
Subcellular Trafficking and Functional Relationship of the HSV-1 Glycoproteins N and M

Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells and Viruses
2.2. Yeast 2-Hybrid System
2.3. Plasmids
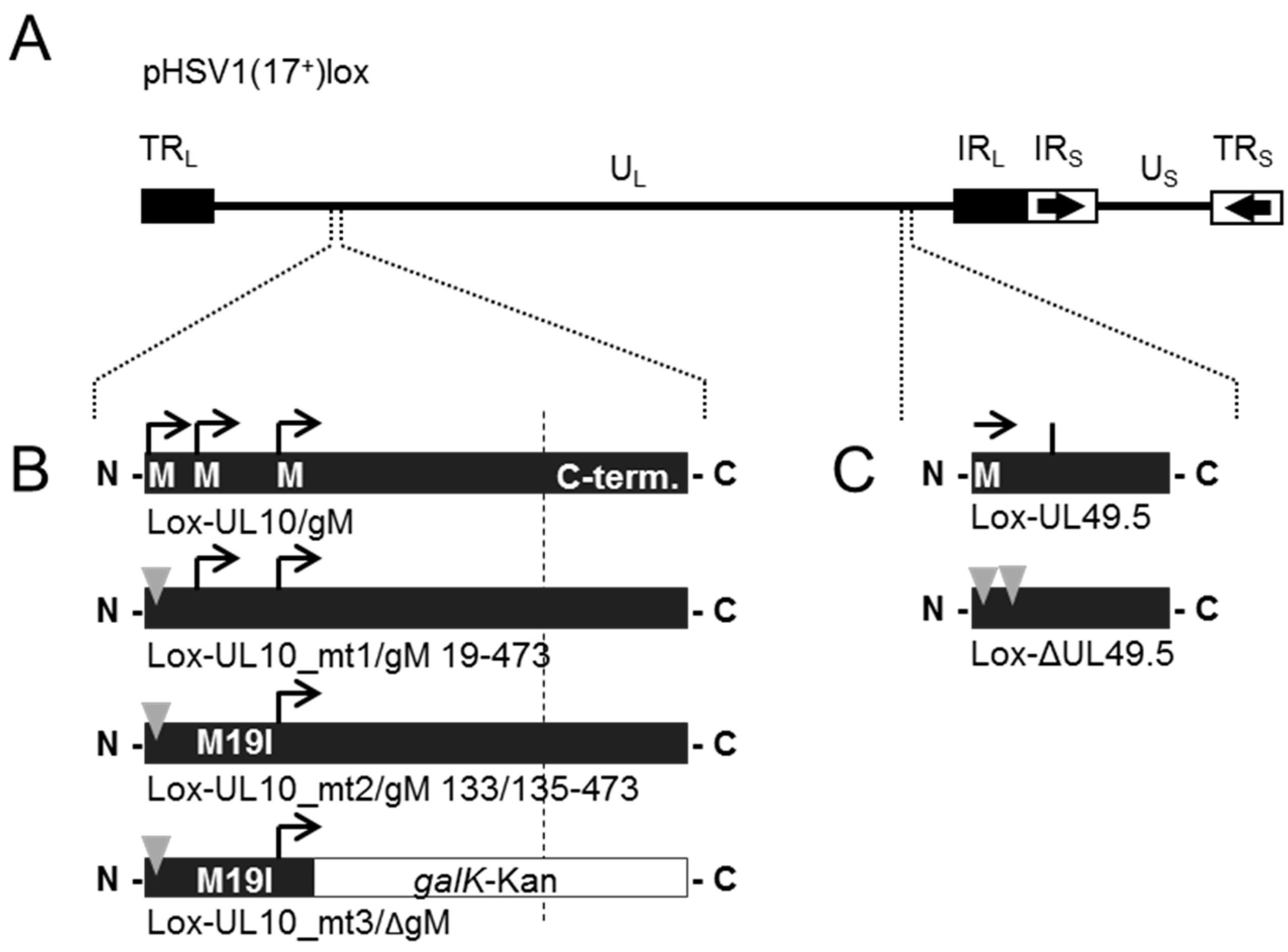
2.4. BAC Mutagenesis
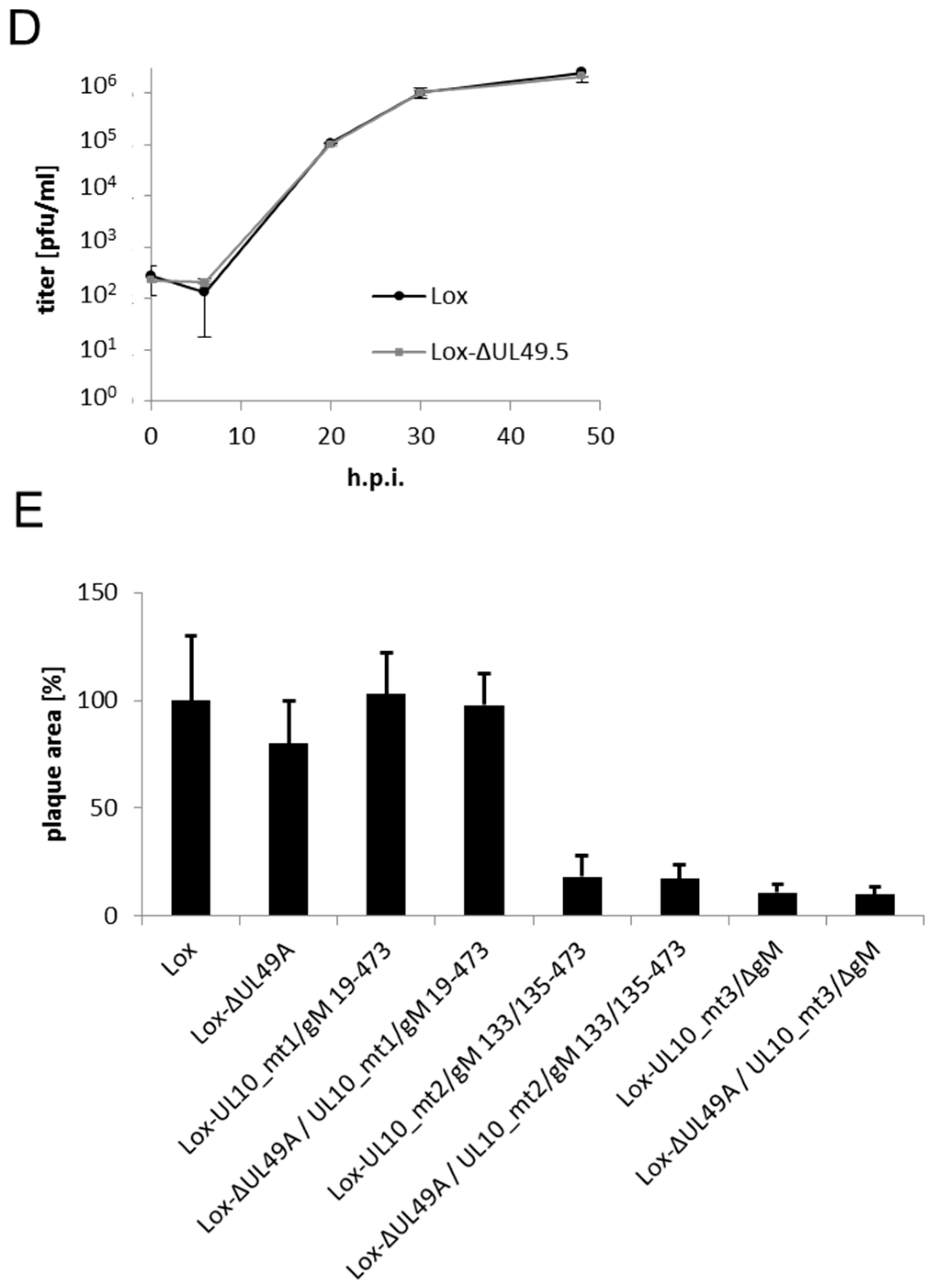
