
Glutamic Acid Residues in HIV-1 p6 Regulate Virus Budding and Membrane Association of Gag

 ,
,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Transfection
2.2. Cultivation and Preparation of Primary Cells
2.3. Investigation of Gag Processing by Steady State Analyses
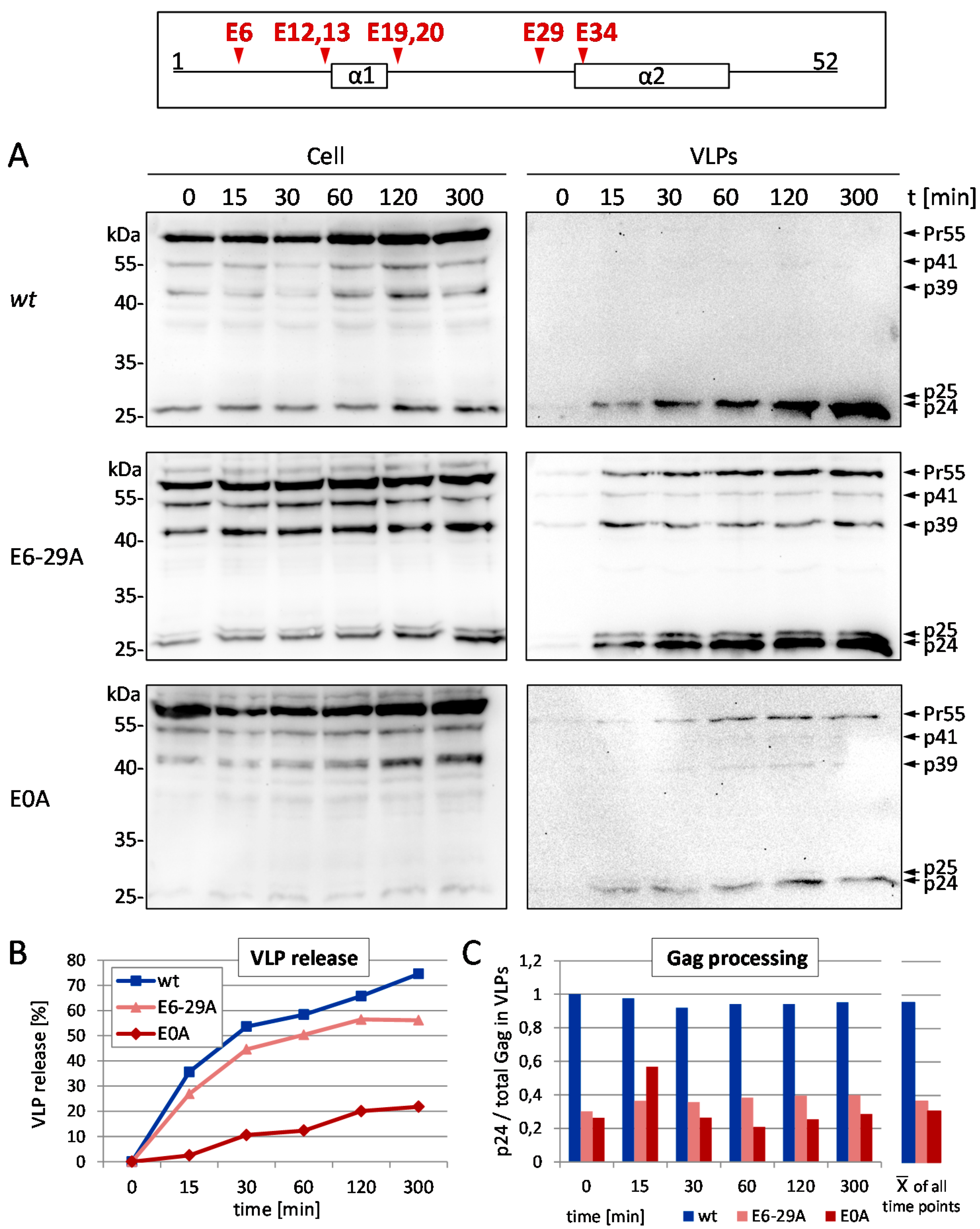
2.4. Time Course Analyses of VLP Release
2.5. Detection of Ubiquitinated Gag
2.6. SDS-PAGE and Western Blotting
2.7. Expression Plasmids
2.8. Flow Cytometry
2.9. Single Round Infection Assay
2.10. Infection of HLA Cultures
2.11. Determination of the Replication Capacity
2.12. Membrane Flotation
2.13. Peptide Synthesis
2.14. NMR Spectroscopy
3. Results
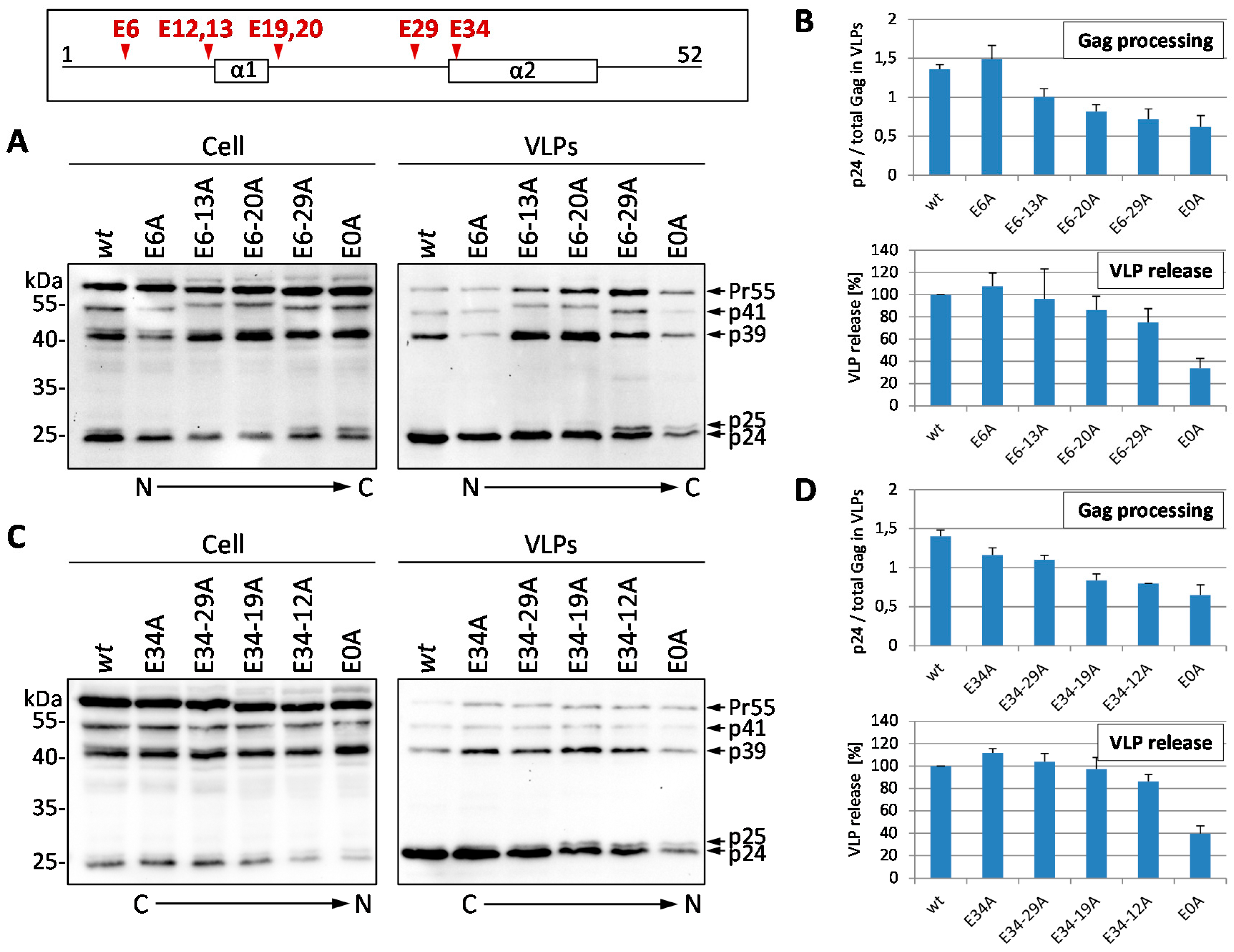
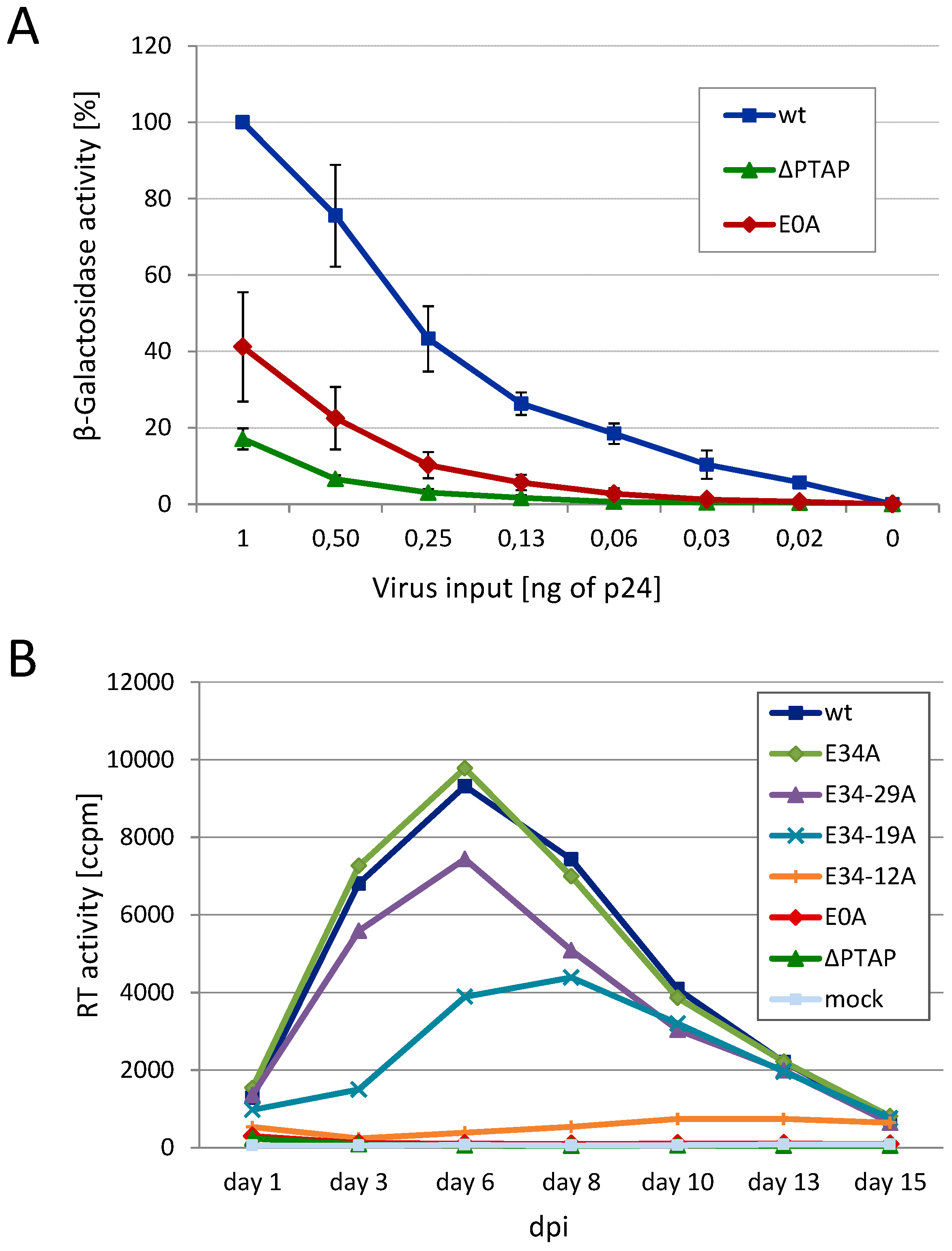
3.1. Mutation of the Glutamic Acids in p6 Impairs Virus Budding and Gag Processing
3.2. Effect of Glu Mutants on Viral Infectivity and Replication Capacity
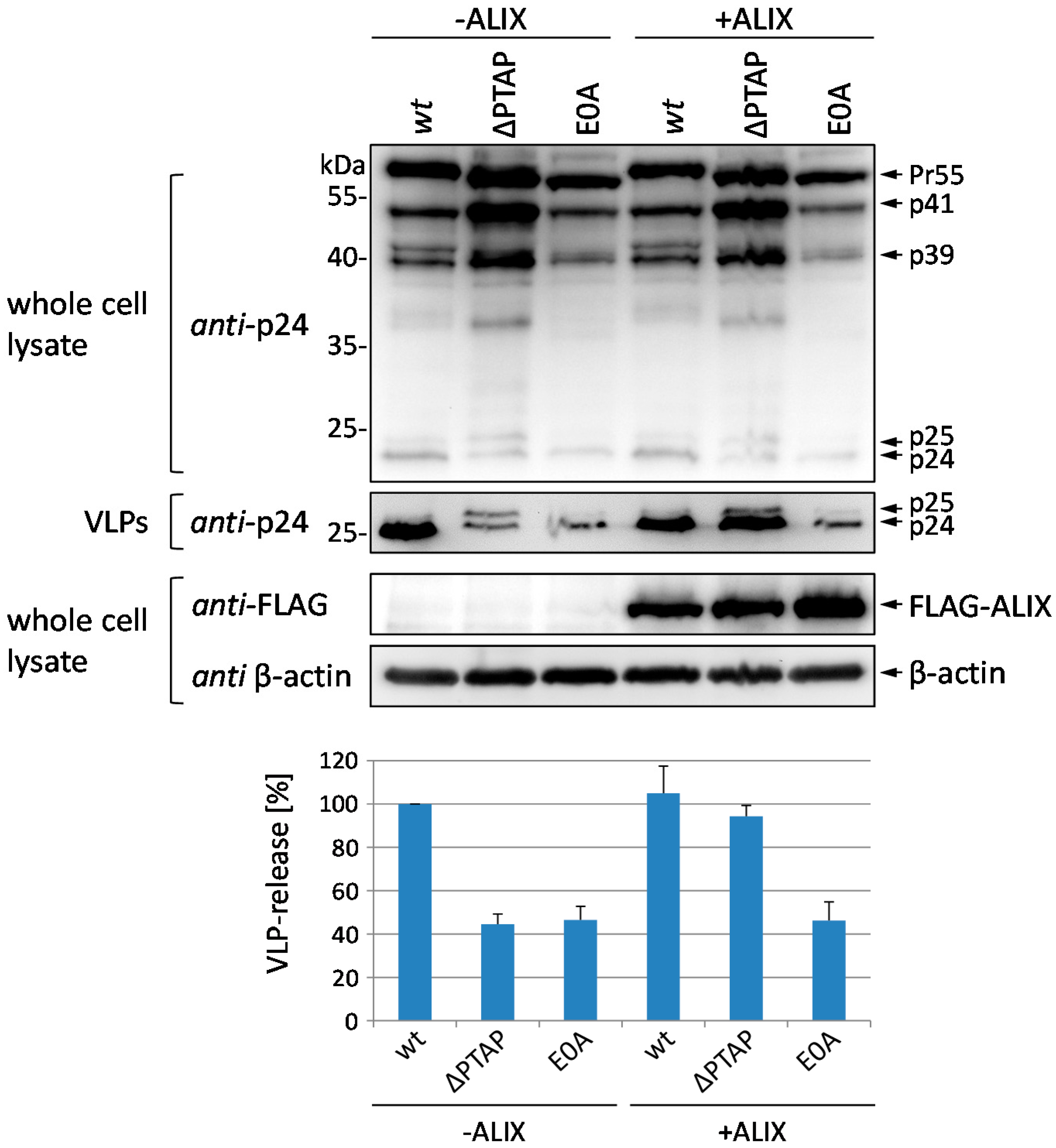
3.3. Budding Defect of the E0A Mutant Cannot Be Rescued by Overexpression of ALIX
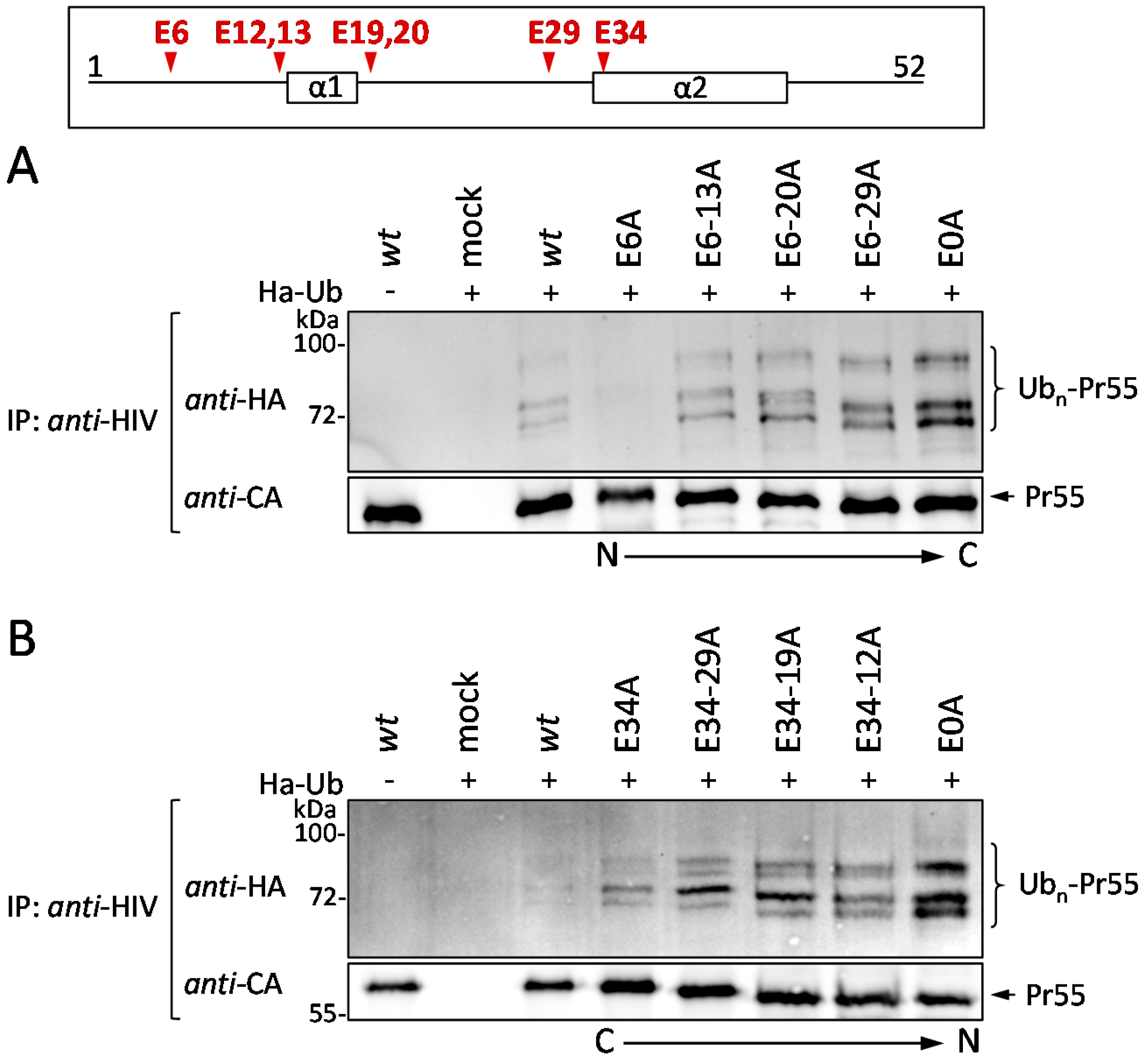
3.4. The Glu Residues in p6 Contribute to the Entry of Gag into the UPS
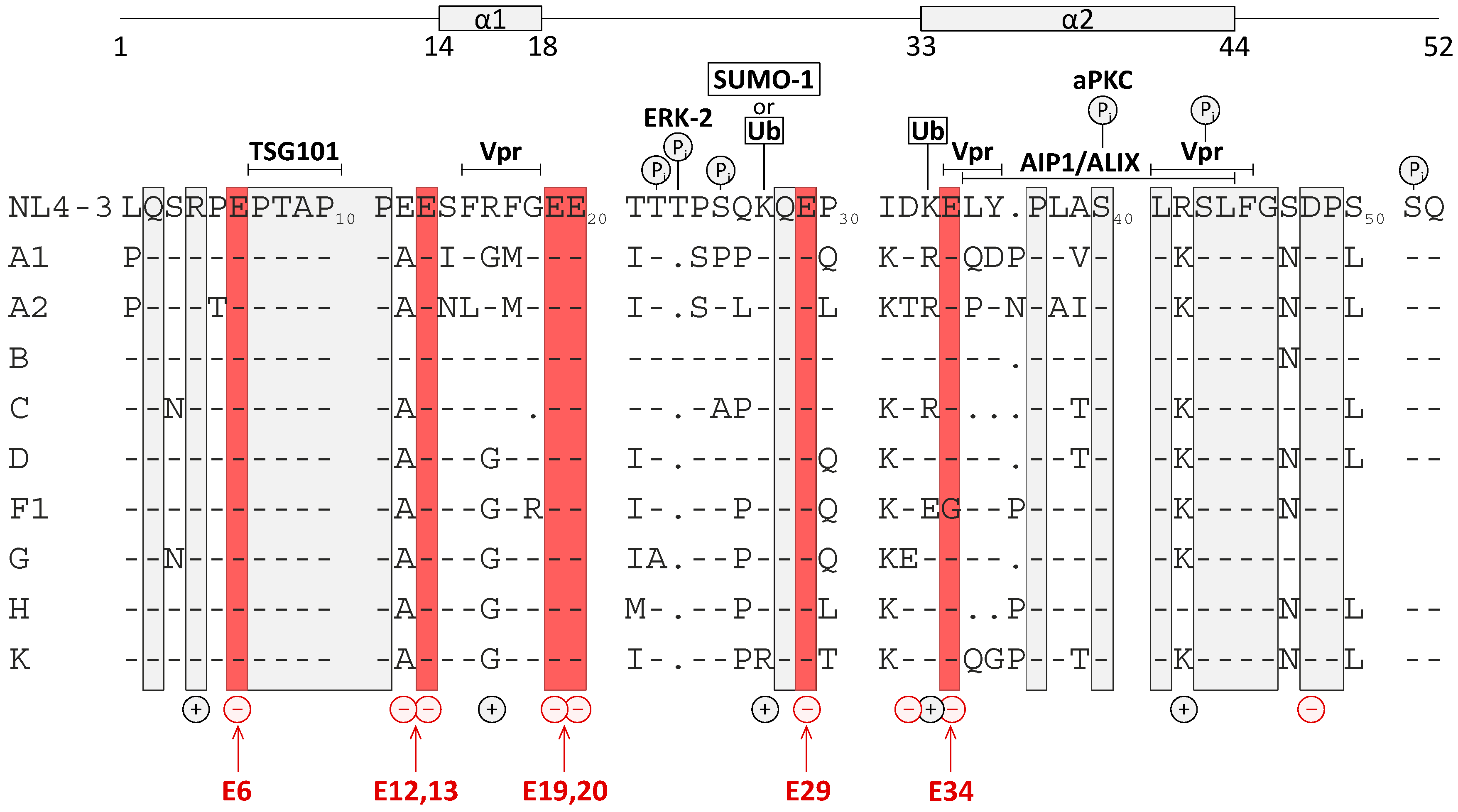
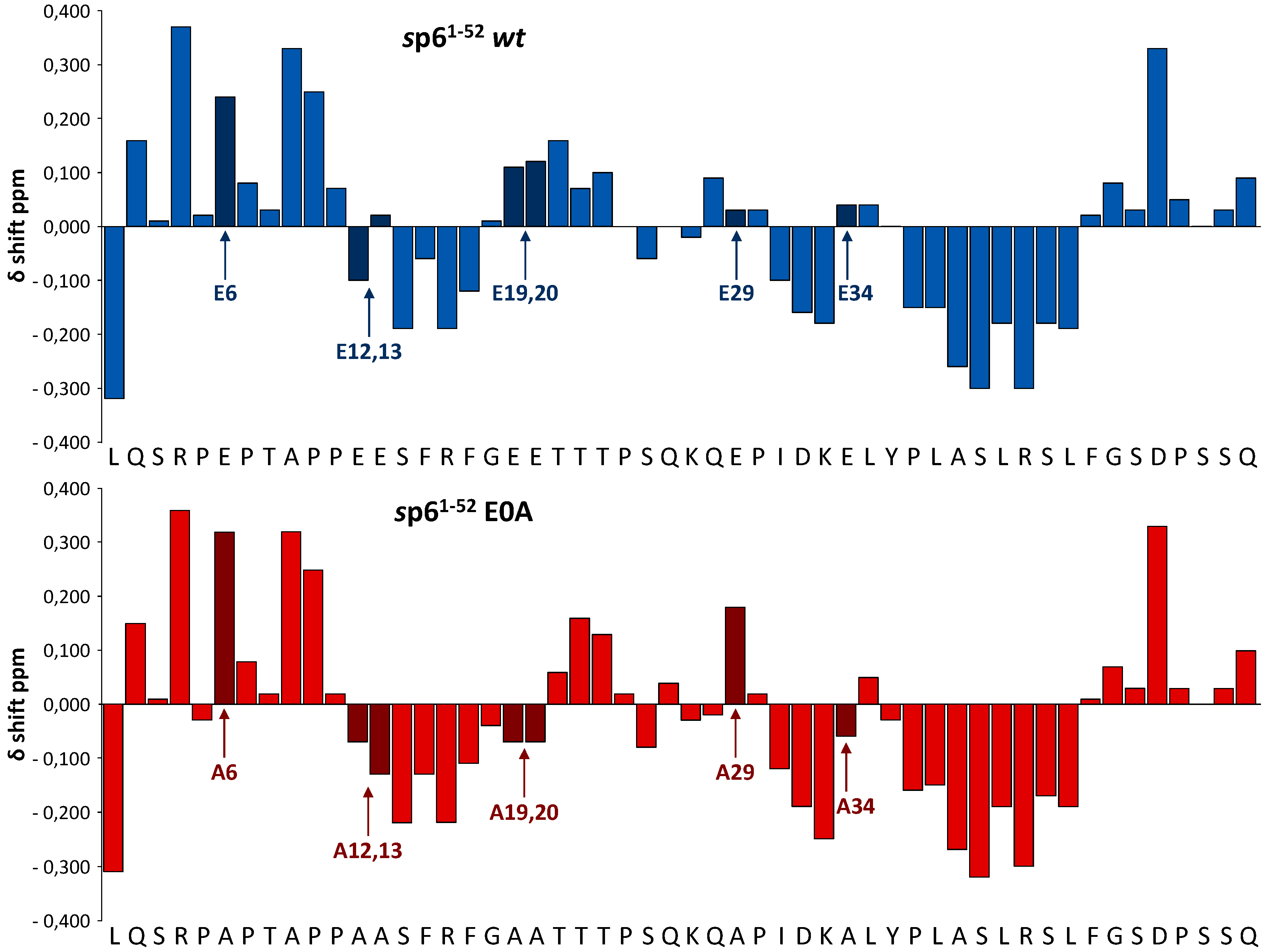
3.5. Effect of Glu Mutations on the Structure of p6
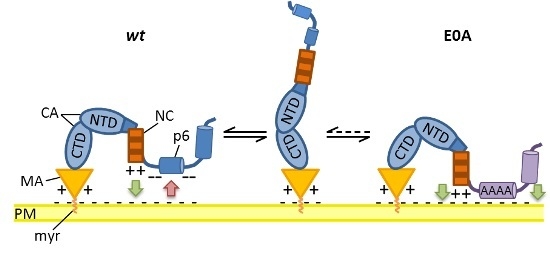
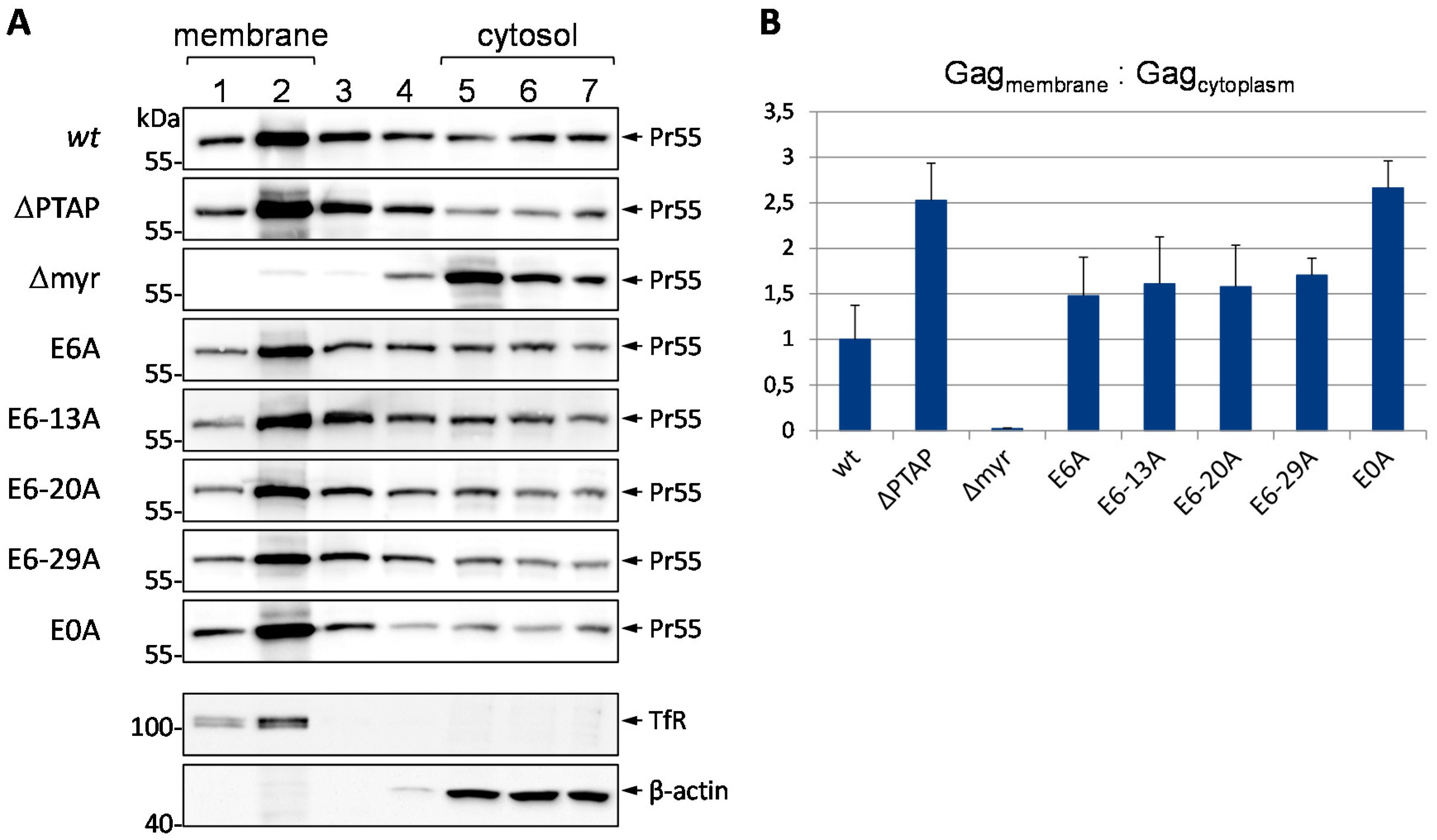
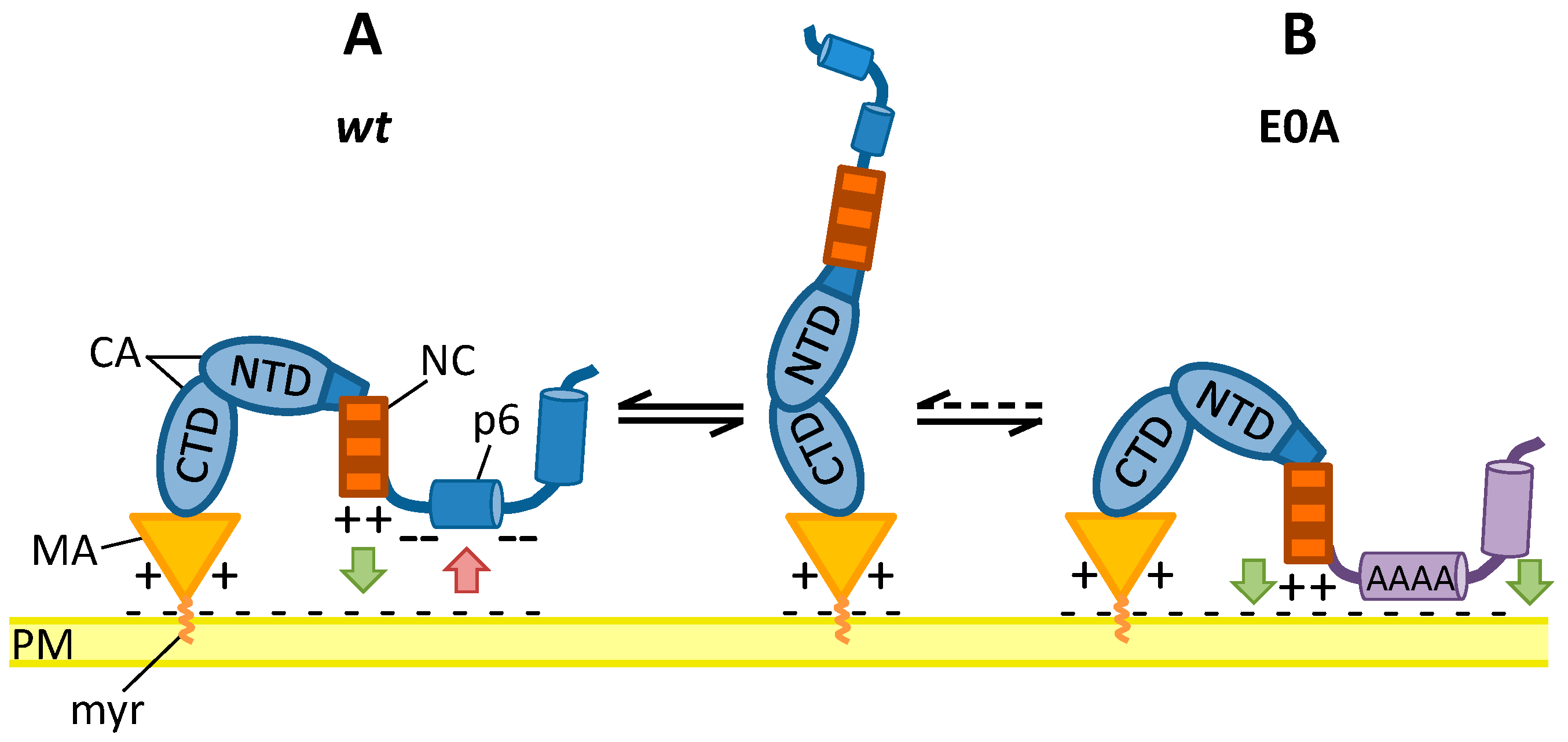
3.6. Mutation of Glu Residues Enhances Membrane Association of Gag
4. Discussion
5. Conclusions
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Sundquist, W.I.; Krausslich, H.G. HIV-1 assembly, budding, and maturation. Cold Spring Harb. Perspect. Med. 2012, 2. [Google Scholar] [CrossRef]
- Freed, E.O. HIV-1 assembly, release and maturation. Nat. Rev. Microbiol. 2015, 13, 484–496. [Google Scholar] [CrossRef] [PubMed]
