
Characterization of Intra-Type Variants of Oncogenic Human Papillomaviruses by Next-Generation Deep Sequencing of the E6/E7 Region

 ,
, 
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Primer Design
2.2. Samples
2.2.1. HPV-Positive Cell Lines
2.2.2. High-Risk HPV-Positive Genital Swabs
2.3. Primer Validation
2.4. Data Processing and Analysis
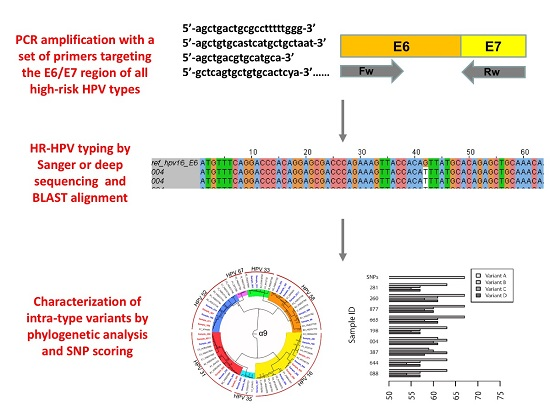
3. Results
3.1. Multiplex Polymerase Chain Reaction (PCR) Primers for Characterization of High-Risk Human Papillomavirus (HR_HPV) Intra-Type Variants
3.2. HPV Typing and Intra-Type Variant Characterization in Single and Multiple Infections
3.2.1. Sanger Sequencing
3.2.2. 454 deep sequencing
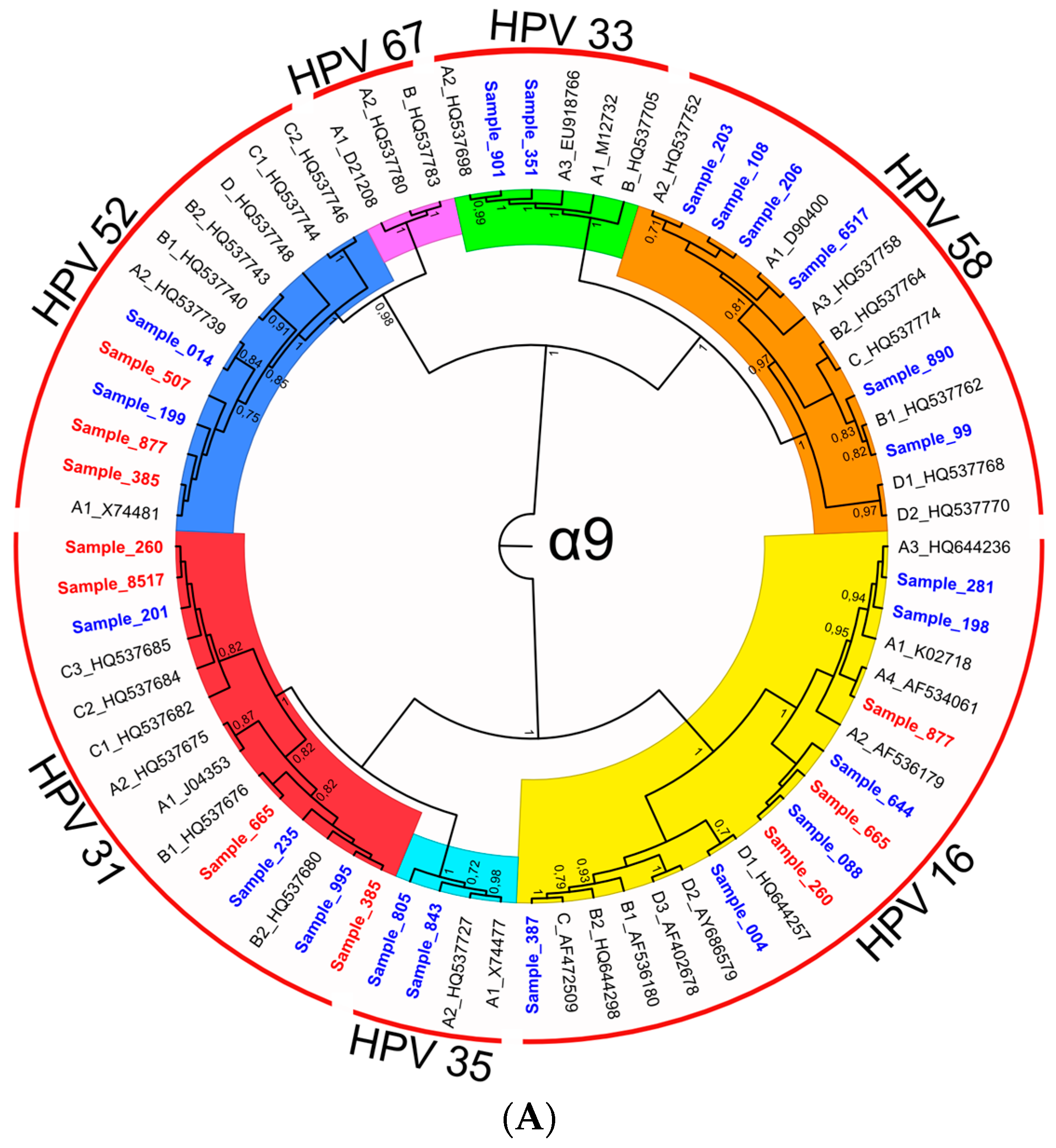
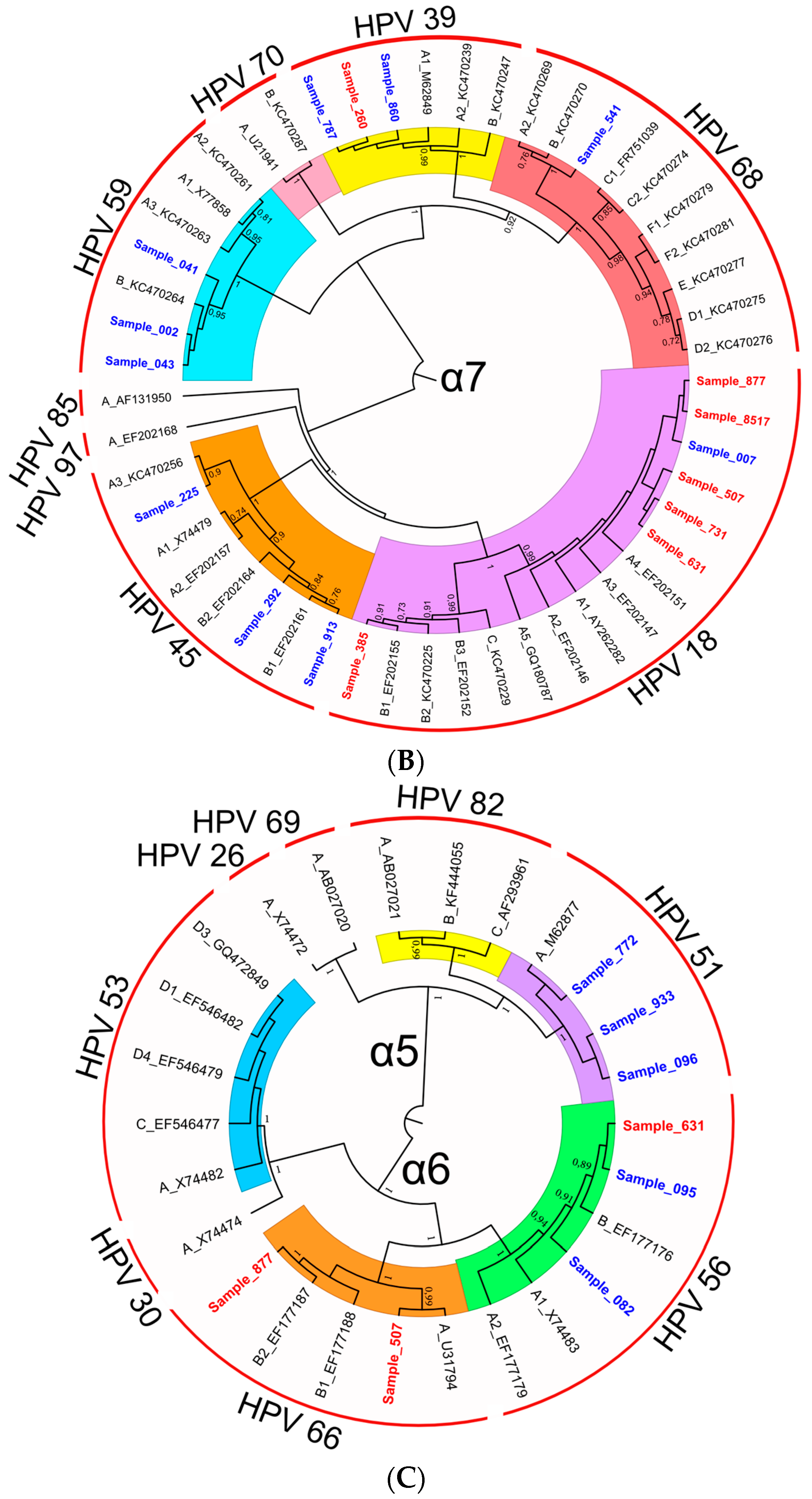
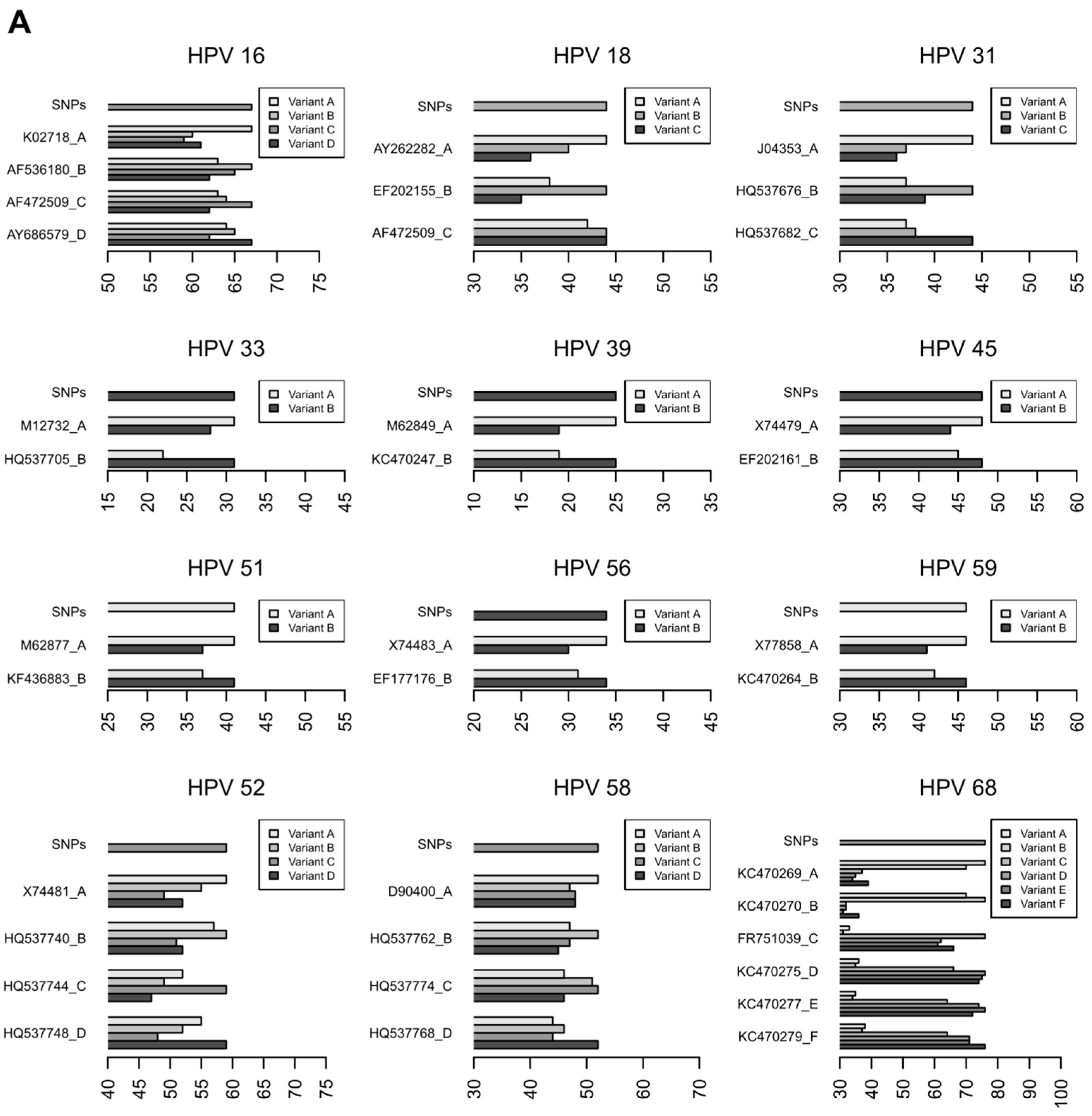
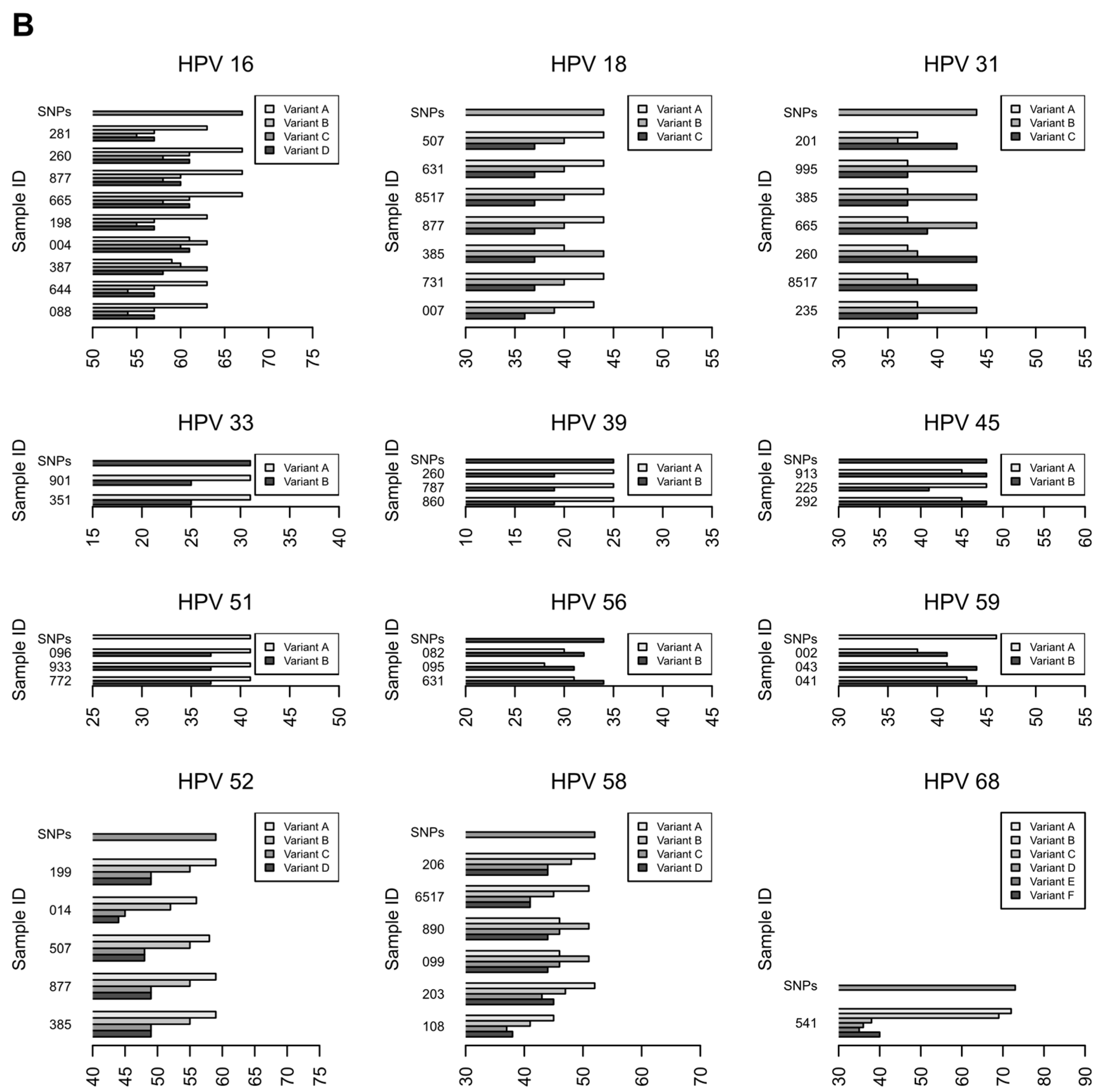
3.2.3. Characterization of HR-HPV intra-type variants
4. Discussion
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bernard, H.U. Taxonomy and phylogeny of papillomaviruses: An overview and recent developments. Infect. Genet. Evol. 2013, 18, 357–361. [Google Scholar] [CrossRef] [PubMed]
- Bzhalava, D.; Eklund, C.; Dillner, J. International standardization and classification of human papillomavirus types. Virology 2015, 476, 341–344. [Google Scholar] [CrossRef] [PubMed]
- Kocjan, B.J.; Bzhalava, D.; Forslund, O.; Dillner, J.; Poljak, M. Molecular methods for identification and characterization of novel papillomaviruses. Clin. Microbiol. Infect. 2015, 21, 808–816. [Google Scholar] [CrossRef] [PubMed]
- Bouvard, V.; Baan, R.; Straif, K.; Grosse, Y.; Secretan, B.; El Ghissassi, F.; Benbrahim-Tallaa, L.; Guha, N.; Freeman, C.; Galichet, L.; et al. A review of human carcinogens-part B: Biological agents. Lancet Oncol. 2009, 10, 321–322. [Google Scholar] [CrossRef]
- Bernard, H.U.; Burk, R.D.; Chen, Z.; van Doorslaer, K.; zur Hausen, H.; de Villiers, E.M. Classification of papillomaviruses (PVs) based on 189 PV types and proposal of taxonomic amendments. Virology 2010, 401, 70–79. [Google Scholar] [CrossRef] [PubMed]