2.5. Analysis of Virus Growth
2.6. Cell Lysis, Reducing and Non-Reducing Gel Electrophoreses and Western Blotting
2.7. Indirect Immunofluorescence Analysis and Microscopy
3. Results
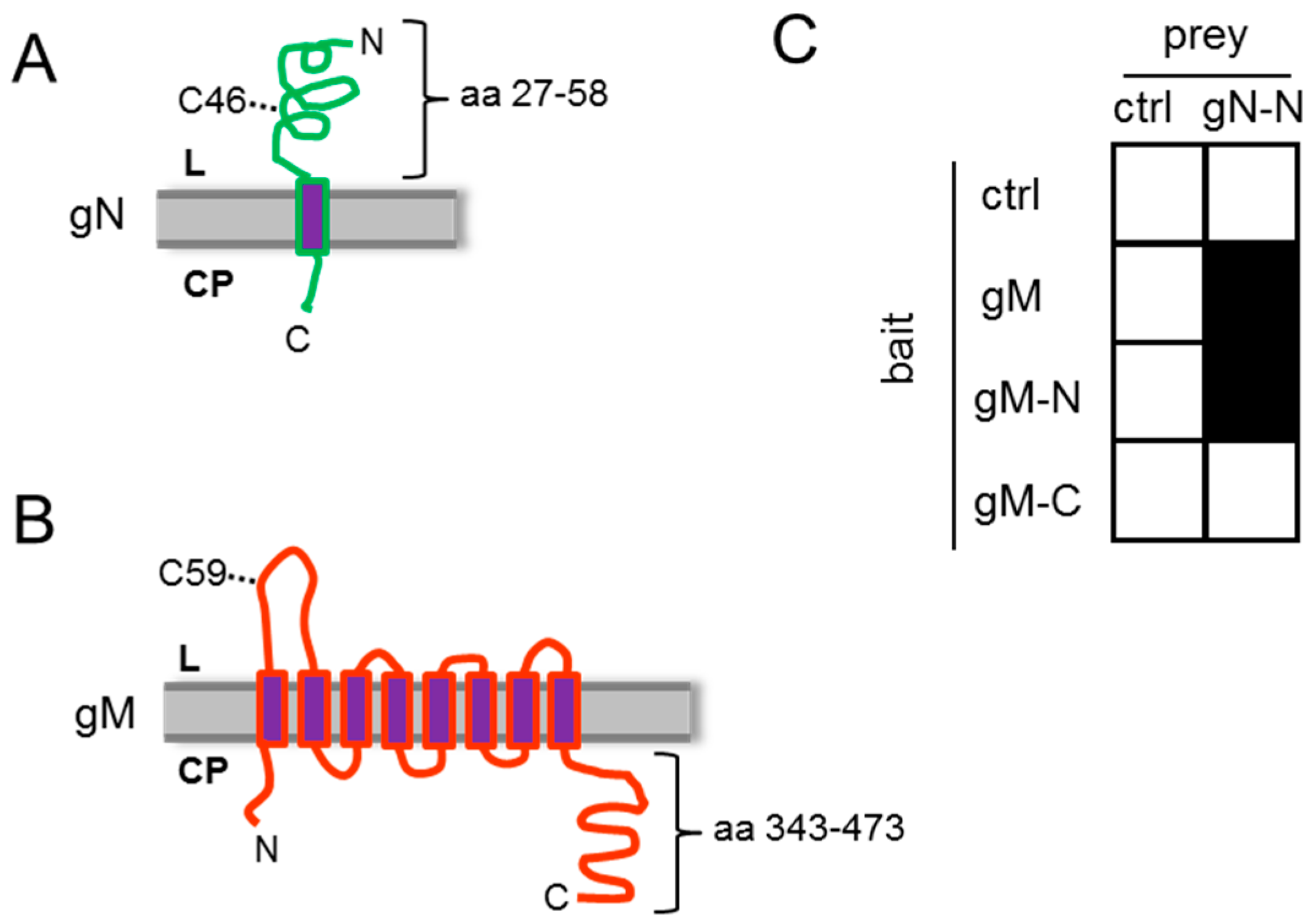
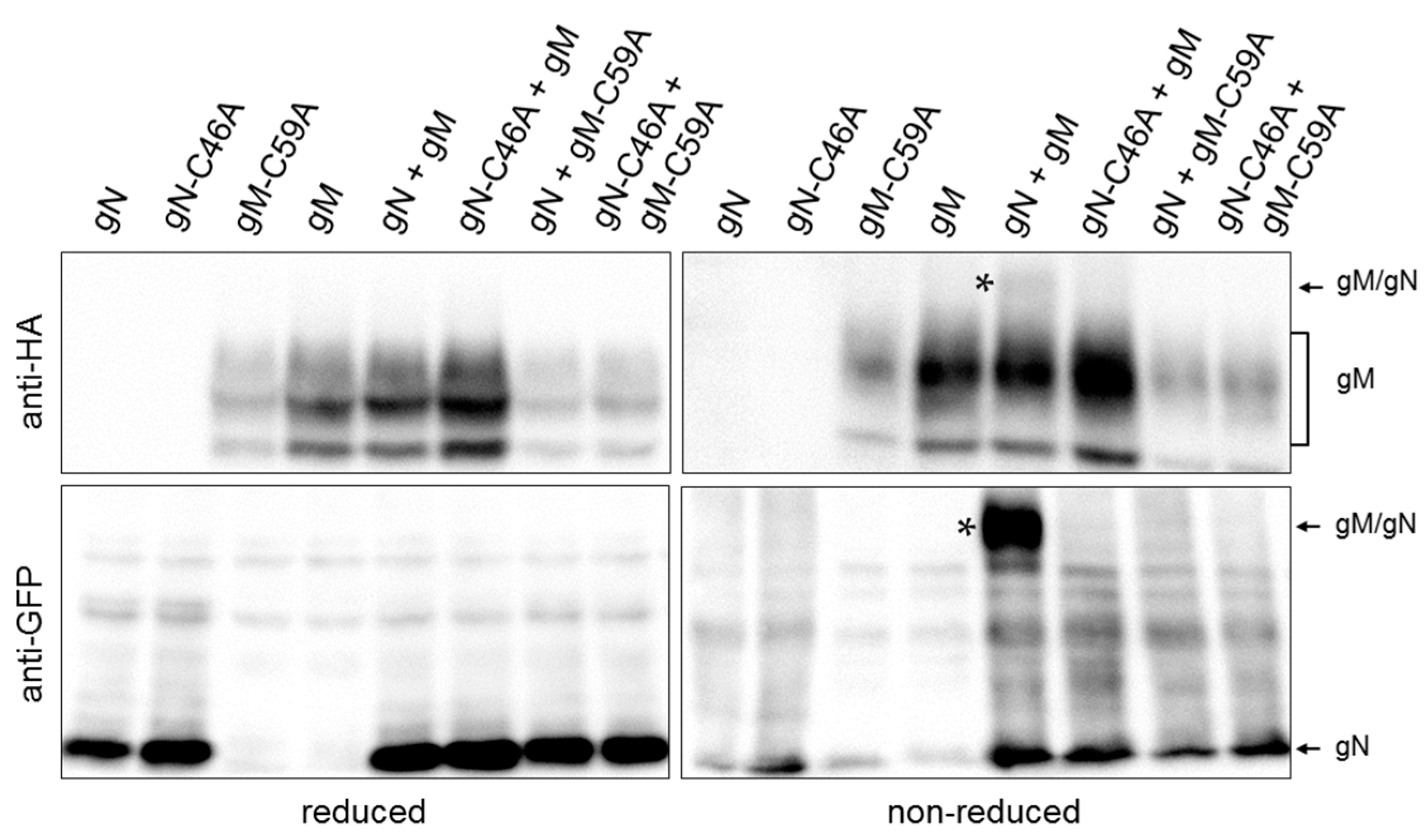
3.1. HSV-1 gN and gM Interact via Their N-Terminal Domains