- Wills, J.W.; Craven, R.C. Form, function, and use of retroviral Gag proteins. AIDS 1991, 5, 639–654. [Google Scholar] [CrossRef] [PubMed]
- Gottlinger, H.G.; Sodroski, J.G.; Haseltine, W.A. Role of capsid precursor processing and myristoylation in morphogenesis and infectivity of human immunodeficiency virus type 1. Proc. Natl. Acad. Sci. USA 1989, 86, 5781–5785. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Parent, L.J.; Wills, J.W.; Resh, M.D. Identification of a membrane-binding domain within the amino-terminal region of human immunodeficiency virus type 1 Gag protein which interacts with acidic phospholipids. J. Virol. 1994, 68, 2556–2569. [Google Scholar] [PubMed]
- Darlix, J.L.; Godet, J.; Ivanyi-Nagy, R.; Fosse, P.; Mauffret, O.; Mely, Y. Flexible nature and specific functions of the HIV-1 nucleocapsid protein. J. Mol. Biol. 2011, 410, 565–581. [Google Scholar] [CrossRef] [PubMed]
- Lu, K.; Heng, X.; Summers, M.F. Structural determinants and mechanism of HIV-1 genome packaging. J. Mol. Biol. 2011, 410, 609–633. [Google Scholar] [CrossRef] [PubMed]
- Garrus, J.E.; von Schwedler, U.K.; Pornillos, O.W.; Morham, S.G.; Zavitz, K.H.; Wang, H.E.; Wettstein, D.A.; Stray, K.M.; Cote, M.; Rich, R.L.; et al. Tsg101 and the vacuolar protein sorting pathway are essential for HIV-1 budding. Cell 2001, 107, 55–65. [Google Scholar] [CrossRef]
- VerPlank, L.; Bouamr, F.; LaGrassa, T.J.; Agresta, B.; Kikonyogo, A.; Leis, J.; Carter, C.A. Tsg101, a homologue of ubiquitin-conjugating (E2) enzymes, binds the L domain in HIV type 1 Pr55(Gag). Proc. Natl. Acad. Sci. USA 2001, 98, 7724–7729. [Google Scholar] [CrossRef] [PubMed]
- Martin-Serrano, J.; Zang, T.; Bieniasz, P.D. HIV-1 and Ebola virus encode small peptide motifs that recruit Tsg101 to sites of particle assembly to facilitate egress. Nat. Med. 2001, 7, 1313–1319. [Google Scholar] [CrossRef] [PubMed]