- Burk, R.D.; Harari, A.; Chen, Z. Human papillomavirus genome variants. Virology 2013, 445, 232–243. [Google Scholar] [CrossRef] [PubMed]
- Poljak, M.; Cuzick, J.; Kocjan, B.J.; Iftner, T.; Dillner, J.; Arbyn, M. Nucleic acid tests for the detection of alpha human papillomaviruses. Vaccine 2012, 30, F100–F106. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, O.; Veeraraghavalu, K.; Tergaonkar, V.; Liu, Y.; Androphy, E.J.; Stanley, M.A.; Krishna, S. Human papillomavirus type 16 E6 amino acid 83 variants enhance E6-mediated MAPK signaling and differentially regulate tumorigenesis by notch signaling and oncogenic Ras. J. Virol. 2004, 78, 5934–5945. [Google Scholar] [CrossRef] [PubMed]
- Xi, L.F.; Schiffman, M.; Koutsky, L.A.; Hughes, J.P.; Winer, R.L.; Mao, C.; Hulbert, A.; Lee, S.K.; Shen, Z.P.; Kiviat, N.B. Lineages of oncogenic human papillomavirus types other than type 16 and 18 and risk for cervical intraepithelial neoplasia. JNCI J. Natl. Cancer Inst. 2014, 106. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.A.; Heideman, D.A.; Boon, D.; Gheit, T.; Snijders, P.J.; Tommasino, M.; Franceschi, S.; Clifford, G.M. Human papillomavirus 45 genetic variation and cervical cancer risk worldwide. J. Virol. 2014, 88, 4514–4521. [Google Scholar] [CrossRef] [PubMed]
- Schiffman, M.; Rodriguez, A.C.; Chen, Z.; Wacholder, S.; Herrero, R.; Hildesheim, A.; Desalle, R.; Befano, B.; Yu, K.; Safaeian, M.; et al. A population-based prospective study of carcinogenic human papillomavirus variant lineages, viral persistence, and cervical neoplasia. Cancer Res. 2010, 70, 3159–3169. [Google Scholar] [CrossRef] [PubMed]
- Villa, L.L.; Sichero, L.; Rahal, P.; Caballero, O.; Ferenczy, A.; Rohan, T.; Franco, E.L. Molecular variants of human papillomavirus types 16 and 18 preferentially associated with cervical neoplasia. J. Gen. Virol. 2000, 81, 2959–2968. [Google Scholar] [CrossRef] [PubMed]
- Berumen, J.; Ordonez, R.M.; Lazcano, E.; Salmeron, J.; Galvan, S.C.; Estrada, R.A.; Yunes, E.; Garcia-Carranca, A.; Gonzalez-Lira, G.; Madrigal-de la Campa, A. Asian-american variants of human papillomavirus 16 and risk for cervical cancer: A case-control study. JNCI J. Natl. Cancer Inst. 2001, 93, 1325–1330. [Google Scholar] [CrossRef] [PubMed]
