoming restriction by tetherin/BST2. PLoS Pathog. 2009, 5, e1000429. [Google Scholar] [CrossRef] [PubMed]
- Le Tortorec, A.; Neil, S.J. Antagonism to and intracellular sequestration of human tetherin by the human immunodeficiency virus type 2 envelope glycoprotein. J. Virol. 2009, 83, 11966–11978. [Google Scholar] [CrossRef] [PubMed]
- Bakouche, N.; Vandenbroucke, A.T.; Goubau, P.; Ruelle, J. Study of the HIV-2 env cytoplasmic tail variability and its impact on Tat, Rev and Nef. PLoS ONE 2013, 8, e79129. [Google Scholar] [CrossRef] [PubMed]
- Exline, C.M.; Yang, S.J.; Haworth, K.G.; Rengarajan, S.; Lopez, L.A.; Droniou, M.E.; Seclen, E.; Cannon, P.M. Determinants in HIV-2 Env and tetherin required for functional interaction. Retrovirology 2015, 12, 67. [Google Scholar] [CrossRef] [PubMed]
- Bour, S.; Akari, H.; Miyagi, E.; Strebel, K. Naturally occurring amino acid substitutions in the HIV-2 ROD envelope glycoprotein regulate its ability to augment viral particle release. Virology 2003, 309, 85–98. [Google Scholar] [CrossRef]
- Bour, S.; Aberham, C.; Perrin, C.; Strebel, K. Lack of effect of cytoplasmic tail truncations on human immunodeficiency virus type 2 ROD Env particle release activity. J. Virol. 1999, 73, 778–782. [Google Scholar] [PubMed]
- Ribeiro, A.C.; Maia e Silva, A.; Santa-Marta, M.; Pombo, A.; Moniz-Pereira, J.; Goncalves, J.; Barahona, I. Functional analysis of Vif protein shows less restriction of human immunodeficiency virus type 2 by APOBEC3G. J. Virol. 2005, 79, 823–833. [Google Scholar] [CrossRef] [PubMed]
- Bour, S.; Schubert, U.; Peden, K.; Strebel, K. The envelope glycoprotein of human immunodeficiency virus type 2 enhances viral particle release: A Vpu-like factor? J. Virol. 1996, 70, 820–829. [Google Scholar] [PubMed]
- Serra-Moreno, R.; Jia, B.; Breed, M.; Alvarez, X.; Evans, D.T. Compensatory changes in the cytoplasmic tail of gp41 confer resistance to tetherin/BST-2 in a pathogenic Nef-deleted SIV. Cell Host Microbe 2011, 9, 46–57. [Google Scholar] [CrossRef] [PubMed]
- Serra-Moreno, R. The end of nef’s tether. Trends Microbiol. 2014, 22, 662–664. [Google Scholar] [CrossRef] [PubMed]
- Postler, T.S.; Desrosiers, R.C. The tale of the long tail: The cytoplasmic domain of HIV-1 gp41. J. Virol. 2013, 87, 2–15. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, K.; Chen, C.Y.; Whitted, S.; Chertova, E.; Roser, D.J.; Wu, F.; Plishka, R.J.; Ourmanov, I.; Buckler-White, A.; Lifson, J.D.; et al. Enhanced antagonism of BST-2 by a neurovirulent SIV envelope. J. Clin. Investig. 2016, 126, 2295–2307. [Google Scholar] [CrossRef] [PubMed]
- Lam, A.J.; St-Pierre, F.; Gong, Y.; Marshall, J.D.; Cranfill, P.J.; Baird, M.A.; McKeown, M.R.; Wiedenmann, J.; Davidson, M.W.; Schnitzer, M.J.; et al. Improving FRET dynamic range with bright green and red fluorescent proteins. Nat. Methods 2012, 9, 1005–1012. [Google Scholar] [CrossRef] [PubMed]
- Heckl, D.; Kowalczyk, M.S.; Yudovich, D.; Belizaire, R.; Puram, R.V.; McConkey, M.E.; Thielke, A.; Aster, J.C.; Regev, A.; Ebert, B.L. Generation of mouse models of myeloid malignancy with combinatorial genetic lesions using CRISPR-Cas9 genome editing. Nat. Biotechnol. 2014, 32, 941–946. [Google Scholar] [CrossRef] [PubMed]
- Ruelle, J.; Yfantis, V.; Duquenne, A.; Goubau, P. Validation of an ultrasensitive digital droplet pcr assay for hiv-2 plasma rna quantification. J. Int. AIDS Soc. 2014, 17, 19675. [Google Scholar] [CrossRef] [PubMed]