- Demirov, D.G.; Ono, A.; Orenstein, J.M.; Freed, E.O. Overexpression of the N-terminal domain of TSG101 inhibits HIV-1 budding by blocking late domain function. Proc. Natl. Acad. Sci. USA 2002, 99, 955–960. [Google Scholar] [CrossRef] [PubMed]
- Strack, B.; Calistri, A.; Craig, S.; Popova, E.; Gottlinger, H.G. AIP1/ALIX is a binding partner for HIV-1 p6 and EIAV p9 functioning in virus budding. Cell 2003, 114, 689–699. [Google Scholar] [CrossRef]
- Usami, Y.; Popov, S.; Gottlinger, H.G. Potent rescue of human immunodeficiency virus type 1 late domain mutants by ALIX/AIP1 depends on its CHMP4 binding site. J. Virol. 2007, 81, 6614–6622. [Google Scholar] [CrossRef] [PubMed]
- Fisher, R.D.; Chung, H.Y.; Zhai, Q.; Robinson, H.; Sundquist, W.I.; Hill, C.P. Structural and biochemical studies of ALIX/AIP1 and its role in retrovirus budding. Cell 2007, 128, 841–852. [Google Scholar] [CrossRef] [PubMed]
- Von Schwedler, U.K.; Stuchell, M.; Muller, B.; Ward, D.M.; Chung, H.Y.; Morita, E.; Wang, H.E.; Davis, T.; He, G.P.; Cimbora, D.M.; et al. The protein network of HIV budding. Cell 2003, 114, 701–713. [Google Scholar] [CrossRef]
- McCullough, J.; Colf, L.A.; Sundquist, W.I. Membrane fission reactions of the mammalian ESCRT pathway. Annu. Rev. Biochem. 2013, 82, 663–692. [Google Scholar] [CrossRef] [PubMed]
- Caballe, A.; Martin-Serrano, J. ESCRT machinery and cytokinesis: The road to daughter cell separation. Traffic 2011, 12, 1318–1326. [Google Scholar] [CrossRef] [PubMed]
- Carlton, J.G.; Caballe, A.; Agromayor, M.; Kloc, M.; Martin-Serrano, J. ESCRT-III governs the Aurora B-mediated abscission checkpoint through CHMP4C. Science 2012, 336, 220–225. [Google Scholar] [CrossRef] [PubMed]