- Cornet, I.; Gheit, T.; Iannacone, M.R.; Vignat, J.; Sylla, B.S.; Del Mistro, A.; Franceschi, S.; Tommasino, M.; Clifford, G.M. HPV16 genetic variation and the development of cervical cancer worldwide. Br. J. Cancer 2013, 108, 240–244. [Google Scholar] [CrossRef] [PubMed]
- Chan, P.K.; Zhang, C.; Park, J.S.; Smith-McCune, K.K.; Palefsky, J.M.; Giovannelli, L.; Coutlee, F.; Hibbitts, S.; Konno, R.; Settheetham-Ishida, W.; et al. Geographical distribution and oncogenic risk association of human papillomavirus type 58 E6 and E7 sequence variations. Int. J. Cancer 2013, 132, 2528–2536. [Google Scholar] [CrossRef] [PubMed]
- De Boer, M.A.; Peters, L.A.W.; Aziz, M.F.; Siregar, B.; Cornain, S.; Vrede, M.A.; Jordanova, E.S.; Fleuren, G.J. Human papillomavirus type 18 variants: Histopathology and E6/E7 polymorphisms in three countries. Int. J. Cancer 2005, 114, 422–425. [Google Scholar] [CrossRef] [PubMed]
- Chan, P.K.; Luk, A.C.; Park, J.S.; Smith-McCune, K.K.; Palefsky, J.M.; Konno, R.; Giovannelli, L.; Coutlee, F.; Hibbitts, S.; Chu, T.Y.; et al. Identification of human papillomavirus type 58 lineages and the distribution worldwide. J. Infect. Dis. 2011, 203, 1565–1573. [Google Scholar] [CrossRef] [PubMed]
- Barzon, L.; Militello, V.; Pagni, S.; Franchin, E.; Dal Bello, F.; Mengoli, C.; Palu, G. Distribution of human papillomavirus types in the anogenital tract of females and males. J. Med. Virol. 2010, 82, 1424–1430. [Google Scholar] [CrossRef] [PubMed]
- Barzon, L.; Militello, V.; Pagni, S.; Palu, G. Comparison of INNO-LiPA genotyping extra and hybrid capture 2 assays for detection of carcinogenic human papillomavirus genotypes. J. Clin. Virol. 2012, 55, 256–261. [Google Scholar] [CrossRef] [PubMed]
- Wood, H.M.; Bolt, R.; Hunter, K.D. Is next-generation sequencing an important tool in HPV subtype diagnosis? Expert Rev. Mol. Diagn. 2012, 12, 663–665. [Google Scholar] [CrossRef] [PubMed]