- Ruelle, J.; Mukadi, B.K.; Schutten, M.; Goubau, P. Quantitative real-time PCR on Lightcycler for the detection of human immunodeficiency virus type 2 (HIV-2). J. Virol. Methods. 2004, 117, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Santos da Silva, E.; Mulinge, M.; Perez Bercoff, D. The frantic play of the concealed HIV envelope cytoplasmic tail. Retrovirology 2013, 10, 54. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, D.; Jenei, A.; Nagy, P.; Vereb, G.; Szollosi, J. Understanding FRET as a research tool for cellular studies. Int. J. Mol. Sci. 2015, 16, 6718–6756. [Google Scholar] [CrossRef] [PubMed]
- Banning, C.; Votteler, J.; Hoffmann, D.; Koppensteiner, H.; Warmer, M.; Reimer, R.; Kirchhoff, F.; Schubert, U.; Hauber, J.; Schindler, M. A flow cytometry-based FRET assay to identify and analyse protein-protein interactions in living cells. PLoS ONE 2010, 5, e9344. [Google Scholar] [CrossRef] [PubMed]
- Abada, P.; Noble, B.; Cannon, P.M. Functional domains within the human immunodeficiency virus type 2 envelope protein required to enhance virus production. J. Virol. 2005, 79, 3627–3638. [Google Scholar] [CrossRef] [PubMed]
- Behrendt, R.; Fiebig, U.; Kurth, R.; Denner, J. Induction of antibodies binding to the membrane proximal external region of gp36 of HIV-2. Intervirology 2012, 55, 252–256. [Google Scholar] [CrossRef] [PubMed]
- Denner, J. Immunising with the transmembrane envelope proteins of different retroviruses including HIV-1. Hum. Vaccines Immunother. 2014, 9, 462–470. [Google Scholar] [CrossRef]
- Munoz-Barroso, I.; Salzwedel, K.; Hunter, E.; Blumenthal, R. Role of the membrane-proximal domain in the initial stages of human immunodeficiency virus type 1 envelope glycoprotein-mediated membrane fusion. J. Virol. 1999, 73, 6089–6092. [Google Scholar] [PubMed]
- Montero, M.; van Houten, N.E.; Wang, X.; Scott, J.K. The membrane-proximal external region of the human immunodeficiency virus type 1 envelope: Dominant site of antibody neutralization and target for vaccine design. Microbiol. Mol. Biol. Rev. 2008, 72, 54–84. [Google Scholar] [CrossRef] [PubMed]
- Salzwedel, K.; West, J.T.; Hunter, E. A conserved tryptophan-rich motif in the membrane-proximal region of the human immunodeficiency virus type 1 gp41 ectodomain is important for Env-mediated fusion and virus infectivity. J. Virol. 1999, 73, 2469–2480. [Google Scholar] [PubMed]







© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dufrasne, F.E.; Lombard, C.; Goubau, P.; Ruelle, J. Single Amino Acid Substitution N659D in HIV-2 Envelope Glycoprotein (Env) Impairs Viral Release and Hampers BST-2 Antagonism. Viruses 2016, 8, 285. https://0-doi-org.brum.beds.ac.uk/10.3390/v8100285
Dufrasne FE, Lombard C, Goubau P, Ruelle J. Single Amino Acid Substitution N659D in HIV-2 Envelope Glycoprotein (Env) Impairs Viral Release and Hampers BST-2 Antagonism. Viruses. 2016; 8(10):285. https://0-doi-org.brum.beds.ac.uk/10.3390/v8100285
Chicago/Turabian StyleDufrasne, François E., Catherine Lombard, Patrick Goubau, and Jean Ruelle. 2016. "Single Amino Acid Substitution N659D in HIV-2 Envelope Glycoprotein (Env) Impairs Viral Release and Hampers BST-2 Antagonism" Viruses 8, no. 10: 285. https://0-doi-org.brum.beds.ac.uk/10.3390/v8100285