- Hanson, P.I.; Cashikar, A. Multivesicular body morphogenesis. Annu. Rev. Cell Dev. Biol. 2012, 28, 337–362. [Google Scholar] [CrossRef] [PubMed]
- Raiborg, C.; Stenmark, H. The ESCRT machinery in endosomal sorting of ubiquitylated membrane proteins. Nature 2009, 458, 445–452. [Google Scholar] [CrossRef] [PubMed]
- Ott, D.E.; Coren, L.V.; Copeland, T.D.; Kane, B.P.; Johnson, D.G.; Sowder, R.C., 2nd; Yoshinaka, Y.; Oroszlan, S.; Arthur, L.O.; Henderson, L.E. Ubiquitin is covalently attached to the p6Gag proteins of human immunodeficiency virus type 1 and simian immunodeficiency virus and to the p12Gag protein of Moloney murine leukemia virus. J. Virol. 1998, 72, 2962–2968. [Google Scholar] [PubMed]
- Gurer, C.; Berthoux, L.; Luban, J. Covalent modification of human immunodeficiency virus type 1 p6 by SUMO-1. J. Virol. 2005, 79, 910–917. [Google Scholar] [CrossRef] [PubMed]
- Gottwein, E.; Krausslich, H.G. Analysis of human immunodeficiency virus type 1 Gag ubiquitination. J. Virol. 2005, 79, 9134–9144. [Google Scholar] [CrossRef] [PubMed]
- Muller, B.; Patschinsky, T.; Krausslich, H.G. The late-domain-containing protein p6 is the predominant phosphoprotein of human immunodeficiency virus type 1 particles. J. Virol. 2002, 76, 1015–1024. [Google Scholar] [CrossRef] [PubMed]
- Radestock, B.; Morales, I.; Rahman, S.A.; Radau, S.; Glass, B.; Zahedi, R.P.; Muller, B.; Krausslich, H.G. Comprehensive mutational analysis reveals p6Gag phosphorylation to be dispensable for HIV-1 morphogenesis and replication. J. Virol. 2013, 87, 724–734. [Google Scholar] [CrossRef] [PubMed]
- Ott, D.E.; Coren, L.V.; Chertova, E.N.; Gagliardi, T.D.; Schubert, U. Ubiquitination of HIV-1 and MuLV Gag. Virology 2000, 278, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Orenstein, J.M.; Martin, M.A.; Freed, E.O. p6Gag is required for particle production from full-length human immunodeficiency virus type 1 molecular clones expressing protease. J. Virol. 1995, 69, 6810–6818. [Google Scholar] [PubMed]