- Ekstrom, J.; Bzhalava, D.; Svenback, D.; Forslund, O.; Dillner, J. High throughput sequencing reveals diversity of human papillomaviruses in cutaneous lesions. Int. J. Cancer 2011, 129, 2643–2650. [Google Scholar] [CrossRef] [PubMed]
- Barzon, L.; Militello, V.; Lavezzo, E.; Franchin, E.; Peta, E.; Squarzon, L.; Trevisan, M.; Pagni, S.; Dal Bello, F.; Toppo, S.; et al. Human papillomavirus genotyping by 454 next generation sequencing technology. J. Clin. Virol. 2011, 52, 93–97. [Google Scholar] [CrossRef] [PubMed]
- Militello, V.; Lavezzo, E.; Costanzi, G.; Franchin, E.; Di Camillo, B.; Toppo, S.; Palu, G.; Barzon, L. Accurate human papillomavirus genotyping by 454 pyrosequencing. Clin. Microbiol. Infect. 2013, 19, E428–E434. [Google Scholar] [CrossRef] [PubMed]
- Arroyo, L.S.; Smelov, V.; Bzhalava, D.; Eklund, C.; Hultin, E.; Dillner, J. Next generation sequencing for human papillomavirus genotyping. J. Clin. Virol. 2013, 58, 437–442. [Google Scholar] [CrossRef] [PubMed]
- Kukimoto, I.; Maehama, T.; Sekizuka, T.; Ogasawara, Y.; Kondo, K.; Kusumoto-Matsuo, R.; Mori, S.; Ishii, Y.; Takeuchi, T.; Yamaji, T.; et al. Genetic variation of human papillomavirus type 16 in individual clinical specimens revealed by deep sequencing. PLoS ONE 2013, 8, e80583. [Google Scholar] [CrossRef] [PubMed]
- Conway, C.; Chalkley, R.; High, A.; Maclennan, K.; Berri, S.; Chengot, P.; Alsop, M.; Egan, P.; Morgan, J.; Taylor, G.R.; et al. Next-generation sequencing for simultaneous determination of human papillomavirus load, subtype, and associated genomic copy number changes in tumors. J. Mol. Diagn. 2012, 14, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Meiring, T.L.; Salimo, A.T.; Coetzee, B.; Maree, H.J.; Moodley, J.; Hitzeroth, I.I.; Freeborough, M.J.; Rybicki, E.P.; Williamson, A.L. Next-generation sequencing of cervical DNA detects human papillomavirus types not detected by commercial kits. Virol. J. 2012, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garnaes, E.; Kiss, K.; Andersen, L.; Therkildsen, M.H.; Franzmann, M.B.; Filtenborg-Barnkob, B.; Hoegdall, E.; Krenk, L.; Josiassen, M.; Lajer, C.B.; et al. A high and increasing HPV prevalence in tonsillar cancers in Eastern Denmark, 2000–2010: The largest registry-based study to date. Int. J. Cancer 2015, 136, 2196–2203. [Google Scholar] [CrossRef] [PubMed]