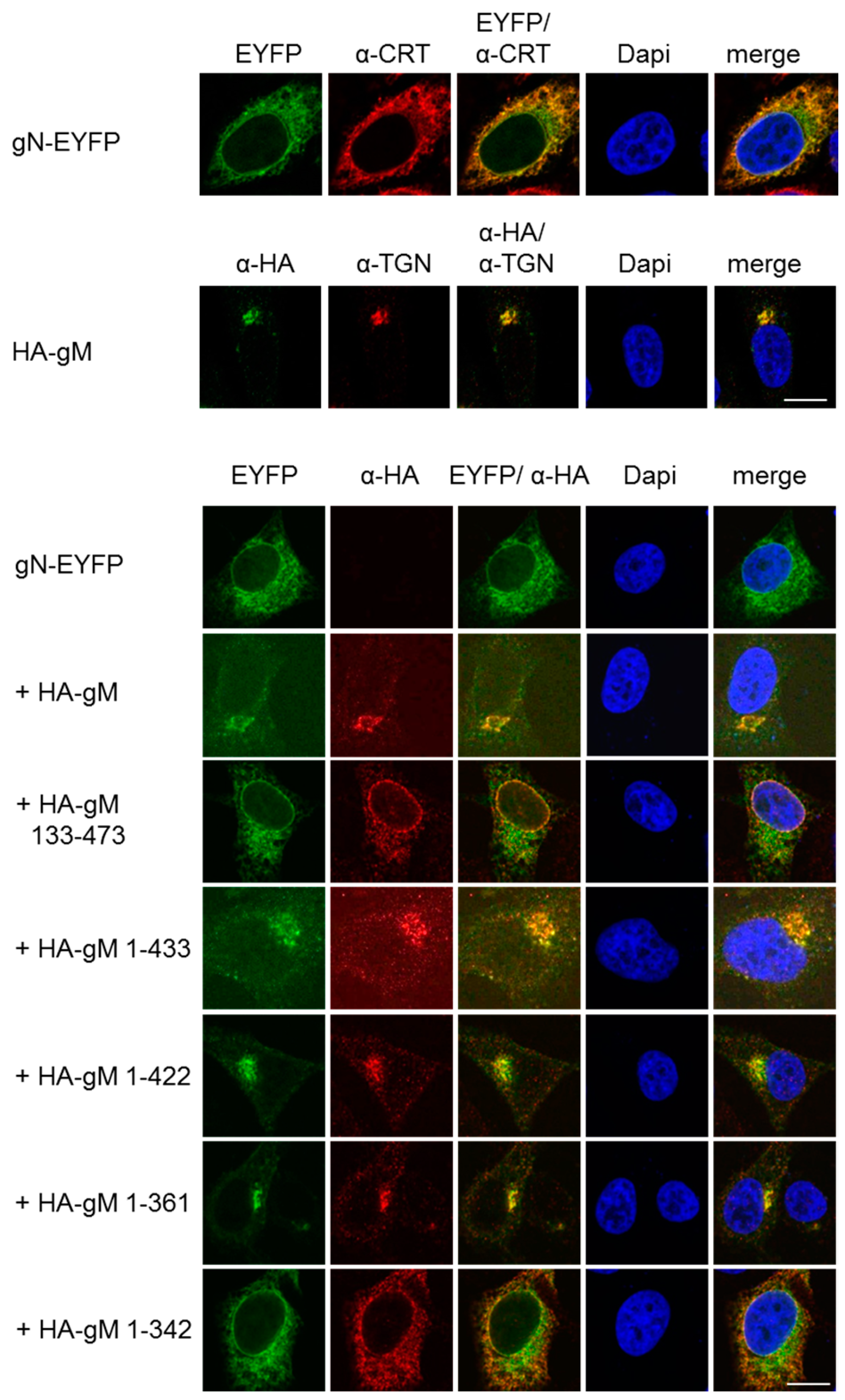
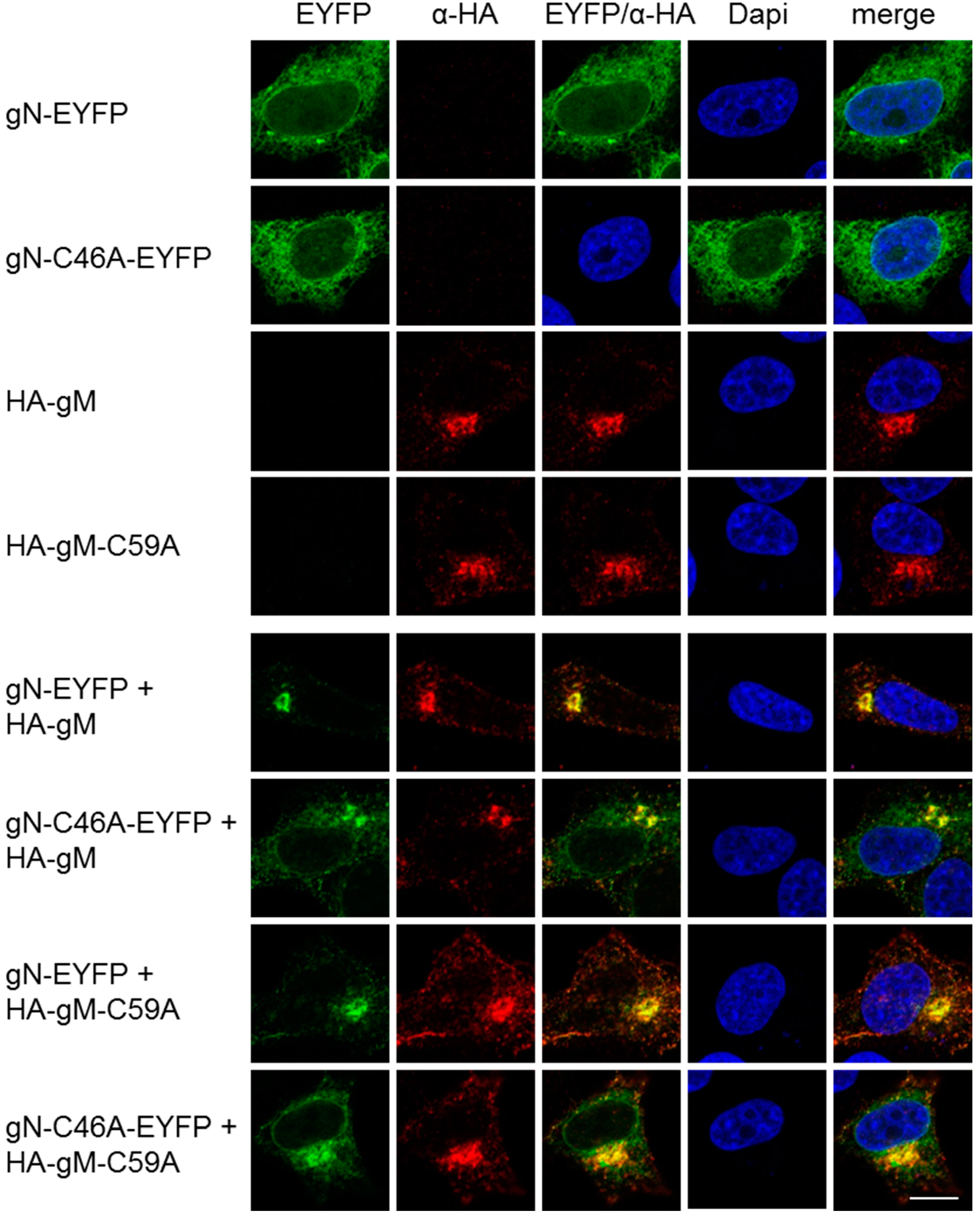
3.2. HSV-1 gN is an ER Resident that Requires the Hydrophobic Core of gM for Transport to the TGN
3.3. A Non-Covalent Interaction of HSV-1 gN with gM is Sufficient for its Transport to the TGN
3.4. During Infection, HSV-1 gN is Nonessential and Redundant in Association with gM
4. Discussion
5. Conclusions
- •
- The evolutionarily conserved transmembrane protein gN of HSV-1 is an ER resident.
- •
- gM and gN are covalently linked by a single disulphide bond formed between cysteine 46 of gN and cysteine 59 of gM
- •
- HSV-1 gN and gM interact via their N-terminal halves, an interaction sufficient for transport of gN to the TGN.
- •
- Non-covalent interaction of HSV-1 gN with gM is sufficient for its transport to the TGN.
- •
- HSV-1 gN/UL49.5 is nonessential in vitro.
- •
- HSV-1 gN/UL49.5 likely functions in gM-dependent as well as gM-independent processes.