- Gottlinger, H.G.; Dorfman, T.; Sodroski, J.G.; Haseltine, W.A. Effect of mutations affecting the p6 gag protein on human immunodeficiency virus particle release. Proc. Natl. Acad. Sci. USA 1991, 88, 3195–3199. [Google Scholar] [CrossRef] [PubMed]
- Votteler, J.; Neumann, L.; Hahn, S.; Hahn, F.; Rauch, P.; Schmidt, K.; Studtrucker, N.; Solbak, S.M.; Fossen, T.; Henklein, P.; et al. Highly conserved serine residue 40 in HIV-1 p6 regulates capsid processing and virus core assembly. Retrovirology 2011, 8. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, S.M.; Chen, M.H.; Khan, M.; Ehrlich, L.; Kemal, K.S.; Weiser, B.; Shi, B.; Chen, C.; Powell, M.; Anastos, K.; et al. The S40 residue in HIV-1 Gag p6 impacts local and distal budding determinants, revealing additional late domain activities. Retrovirology 2013, 10. [Google Scholar] [CrossRef] [PubMed]
- Radestock, B.; Burk, R.; Muller, B.; Krausslich, H.G. Re-visiting the functional Relevance of the highly conserved Serine 40 Residue within HIV-1 p6(Gag). Retrovirology 2014, 11. [Google Scholar] [CrossRef] [PubMed]
- Hahn, F.; Setz, C.; Friedrich, M.; Rauch, P.; Solbak, S.M.; Froystein, N.A.; Henklein, P.; Votteler, J.; Fossen, T.; Schubert, U. Mutation of the highly conserved Ser-40 of the HIV-1 p6 gag protein to Phe causes the formation of a hydrophobic patch, enhances membrane association, and polyubiquitination of Gag. Viruses 2014, 6, 3738–3765. [Google Scholar] [CrossRef] [PubMed]
- Hahn, S.; Setz, C.; Wild, J.; Schubert, U. The PTAP sequence within the p6 domain of human immunodeficiency virus type 1 Gag regulates its ubiquitination and MHC class I antigen presentation. J. Immunol. 2011, 186, 5706–5718. [Google Scholar] [CrossRef] [PubMed]