- Yi, X.; Zou, J.; Xu, J.; Liu, T.; Hua, S.; Xi, F.; Nie, X.; Ye, L.; Luo, Y.; Xu, L.; et al. Development and validation of a new HPV genotyping assay based on next-generation sequencing. Am. J. Clin. Pathol. 2014, 141, 796–804. [Google Scholar] [CrossRef] [PubMed]
- Bzhalava, D.; Muhr, L.S.; Lagheden, C.; Ekstrom, J.; Forslund, O.; Dillner, J.; Hultin, E. Deep sequencing extends the diversity of human papillomaviruses in human skin. Sci. Rep. 2014, 4. [Google Scholar] [CrossRef] [PubMed]
- Johansson, H.; Bzhalava, D.; Ekstrom, J.; Hultin, E.; Dillner, J.; Forslund, O. Metagenomic sequencing of “HPV-negative” condylomas detects novel putative HPV types. Virology 2013, 440, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Tang, K.W.; Alaei-Mahabadi, B.; Samuelsson, T.; Lindh, M.; Larsson, E. The landscape of viral expression and host gene fusion and adaptation in human cancer. Nat. Commun. 2013, 4. [Google Scholar] [CrossRef] [PubMed]
- Chandrani, P.; Kulkarni, V.; Iyer, P.; Upadhyay, P.; Chaubal, R.; Das, P.; Mulherkar, R.; Singh, R.; Dutt, A. NGS-based approach to determine the presence of HPV and their sites of integration in human cancer genome. Br. J. Cancer 2015, 112, 1958–1965. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.; Clamp, M.; Barton, G.J. Jalview Version 2—A multiple sequence alignment editor and analysis workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef] [PubMed]
- Linhart, C.; Shamir, R. The degenerate primer design problem. Bioinformatics 2002, 18 (Suppl. 1), S172–S181. [Google Scholar] [CrossRef] [PubMed]
- WHO. Human Papillomavirus Laboratory Manual, 1st ed.; Available online: http://www.who.int/immunization/hpv/learn/hpv_laboratory_manual__who_ivb_2009_2010.pdf (accessed on 8 March 2016).
- Papillomavirus Episteme. Available online: http://pave.niaid.nih.gov/ (accessed on 31 October 2015).