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Mettenleiter, T.C.; Klupp, B.G.; Granzow, H. Herpesvirus assembly: An update. Virus Res. 2009, 143, 222–234. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.C.; Baines, J.D. Herpesviruses remodel host membranes for virus egress. Nat. Rev. Microbiol. 2011, 9, 382–394. [Google Scholar] [CrossRef] [PubMed]
- Mach, M.; Kropff, B.; Kryzaniak, M.; Britt, W. Complex formation by glycoproteins M and N of human cytomegalovirus: Structural and functional aspects. J. Virol. 2005, 79, 2160–2170. [Google Scholar] [CrossRef] [PubMed]
- MacLean, C.A.; Efstathiou, S.; Elliott, M.L.; Jamieson, F.E.; McGeoch, D.J. Investigation of herpes simplex virus type 1 genes encoding multiply inserted membrane proteins. J. Gen. Virol. 1991, 72, 897–906. [Google Scholar] [CrossRef] [PubMed]
- Baines, J.D.; Roizman, B. The open reading frames UL3, UL4, UL10, and UL16 are dispensable for the replication of herpes simplex virus 1 in cell culture. J. Virol. 1991, 65, 938–944. [Google Scholar] [PubMed]
- MacLean, C.A.; Robertson, L.M.; Jamieson, F.E. Characterization of the UL10 gene product of herpes simplex virus type 1 and investigation of its role in vivo. J. Gen. Virol. 1993, 74, 975–983. [Google Scholar] [CrossRef] [PubMed]
- Dijkstra, J.M.; Visser, N.; Mettenleiter, T.C.; Klupp, B.G. Identification and characterization of pseudorabies virus glycoprotein gM as a nonessential virion component. J. Virol. 1996, 70, 5684–5688. [Google Scholar] [PubMed]
- Osterrieder, N.; Neubauer, A.; Brandmuller, C.; Braun, B.; Kaaden, O.R.; Baines, J.D. The equine herpesvirus 1 glycoprotein gp21/22a, the herpes simplex virus type 1 gM homolog, is involved in virus penetration and cell-to-cell spread of virions. J. Virol. 1996, 70, 4110–4115. [Google Scholar] [PubMed]
- Fuchs, W.; Mettenleiter, T.C. DNA sequence of the UL6 to UL20 genes of infectious laryngotracheitis virus and characterization of the UL10 gene product as a nonglycosylated and nonessential virion protein. J. Gen. Virol. 1999, 80, 2173–2182. [Google Scholar] [CrossRef] [PubMed]
- Tischer, B.K.; Schumacher, D.; Messerle, M.; Wagner, M.; Osterrieder, N. The products of the UL10 (gM) and the UL49.5 genes of Marek’s disease virus serotype 1 are essential for virus growth in cultured cells. J. Gen. Virol. 2002, 83, 997–1003. [Google Scholar] [CrossRef] [PubMed]
- Browne, H.; Bell, S.; Minson, T. Analysis of the requirement for glycoprotein m in herpes simplex virus type 1 morphogenesis. J. Virol. 2004, 78, 1039–1041. [Google Scholar] [CrossRef] [PubMed]
- Leege, T.; Fuchs, W.; Granzow, H.; Kopp, M.; Klupp, B.G.; Mettenleiter, T.C. Effects of simultaneous deletion of pUL11 and glycoprotein M on virion maturation of herpes simplex virus type 1. J. Virol. 2009, 83, 896–907. [Google Scholar] [CrossRef] [PubMed]
- Sadaoka, T.; Yanagi, T.; Yamanishi, K.; Mori, Y. Characterization of the varicella-zoster virus ORF50 gene, which encodes glycoprotein M. J. Virol. 2010, 84, 3488–3502. [Google Scholar] [CrossRef] [PubMed]
- Striebinger, H.; Zhang, J.; Ott, M.; Funk, C.; Radtke, K.; Duron, J.; Ruzsics, Z.; Haas, J.; Lippe, R.; Bailer, S.M. Subcellular trafficking and functional importance of Herpes simplex virus type 1 Glycoprotein M domains. J. Gen. Virol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Crump, C.M.; Bruun, B.; Bell, S.; Pomeranz, L.E.; Minson, T.; Browne, H.M. Alphaherpesvirus glycoprotein M causes the relocalization of plasma membrane proteins. J. Gen. Virol. 2004, 85, 3517–3527. [Google Scholar] [CrossRef] [PubMed]
- Baines, J.D.; Roizman, B. The UL10 gene of herpes simplex virus 1 encodes a novel viral glycoprotein, gM, which is present in the virion and in the plasma membrane of infected cells. J. Virol. 1993, 67, 1441–1452. [Google Scholar] [PubMed]