- Demirov, D.G.; Orenstein, J.M.; Freed, E.O. The late domain of human immunodeficiency virus type 1 p6 promotes virus release in a cell type-dependent manner. J. Virol. 2002, 76, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Hemonnot, B.; Cartier, C.; Gay, B.; Rebuffat, S.; Bardy, M.; Devaux, C.; Boyer, V.; Briant, L. The host cell MAP kinase ERK-2 regulates viral assembly and release by phosphorylating the p6gag protein of HIV-1. J. Biol. Chem. 2004, 279, 32426–32434. [Google Scholar] [CrossRef] [PubMed]
- Kudoh, A.; Takahama, S.; Sawasaki, T.; Ode, H.; Yokoyama, M.; Okayama, A.; Ishikawa, A.; Miyakawa, K.; Matsunaga, S.; Kimura, H.; et al. The phosphorylation of HIV-1 Gag by atypical protein kinase C facilitates viral infectivity by promoting Vpr incorporation into virions. Retrovirology 2014, 11. [Google Scholar] [CrossRef] [PubMed]
- Glushakova, S.; Baibakov, B.; Margolis, L.B.; Zimmerberg, J. Infection of human tonsil histocultures: A model for HIV pathogenesis. Nat. Med. 1995, 1, 1320–1322. [Google Scholar] [CrossRef] [PubMed]
- Glushakova, S.; Baibakov, B.; Zimmerberg, J.; Margolis, L.B. Experimental HIV infection of human lymphoid tissue: Correlation of CD4+ T cell depletion and virus syncytium-inducing/non-syncytium-inducing phenotype in histocultures inoculated with laboratory strains and patient isolates of HIV type 1. AIDS Res. Hum. Retrovir. 1997, 13, 461–471. [Google Scholar] [CrossRef] [PubMed]
- Jager, S.; Gottwein, E.; Krausslich, H.G. Ubiquitination of human immunodeficiency virus type 1 Gag is highly dependent on Gag membrane association. J. Virol. 2007, 81, 9193–9201. [Google Scholar] [CrossRef] [PubMed]
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef] [PubMed]
- AIDA, version 4.22.034; Raytest: Straubenhardt, Germany, 2008.