- Altschul, S.F.; Madden, T.L.; Schaffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. Mega6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed]
- Schlecht, N.F.; Burk, R.D.; Palefsky, J.M.; Minkoff, H.; Xue, X.A.; Massad, L.S.; Bacon, M.; Levine, A.M.; Anastos, K.; Gange, S.J.; et al. Variants of human papillomaviruses 16 and 18 and their natural history in human immunodeficiency virus-positive women. J. Gen. Virol. 2005, 86, 2709–2720. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Yang, H.; Wu, K.; Shi, X.; Ma, S.; Sun, Q. Prevalence of HPV and variation of HPV 16/HPV 18 E6/E7 genes in cervical cancer in women in South West China. J. Med. Virol. 2014, 86, 1926–1936. [Google Scholar] [CrossRef] [PubMed]
- Perez, S.; Cid, A.; Inarrea, A.; Pato, M.; Lamas, M.J.; Couso, B.; Gil, M.; Alvarez, M.J.; Rey, S.; Lopez-Miragaya, I.; et al. Prevalence of HPV 16 and HPV 18 lineages in Galicia, Spain. PLoS ONE 2014, 9, e104678. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Schiffman, M.; Herrero, R.; DeSalle, R.; Anastos, K.; Segondy, M.; Sahasrabuddhe, V.V.; Gravitt, P.E.; Hsing, A.W.; Burk, R.D. Evolution and taxonomic classification of alphapapillomavirus 7 complete genomes: HPV18, HPV39, HPV45, HPV59, HPV68 and HPV70. PLoS ONE 2013, 8, e72565. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Liao, H.; Yang, B.; Geffre, C.P.; Zhang, A.; Zhou, A.; Cao, H.; Wang, J.; Zhang, Z.; Zheng, W. Variants of human papillomavirus type 16 predispose toward persistent infection. Int. J. Clin. Exp. Pathol. 2015, 8, 8453–8459. [Google Scholar] [PubMed]
- Arroyo, S.L.; Basaras, M.; Arrese, E.; Hernaez, S.; Andia, D.; Esteban, V.; Garcia-Etxebarria, K.; Jugo, B.M.; Cisterna, R. Human papillomavirus (HPV) genotype 18 variants in patients with clinical manifestations of HPV related infections in Bilbao, Spain. Virol. J. 2012, 9. [Google Scholar] [CrossRef] [PubMed]
- Mosmann, J.P.; Monetti, M.S.; Frutos, M.C.; Kiguen, A.X.; Venezuela, R.F.; Cuffini, C.G. Mutation detection of E6 and LCR genes from HPV 16 associated with carcinogenesis. Asian Pac. J. Cancer Prev. 2015, 16, 1151–1157. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.A.; Gheit, T.; Franceschi, S.; Tommasino, M.; Clifford, G.M. Human papillomavirus 18 genetic variation and cervical cancer risk worldwide. J. Virol. 2015, 89, 10680–10687. [Google Scholar] [CrossRef] [PubMed]
- Eklund, C.; Forslund, O.; Wallin, K.L.; Dillner, J. Global improvement in genotyping of human papillomavirus DNA: The 2011 HPV LabNet International Proficiency Study. J. Clin. Microbiol. 2014, 52, 449–459. [Google Scholar] [CrossRef] [PubMed]
- Eklund, C.; Forslund, O.; Wallin, K.L.; Zhou, T.; Dillner, J. The 2010 global proficiency study of human papillomavirus genotyping in vaccinology. J. Clin. Microbiol. 2012, 50, 2289–2298. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K. Estimation of the number of nucleotide substitutions when there are strong transition-transversion and G+C-content biases. Mol. Biol. Evol. 1992, 9, 678–687. [Google Scholar] [PubMed]
- Hasegawa, M.; Kishino, H.; Yano, T. Dating of the human-ape splitting by a molecular clock of mitochondrial DNA. J. Mol. Evol. 1985, 22, 160–174. [Google Scholar] [CrossRef] [PubMed]
- Molecular evolution, phylogenetics and epidemiology. Available online at: http://tree.bio.ed.ac.uk/software/figtree/ (accessed on 10 November 2015).
- Geraets, D.T.; van Doorn, L.J.; Kleter, B.; Colau, B.; Harper, D.M.; Quint, W.G. Long-term follow-up of HPV16-positive women: Persistence of the same genetic variant and low prevalence of variant co-infections. PLoS ONE 2013, 8, e80382. [Google Scholar] [CrossRef] [PubMed]
- Meza-Menchaca, T.; Williams, J.; Rodriguez-Estrada, R.B.; Garcia-Bravo, A.; Ramos-Ligonio, A.; Lopez-Monteon, A.; Zepeda, R.C. A low density microarray method for the identification of human papillomavirus type 18 variants. Sensors 2013, 13, 12975–12993. [Google Scholar] [CrossRef] [PubMed]
- Larsson, G.L.; Helenius, G.; Andersson, S.; Elgh, F.; Sorbe, B.; Karlsson, M.G. Human papillomavirus (HPV) and HPV 16-variant distribution in vulvar squamous cell carcinoma in Sweden. Int. J. Gynecol. Cancer 2012, 22, 1413–1419. [Google Scholar] [CrossRef] [PubMed]