- Baines, J.D.; Wills, E.; Jacob, R.J.; Pennington, J.; Roizman, B. Glycoprotein M of herpes simplex virus 1 is incorporated into virions during budding at the inner nuclear membrane. J. Virol. 2007, 81, 800–812. [Google Scholar] [CrossRef] [PubMed]
- Wills, E.; Mou, F.; Baines, J.D. The U(L)31 and U(L)34 gene products of herpes simplex virus 1 are required for optimal localization of viral glycoproteins D and M to the inner nuclear membranes of infected cells. J. Virol. 2009, 83, 4800–4809. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Nagel, C.H.; Sodeik, B.; Lippe, R. Early, active, and specific localization of herpes simplex virus type 1 gM to nuclear membranes. J. Virol. 2009, 83, 12984–12997. [Google Scholar] [CrossRef] [PubMed]
- Klupp, B.G.; Nixdorf, R.; Mettenleiter, T.C. Pseudorabies virus glycoprotein M inhibits membrane fusion. J. Virol. 2000, 74, 6760–6768. [Google Scholar] [CrossRef] [PubMed]
- Koyano, S.; Mar, E.C.; Stamey, F.R.; Inoue, N. Glycoproteins M and N of human herpesvirus 8 form a complex and inhibit cell fusion. J. Gen. Virol. 2003, 84, 1485–1491. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.J.; Chouljenko, V.N.; Walker, J.D.; Kousoulas, K.G. Herpes simplex virus 1 glycoprotein M and the membrane-associated protein UL11 are required for virus-induced cell fusion and efficient virus entry. J. Virol. 2013, 87, 8029–8037. [Google Scholar] [CrossRef] [PubMed]
- Lau, S.Y.; Crump, C.M. HSV-1 gM and the gK/pUL20 complex are important for the localization of gD and gH/L to viral assembly sites. Viruses 2015, 7, 915–938. [Google Scholar] [CrossRef] [PubMed]
- El Kasmi, I.; Lippe, R. HSV-1 gN partners with gM to modulate the viral fusion machinery. J. Virol. 2014, 89, 2313–2323. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Chow, B.; Raggo, C.; Babiuk, L.A. Bovine herpesvirus 1 UL49.5 homolog gene encodes a novel viral envelope protein that forms a disulfide-linked complex with a second virion structural protein. J. Virol. 1996, 70, 1448–1454. [Google Scholar] [PubMed]
- Jons, A.; Dijkstra, J.M.; Mettenleiter, T.C. Glycoproteins M and N of pseudorabies virus form a disulfide-linked complex. J. Virol. 1998, 72, 550–557. [Google Scholar] [PubMed]
- Wu, S.X.; Zhu, X.P.; Letchworth, G.J. Bovine herpesvirus 1 glycoprotein M forms a disulfide-linked heterodimer with the U(L)49.5 protein. J. Virol. 1998, 72, 3029–3036. [Google Scholar] [PubMed]
- Ott, M.; Tascher, G.; Hassdenteufel, S.; Zimmermann, R.; Haas, J.; Bailer, S.M. Functional characterization of the essential tail anchor of the herpes simplex virus type 1 nuclear egress protein pUL34. J. Gen. Virol. 2011, 92, 2734–2745. [Google Scholar] [CrossRef] [PubMed]
- Loret, S.; Guay, G.; Lippe, R. Comprehensive characterization of extracellular herpes simplex virus type 1 virions. J. Virol. 2008, 82, 8605–8618. [Google Scholar] [CrossRef] [PubMed]
- Jons, A.; Granzow, H.; Kuchling, R.; Mettenleiter, T.C. The UL49.5 gene of pseudorabies virus codes for an O-glycosylated structural protein of the viral envelope. J. Virol. 1996, 70, 1237–1241. [Google Scholar] [PubMed]
- Verweij, M.C.; Lipinska, A.D.; Koppers-Lalic, D.; van Leeuwen, W.F.; Cohen, J.I.; Kinchington, P.R.; Messaoudi, I.; Bienkowska-Szewczyk, K.; Ressing, M.E.; Rijsewijk, F.A.; et al. The capacity of UL49.5 proteins to inhibit TAP is widely distributed among members of the genus Varicellovirus. J. Virol. 2011, 85, 2351–2363. [Google Scholar] [CrossRef] [PubMed]
- Verweij, M.C.; Lipinska, A.D.; Koppers-Lalic, D.; Quinten, E.; Funke, J.; van Leeuwen, H.C.; Bienkowska-Szewczyk, K.; Koch, J.; Ressing, M.E.; Wiertz, E.J. Structural and functional analysis of the TAP-inhibiting UL49.5 proteins of varicelloviruses. Mol. Immun. 2011, 48, 2038–2051. [Google Scholar] [CrossRef] [PubMed]