- Votteler, J.; Iavnilovitch, E.; Fingrut, O.; Shemesh, V.; Taglicht, D.; Erez, O.; Sorgel, S.; Walther, T.; Bannert, N.; Schubert, U.; et al. Exploring the functional interaction between POSH and ALIX and the relevance to HIV-1 release. BMC Biochem. 2009, 10. [Google Scholar] [CrossRef] [PubMed]
- Adachi, A.; Gendelman, H.E.; Koenig, S.; Folks, T.; Willey, R.; Rabson, A.; Martin, M.A. Production of acquired immunodeficiency syndrome-associated retrovirus in human and nonhuman cells transfected with an infectious molecular clone. J. Virol. 1986, 59, 284–291. [Google Scholar] [PubMed]
- Khan, M.A.; Aberham, C.; Kao, S.; Akari, H.; Gorelick, R.; Bour, S.; Strebel, K. Human immunodeficiency virus type 1 Vif protein is packaged into the nucleoprotein complex through an interaction with viral genomic RNA. J. Virol. 2001, 75, 7252–7265. [Google Scholar] [CrossRef] [PubMed]
- Goldwich, A.; Hahn, S.S.; Schreiber, S.; Meier, S.; Kampgen, E.; Wagner, R.; Lutz, M.B.; Schubert, U. Targeting HIV-1 Gag into the defective ribosomal product pathway enhances MHC class I antigen presentation and CD8+ T cell activation. J. Immunol. 2008, 180, 372–382. [Google Scholar] [CrossRef] [PubMed]
- FCS Express, version 3.00.0819; Professional Network; De novo Software: Los Angeles, CA, USA, 2001.
- Willey, R.L.; Smith, D.H.; Lasky, L.A.; Theodore, T.S.; Earl, P.L.; Moss, B.; Capon, D.J.; Martin, M.A. In vitro mutagenesis identifies a region within the envelope gene of the human immunodeficiency virus that is critical for infectivity. J. Virol. 1988, 62, 139–147. [Google Scholar] [PubMed]
- Solbak, S.M.; Reksten, T.R.; Hahn, F.; Wray, V.; Henklein, P.; Halskau, O.; Schubert, U.; Fossen, T. HIV-1 p6—A structured to flexible multifunctional membrane-interacting protein. Biochim. Biophys. Acta 2013, 1828, 816–823. [Google Scholar] [CrossRef] [PubMed]
- Kondo, E.; Gottlinger, H.G. A conserved LXXLF sequence is the major determinant in p6gag required for the incorporation of human immunodeficiency virus type 1 Vpr. J. Virol. 1996, 70, 159–164. [Google Scholar] [PubMed]
- Fossen, T.; Wray, V.; Bruns, K.; Rachmat, J.; Henklein, P.; Tessmer, U.; Maczurek, A.; Klinger, P.; Schubert, U. Solution structure of the human immunodeficiency virus type 1 p6 protein. J. Biol. Chem. 2005, 280, 42515–42527. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Cohen, S.N. Tsg101: A novel tumor susceptibility gene isolated by controlled homozygous functional knockout of allelic loci in mammalian cells. Cell 1996, 85, 319–329. [Google Scholar] [CrossRef]
- Los Alamos National Laboratory. Available online: http://www.hiv.lanl.gov (accessed on 1 April 2016).
- Degasperi, A.; Birtwistle, M.R.; Volinsky, N.; Rauch, J.; Kolch, W.; Kholodenko, B.N. Evaluating strategies to normalise biological replicates of western blot data. PLoS ONE 2014, 9, e87293. [Google Scholar] [CrossRef] [PubMed]
- Ott, D.E.; Coren, L.V.; Sowder, R.C., 2nd; Adams, J.; Schubert, U. Retroviruses have differing requirements for proteasome function in the budding process. J. Virol. 2003, 77, 3384–3393. [Google Scholar] [CrossRef] [PubMed]
- Martin-Serrano, J.; Perez-Caballero, D.; Bieniasz, P.D. Context-dependent effects of L domains and ubiquitination on viral budding. J. Virol. 2004, 78, 5554–5563. [Google Scholar] [CrossRef] [PubMed]
- Ciechanover, A. Proteolysis: From the lysosome to ubiquitin and the proteasome. Nat. Rev. Mol. Cell Biol. 2005, 6, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Schubert, U.; Anton, L.C.; Gibbs, J.; Norbury, C.C.; Yewdell, J.W.; Bennink, J.R. Rapid degradation of a large fraction of newly synthesized proteins by proteasomes. Nature 2000, 404, 770–774. [Google Scholar] [PubMed]
- Porgador, A.; Yewdell, J.W.; Deng, Y.; Bennink, J.R.; Germain, R.N. Localization, quantitation, and in situ detection of specific peptide-MHC class I complexes using a monoclonal antibody. Immunity 1997, 6, 715–726. [Google Scholar] [CrossRef]
- Rotzschke, O.; Falk, K.; Stevanovic, S.; Jung, G.; Walden, P.; Rammensee, H.G. Exact prediction of a natural T cell epitope. Eur. J. Immunol. 1991, 21, 2891–2894. [Google Scholar] [CrossRef] [PubMed]
- York, I.A.; Chang, S.C.; Saric, T.; Keys, J.A.; Favreau, J.M.; Goldberg, A.L.; Rock, K.L. The ER aminopeptidase ERAP1 enhances or limits antigen presentation by trimming epitopes to 8–9 residues. Nat. Immunol. 2002, 3, 1177–1184. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Sykes, B.D.; Richards, F.M. The chemical shift index: A fast and simple method for the assignment of protein secondary structure through NMR spectroscopy. Biochemistry 1992, 31, 1647–1651. [Google Scholar] [CrossRef] [PubMed]
- Bruns, K.; Fossen, T.; Wray, V.; Henklein, P.; Tessmer, U.; Schubert, U. Structural characterization of the HIV-1 Vpr N terminus: Evidence of cis/trans-proline isomerism. J. Biol. Chem. 2003, 278, 43188–43201. [Google Scholar] [CrossRef] [PubMed]
- Wei, M.; Zhao, X.; Liu, M.; Huang, Z.; Xiao, Y.; Niu, M.; Shao, Y.; Kleiman, L. Inhibition of HIV-1 assembly by coiled-coil domain containing protein 8 in human cells. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Graf, M.; Bojak, A.; Deml, L.; Bieler, K.; Wolf, H.; Wagner, R. Concerted action of multiple cis-acting sequences is required for Rev dependence of late human immunodeficiency virus type 1 gene expression. J. Virol. 2000, 74, 10822–10826. [Google Scholar] [CrossRef] [PubMed]
- Deml, L.; Bojak, A.; Steck, S.; Graf, M.; Wild, J.; Schirmbeck, R.; Wolf, H.; Wagner, R. Multiple effects of codon usage optimization on expression and immunogenicity of DNA candidate vaccines encoding the human immunodeficiency virus type 1 Gag protein. J. Virol. 2001, 75, 10991–11001. [Google Scholar] [CrossRef] [PubMed]