- Xi, L.F.; Schiffman, M.; Koutsky, L.A.; Hulbert, A.; Lee, S.K.; Defilippis, V.; Shen, Z.; Kiviat, N.B. Association of human papillomavirus type 31 variants with risk of cervical intraepithelial neoplasia grades 2–3. Int. J. Cancer 2012, 131, 2300–2307. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, G.I.; Kleter, B.; Gheit, T.; van Doorn, L.J.; de Koning, M.N.; de Sanjose, S.; Alemany, L.; Bosch, X.F.; Tommasino, M.; Munoz, N.; et al. Clinical evaluation of polymerase chain reaction reverse hybridization assay for detection and identification of human papillomavirus type 16 variants. J. Clin. Virol. 2011, 51, 165–169. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Schiffman, M.; Herrero, R.; Desalle, R.; Anastos, K.; Segondy, M.; Sahasrabuddhe, V.V.; Gravitt, P.E.; Hsing, A.W.; Burk, R.D. Evolution and taxonomic classification of human papillomavirus 16 (HPV16)-related variant genomes: HPV31, HPV33, HPV35, HPV52, HPV58 and HPV67. PLoS ONE 2011, 6, e20183. [Google Scholar] [CrossRef] [PubMed]
- Poljak, M.; Kocjan, B.J.; Ostrbenk, A.; Seme, K. Commercially available molecular tests for human papillomaviruses (HPV): 2015 update. J. Clin. Virol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Ndiaye, C.; Mena, M.; Alemany, L.; Arbyn, M.; Castellsague, X.; Laporte, L.; Bosch, F.X.; de Sanjose, S.; Trottier, H. HPV DNA, E6/E7 mRNA, and p16INK4a detection in head and neck cancers: A systematic review and meta-analysis. Lancet Oncol. 2014, 15, 1319–1331. [Google Scholar] [CrossRef]
- Barzon, L.; Cappellesso, R.; Peta, E.; Militello, V.; Sinigaglia, A.; Fassan, M.; Simonato, F.; Guzzardo, V.; Ventura, L.; Blandamura, S.; et al. Profiling of expression of human papillomavirus-related cancer miRNAs in penile squamous cell carcinomas. Am. J. Pathol. 2014, 184, 3376–3383. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HPV Species | HPV Type | IARC Group | No. of Sequences in Nucleotide Database Covering the HPV E6/E7 Region |
|---|---|---|---|
| α 5 | 51 | 1 | 6 |
| α 6 | 56 | 1 | 126 |
| α 7 | 18 | 1 | 130 |
| 39 | 1 | 25 | |
| 45 | 1 | 104 | |
| 59 | 1 | 13 | |
| 68 | 2A | 92 | |
| α 9 | 16 | 1 | 1897 |
| 31 | 1 | 276 | |
| 33 | 1 | 82 | |
| 35 | 1 | 98 | |
| 52 | 1 | 363 | |
| 58 | 1 | 795 |
| HPV Species | Primer Type | Primer Sequence * | HPV Type and Variant § | Amplicon Length (HPV Type) |
|---|---|---|---|---|
| α 5 | F | AACCGAAAAGGGTTATGACCGA | 51 (all known variants) | 815 |
| R | TCTGCTGTACAACGCGAAGG | |||
| α 6 | F | AGGCAGCTTATTCTGTGTGGA | 56 (all known variants) | 873 |
| R | CAGAGTGGGCACGTTACTGT | |||
| α 7 | F | AGGGAGTRACCRAAAACGGT | 18, 39 (all known variants) | 814 (HPV18) 807 (HPV39) |
| R | GGAATGCTCGAAGGTCGTCT | 18 (all known variants) | ||
| R | CCCGTGAGGCTTCTACTACC | 39 (all known variants) | ||
| F | TGCAACCAAAAACGGTGCAT | 45 (all known variants) | 849 | |
| R | TTAGTTGCACACCACGGACA | |||
| F | GCATGGCACGCTTTGAGG | 59 (all known variants) | 806 | |
| R | GTTTGCTGCACACAAAGGACA | |||
| F | GGTCACGACCGAAAACGG | 68 (A2, B, D1, D2, E, F1, F2) | 815 | |
| R | AGCAGYTSYAGCTTCCGCA | 68 (C1, C2, D1, D2, E, F1, F2) | ||
| F | GKGACCGRAARCGGTCAT | 68 (C1, C2) | 830 | |
| R | AACAGCTGYTSTAGTGTCCG | 68 (A2, B) | ||
| α 9 | F | AGGGYGTAACCGAAAACGGT | 52, 58 (all known variants) | 801 (HPV52) 828 (HPV58) |
| R | CCGGGGCACACAACTTGTAA | 52 (all known variants) | ||
| R | ACAGCTAGGGCACACAATGG | 58 (A1, A2, A3, B1, B2, D1) | ||
| R | GCTGTAGGGTTCGTSCTTCA | 58 (C, D2) | 785 | |
| F | AGGGCGTAACCGAAATCGGT | 16 (A1, A2, A3, A4, D1, D2, D3) | 830 | |
| R | TGAGAACAGATGGGGCACAC | 16 (A1, A2, A3, B1, B2, C, D1, D2, D3) | ||
| F | TTGMACCGAAACCGGTTAGT | 16 (B1, B2, C) | 807 | |
| R | RCAGATGGGGCACACAATTC | 16 (A4) | ||
| F | GGTGAACCGAAAACGGTTGG | 31 (all known variants) | 793 | |
| R | GGGGCACACGATTCCAAATG | |||
| F | AAGTAGGGTGTAACCGAAAGCG | 33 (all known variants) | 787 | |
| R | TGCTGTATGGTTCGTAGGTCAC | |||
| F | ACGGTTGCCATAAAAGCAGAA | 35 (all known variants) | 827 | |
| R | TCTCTGTGAACAGCCGGGG |
| Sample ID | LiPA | Sanger Sequencing | |
|---|---|---|---|
| HPV Type | HPV Type | Scores (Fw, Rw) * | |
| 004 | HPV16 | HPV16 | 49, 38 |
| 088 | HPV16 | HPV16 | 48, 42 |
| 198 | HPV16 | HPV16 | 45, 42 |
| 281 | HPV16, HPV53 | HPV16 | 46, 45 |
| 387 | HPV16 | HPV16 | 47, 47 |
| 644 | HPV16 | HPV16 | 47, 46 |
| 007 | HPV18 (HPV39) | HPV18 | 36, 15 |
| 201 | HPV31 | HPV31 | 30, 15 |
| 235 | HPV31 (HPV52, HPV54) | HPV31 | 30, 36 |
| 995 | HPV31 (HPV52, HPV54) | HPV31 | 50, 47 |