- Verweij, M.C.; Koppers-Lalic, D.; Loch, S.; Klauschies, F.; de la Salle, H.; Quinten, E.; Lehner, P.J.; Mulder, A.; Knittler, M.R.; Tampe, R.; et al. The varicellovirus UL49.5 protein blocks the transporter associated with antigen processing (TAP) by inhibiting essential conformational transitions in the 6+6 transmembrane TAP core complex. J. Immun. 2008, 181, 4894–4907. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Wang, Y.; Chowdhury, S.I. Bovine herpesvirus type 1 (BHV-1) UL49.5 luminal domain residues 30 to 32 are critical for MHC-I down-regulation in virus-infected cells. PLoS ONE 2011, 6, e25742. [Google Scholar] [CrossRef] [PubMed]
- Jarosinski, K.W.; Hunt, H.D.; Osterrieder, N. Down-regulation of MHC class I by the Marek’s disease virus (MDV) UL49.5 gene product mildly affects virulence in a haplotype-specific fashion. Virology 2010, 405, 457–463. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer Name | Primer Sequence |
|---|---|
| attB_5′ | ggggacaagtttgtacaaaaaagcaggct |
| attB_3′ | ggggaccactttgtacaagaaagctgggt |
| UL49.5–FL_5′ | aaaaagcaggctccgccatgggcccccccagaag |
| UL49.5–FL_3′ | agaaagctgggtctaggcgtgcccggcagc |
| UL49.5–Y2H_3′ | |
| UL49.5–Y2H_5′ | aaaaagcaggctccgccatgccgcgcggggagccg |
| UL49.5–IF_3′ | agaaagctgggtggcgtgcccggcagc |
| UL10–FL_5′ | aaaaagcaggctccgccatgggacgcccggcccccagag |
| UL10–Y2H_5′ | |
| UL10–Y2H_5′–N | |
| UL10–FL_3′ | agaaagctgggtctaccaacggcggacggtgc |
| UL10–Y2H_3′ | |
| UL10–Y2H_3′–C | |
| UL10–∆C_3′ | agaaagctgggtctaggtgcagcggagcacggccatgc |
| UL10–Y2H_3′–N | |
| UL10–C_5′ | aaaaagcaggctccgccatgcgcgcctatctgtatcac |
| UL10–Y2H_5′–C | |
| UL10–M19I_5′ | cagaggatctcccgactccgcgccccccacgaaaggcataaccggggcgcggac |
| UL10stop362aa_5′ | gcgcatgcgcgactagcgacaccgcgcac |
| UL10stop362aa_3′ | gtgcgcggtgtcgctagtcgcgcatgcgc |
| UL10stop423aa_5′ | ggggagccgatttaggacgaggtggcg |
| UL10stop423aa_3′ | cgccacctcgtcctaaatcggctcccc |
| UL10stop434aa_5′ | cgaccaaaccgacgtatagtacgccaagatacaacac |
| UL10stop434aa_3′ | gtgttgtatcttggcgtactatacgtcggtttggtcg |
| gM–C59A_5′ | cacggtttcccgccttttacgccacggcg |
| gM–C59A_3′ | gtggcgtaaaaggcgggaaaccgtgcccg |
| gN–C46A_5′ | gggcgcgcgggccgagacccaaaacactg |
| gN–C46A_3′ | gttttgggtctcggcccgcgcgcccccgatc |
| H5–gM/gk | tccgcgctagcgatacgctcgacgtgtactgttcgcactcgtcgtccccacctgttgacaattaatcatcggca |
| H3–gM/gk | caccacggtcgggttaaacacaaacggtttattaaaacggaaccaaacaggccagtgttacaaccaattaacc |
| UL10–mt1_5′ | gcgctagcgatacgctcgacgtgtactgttcgcactcgtcgtccccaatgggatgaccggcccccagaggatctcccg |
| UL10–mt1_3′ | caccacggtcgggttaaacacaaacggtttattaaaacggaaccaaacagctaccaacggcggacggtgc |
| H5–gM–mt3/gK | gccatgccgcacgccacgctgatcgccggaaacgtctgctcttggttgctcctgttgacaattaatcatcggca |
| H5–UL49.5/gK | cccaacacatagcaggccgcgggcccggcgtccgcgtggagcatgcggagggcctgttgacaattaatcatcggca |
| H3–UL49.5/gK | aagtcctgggacaccctccacccccacccctcaccccacacagggcgggtgccagtgttacaaccaattaacc |
| ΔUL49.5_5′ | cccaacacatagcaggccgcgggcccggcgtccgcgtggagcatgcggagggatgggccccccctagtaggtct |
| ΔUL49.5_3′ | aagtcctgggacaccctccacccccacccctcaccccacacagggcgggttcaggcgtgcccggcagccagt |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Striebinger, H.; Funk, C.; Raschbichler, V.; Bailer, S.M. Subcellular Trafficking and Functional Relationship of the HSV-1 Glycoproteins N and M. Viruses 2016, 8, 83. https://0-doi-org.brum.beds.ac.uk/10.3390/v8030083
Striebinger H, Funk C, Raschbichler V, Bailer SM. Subcellular Trafficking and Functional Relationship of the HSV-1 Glycoproteins N and M. Viruses. 2016; 8(3):83. https://0-doi-org.brum.beds.ac.uk/10.3390/v8030083
Chicago/Turabian StyleStriebinger, Hannah, Christina Funk, Verena Raschbichler, and Susanne M. Bailer. 2016. "Subcellular Trafficking and Functional Relationship of the HSV-1 Glycoproteins N and M" Viruses 8, no. 3: 83. https://0-doi-org.brum.beds.ac.uk/10.3390/v8030083