| 351 | HPV33, HPV11 (HPV52, HPV54) | HPV33 | 31, 46 |
| 901 | HPV33 (HPV52, HPV54) | HPV33 | 29, 45 |
| 805 | HPV35, HPV44 | HPV35 | 46, 45 |
| 843 | HPV35, HPV54, HPV69/71, HPVX | HPV35 | 46, 48 |
| 787 | HPV39, HPV66 | HPV39 | 46, 48 |
| 860 | HPV39 | HPV39 | 49, 49 |
| 225 | HPV45, HPV74 | HPV45 | 45, 31 |
| 292 | HPV45 | HPV45 | 47, 43 |
| 913 | HPV45 | HPV45 | 46, 38 |
| 096 | HPV51 | HPV51 | 37, 43 |
| 772 | HPV51 | HPV51 | 39, 40 |
| 933 | HPV51 | HPV51 | 42, 42 |
| 014 | HPV52 | HPV52 | 37, 41 |
| 199 | HPV52, HPVX | HPV52 | 33, 48 |
| 082 | HPV56 | HPV56 | 15, 30 |
| 095 | HPV56 | HPV56 | 34, 23 |
| 099 | HPV58 | HPV58 | 23, 34 |
| 108 | HPV58 | HPV58 | 16, 26 |
| 203 | HPV58, HPV54 | HPV58 | 43, 35 |
| 206 | HPV58 | HPV58 | 47, 35 |
| 890 | HPV58 (HPV52) | HPV58 | 28, 43 |
| 6517 | HPV58, HPV44 (HPV52) | HPV58 | 26, 14 |
| 002 | HPV59 | HPV59 | 28, 26 |
| 041 | HPV59, HPV54 | HPV59 | 49, 45 |
| 043 | HPV59, HPV43 | HPV59 | 39, 39 |
| 541 | HPV68 (HPV39) | HPV68 | 49, 40 |
| Sample ID | LiPA | 454 Deep Sequencing | |||
|---|---|---|---|---|---|
| HPV Type | HPV Type | Total No. Reads (% of the Total) | No. Forward Reads | No. Reverse Reads | |
| 8517 | HPV18, HPV31, (HPV39, HPV52, HPV54) | HPV31 | 50,660 (93.6) | 25,876 | 24,784 |
| HPV18 | 3435 (6.3) | 1738 | 1697 | ||
| 507 | HPV18, HPV52, HPV66, HPV69/71, HPVX, (HPV39) | HPV52 | 39,860 (72.5) | 19,435 | 20,425 |
| HPV18 | 14,913 (27.1) | 7629 | 7284 | ||
| HPV66 | 151 (0.3) | 81 | 70 | ||
| 665 | HPV16, HPV31, HPV6, HPV69/71, (HPV52, HPV54) | HPV31 | 69,627 (99.4) | 36,230 | 33,397 |
| HPV16 | 408 (0.6) | 214 | 194 | ||
| 731 | HPV16, HPV18, (HPV39) | HPV18 | 42,878 (99.9) | 23115 | 19,763 |
| 631 | HPV18, HPV56, (HPV39, HPV74) | HPV18 | 62,182 (97.4) | 33,380 | 28,802 |
| HPV56 | 1564 (2.4) | 845 | 719 | ||
| 385 | HPV18, HPV31, (HPV39, HPV52, HPV54) | HPV31 | 45,947 (83.7) | 26,291 | 19,656 |
| HPV18 | 8915 (16.2) | 5328 | 3587 | ||
| HPV52 | 22 (0.04) | 11 | 11 | ||
| 877 | HPV18, HPV52, HPV66, HPV11, HPVX, (HPV39) | HPV18 | 32,879 (68.6) | 14,681 | 18,198 |
| HPV34 | 13,895 (29) | 6048 | 7847 | ||
| HPV52 | 1012 (2.1) | 421 | 591 | ||
| HPV16 | 137 (0.3) | 82 | 55 | ||
| 260 | HPV16, HPV39, HPV52, HPV6, HPV69/71, HPVX | HPV39 | 59,838 (72.9) | 29,785 | 30,053 |
| HPV34 | 15,042 (18.3) | 7274 | 7768 | ||
| HPV16 | 6145 (7.5) | 2921 | 3224 | ||
| HPV31 | 942 (1.1) | 423 | 519 | ||
| HPV Type | Bootstrap Value | HPV Intra-Type Variant | Sample IDs | |
|---|---|---|---|---|
| Phylogenetic Analysis | SNP Score | |||
| 16 | 1.00 | A | A | 281, 260, 877, 665, 198, 644, 088 |
| 16 | 1.00 | C | C | 387 |
| 16 | 0.70 | D | A, B, C, D * | 004 |
| 18 | 0.99 | A | A | 507, 631, 665, 8517, 877, 731, 007 |
| 18 | 0.91 | B | B | 385 |
| 31 | 1.00 | B | B | 995, 385, 665, 235 |
| 31 | 1.00 | C | C | 201, 631, 260, 8517 |
| 33 | 1.00 | A | A | 901, 351 |
| 35 | 1.00 | A | -** | 805, 843 |
| 39 | 0.99 | A | A | 260, 787, 860 |
| 45 | 0.90 | A | A | 225 |
| 45 | 0.84 | B | B | 913, 292 |
| 51 | 1.00 | A | A | 096, 933, 772 |
| 52 | 0.75 | A | A | 199, 014, 507, 877, 385 |
| 56 | 0.91 | B | B | 082, 095, 631 |
| 58 | 0.78 | A | A | 206, 6517, 203, 108 |
| 58 | 0.96 | B | B | 890, 099 |
| 59 | 0.95 | B | B | 002, 043, 041 |
| 68 | 1.00 | A, B * | A, B * | 541 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lavezzo, E.; Masi, G.; Toppo, S.; Franchin, E.; Gazzola, V.; Sinigaglia, A.; Masiero, S.; Trevisan, M.; Pagni, S.; Palù, G.; et al. Characterization of Intra-Type Variants of Oncogenic Human Papillomaviruses by Next-Generation Deep Sequencing of the E6/E7 Region. Viruses 2016, 8, 79. https://0-doi-org.brum.beds.ac.uk/10.3390/v8030079
Lavezzo E, Masi G, Toppo S, Franchin E, Gazzola V, Sinigaglia A, Masiero S, Trevisan M, Pagni S, Palù G, et al. Characterization of Intra-Type Variants of Oncogenic Human Papillomaviruses by Next-Generation Deep Sequencing of the E6/E7 Region. Viruses. 2016; 8(3):79. https://0-doi-org.brum.beds.ac.uk/10.3390/v8030079
Chicago/Turabian StyleLavezzo, Enrico, Giulia Masi, Stefano Toppo, Elisa Franchin, Valentina Gazzola, Alessandro Sinigaglia, Serena Masiero, Marta Trevisan, Silvana Pagni, Giorgio Palù, and et al. 2016. "Characterization of Intra-Type Variants of Oncogenic Human Papillomaviruses by Next-Generation Deep Sequencing of the E6/E7 Region" Viruses 8, no. 3: 79. https://0-doi-org.brum.beds.ac.uk/10.3390/v8030079