- Freed, E.O.; Orenstein, J.M.; Buckler-White, A.J.; Martin, M.A. Single amino acid changes in the human immunodeficiency virus type 1 matrix protein block virus particle production. J. Virol. 1994, 68, 5311–5320. [Google Scholar] [PubMed]
- Spearman, P.; Wang, J.J.; Vander Heyden, N.; Ratner, L. Identification of human immunodeficiency virus type 1 Gag protein domains essential to membrane binding and particle assembly. J. Virol. 1994, 68, 3232–3242. [Google Scholar] [PubMed]
- Strack, B.; Calistri, A.; Accola, M.A.; Palu, G.; Gottlinger, H.G. A role for ubiquitin ligase recruitment in retrovirus release. Proc. Natl. Acad. Sci. USA 2000, 97, 13063–13068. [Google Scholar] [CrossRef] [PubMed]
- Schubert, U.; Ott, D.E.; Chertova, E.N.; Welker, R.; Tessmer, U.; Princiotta, M.F.; Bennink, J.R.; Krausslich, H.G.; Yewdell, J.W. Proteasome inhibition interferes with gag polyprotein processing, release, and maturation of HIV-1 and HIV-2. Proc. Natl. Acad. Sci. USA 2000, 97, 13057–13062. [Google Scholar] [CrossRef] [PubMed]
- Patnaik, A.; Chau, V.; Wills, J.W. Ubiquitin is part of the retrovirus budding machinery. Proc. Natl. Acad. Sci. USA 2000, 97, 13069–13074. [Google Scholar] [CrossRef] [PubMed]
- Gottwein, E.; Jager, S.; Habermann, A.; Krausslich, H.G. Cumulative mutations of ubiquitin acceptor sites in human immunodeficiency virus type 1 gag cause a late budding defect. J. Virol. 2006, 80, 6267–6275. [Google Scholar] [CrossRef] [PubMed]
- Sette, P.; Nagashima, K.; Piper, R.C.; Bouamr, F. Ubiquitin conjugation to Gag is essential for ESCRT-mediated HIV-1 budding. Retrovirology 2013, 10. [Google Scholar] [CrossRef] [PubMed]
- Zhadina, M.; McClure, M.O.; Johnson, M.C.; Bieniasz, P.D. Ubiquitin-dependent virus particle budding without viral protein ubiquitination. Proc. Natl. Acad. Sci. USA 2007, 104, 20031–20036. [Google Scholar] [CrossRef] [PubMed]
- Zhadina, M.; Bieniasz, P.D. Functional interchangeability of late domains, late domain cofactors and ubiquitin in viral budding. PLoS Pathog. 2010, 6, e1001153. [Google Scholar] [CrossRef] [PubMed]
- Salgado, G.F.; Vogel, A.; Marquant, R.; Feller, S.E.; Bouaziz, S.; Alves, I.D. The role of membranes in the organization of HIV-1 Gag p6 and Vpr: p6 shows high affinity for membrane bilayers which substantially increases the interaction between p6 and Vpr. J. Med. Chem. 2009, 52, 7157–7162. [Google Scholar] [CrossRef] [PubMed]
- Datta, S.A.; Curtis, J.E.; Ratcliff, W.; Clark, P.K.; Crist, R.M.; Lebowitz, J.; Krueger, S.; Rein, A. Conformation of the HIV-1 Gag protein in solution. J. Mol. Biol. 2007, 365, 812–824. [Google Scholar] [CrossRef] [PubMed]
- Datta, S.A.; Heinrich, F.; Raghunandan, S.; Krueger, S.; Curtis, J.E.; Rein, A.; Nanda, H. HIV-1 Gag extension: Conformational changes require simultaneous interaction with membrane and nucleic acid. J. Mol. Biol. 2011, 406, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Munro, J.B.; Nath, A.; Farber, M.; Datta, S.A.; Rein, A.; Rhoades, E.; Mothes, W. A conformational transition observed in single HIV-1 Gag molecules during in vitro assembly of virus-like particles. J. Virol. 2014, 88, 3577–3585. [Google Scholar] [CrossRef] [PubMed]
- Kempf, N.; Postupalenko, V.; Bora, S.; Didier, P.; Arntz, Y.; de Rocquigny, H.; Mely, Y. The HIV-1 nucleocapsid protein recruits negatively charged lipids to ensure its optimal binding to lipid membranes. J. Virol. 2015, 89, 1756–1767. [Google Scholar] [CrossRef] [PubMed]
- Ko, L.J.; Yu, F.H.; Huang, K.J.; Wang, C.T. HIV-1 matrix domain removal ameliorates virus assembly and processing defects incurred by positive nucleocapsid charge elimination. FEBS Openbio 2015, 5, 283–291. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Gorelick, R.J.; Levin, J.G. Selection of fully processed HIV-1 nucleocapsid protein is required for optimal nucleic acid chaperone activity in reverse transcription. Virus Res. 2014, 193, 52–64. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Naiyer, N.; Mitra, M.; Li, J.; Williams, M.C.; Rouzina, I.; Gorelick, R.J.; Wu, Z.; Musier-Forsyth, K. Distinct nucleic acid interaction properties of HIV-1 nucleocapsid protein precursor NCp15 explain reduced viral infectivity. Nucleic Acids Res. 2014, 42, 7145–7159. [Google Scholar] [CrossRef] [PubMed]
- Martins, A.N.; Waheed, A.A.; Ablan, S.D.; Huang, W.; Newton, A.; Petropoulos, C.J.; Brindeiro, R.M.; Freed, E.O. Elucidation of the molecular mechanism driving duplication of the HIV-1 PTAP late domain. J. Virol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Chukkapalli, V.; Oh, S.J.; Ono, A. Opposing mechanisms involving RNA and lipids regulate HIV-1 Gag membrane binding through the highly basic region of the matrix domain. Proc. Natl. Acad. Sci. USA 2010, 107, 1600–1605. [Google Scholar] [CrossRef] [PubMed]
- Alfadhli, A.; Still, A.; Barklis, E. Analysis of human immunodeficiency virus type 1 matrix binding to membranes and nucleic acids. J. Virol. 2009, 83, 12196–12203. [Google Scholar] [CrossRef] [PubMed]
- Purohit, P.; Dupont, S.; Stevenson, M.; Green, M.R. Sequence-specific interaction between HIV-1 matrix protein and viral genomic RNA revealed by in vitro genetic selection. RNA 2001, 7, 576–584. [Google Scholar] [CrossRef] [PubMed]
- Cimarelli, A.; Luban, J. Translation elongation factor 1-alpha interacts specifically with the human immunodeficiency virus type 1 Gag polyprotein. J. Virol. 1999, 73, 5388–5401. [Google Scholar] [PubMed]
- Hearps, A.C.; Wagstaff, K.M.; Piller, S.C.; Jans, D.A. The N-terminal basic domain of the HIV-1 matrix protein does not contain a conventional nuclear localization sequence but is required for DNA binding and protein self-association. Biochemistry 2008, 47, 2199–2210. [Google Scholar] [CrossRef] [PubMed]
- Yuan, B.; Campbell, S.; Bacharach, E.; Rein, A.; Goff, S.P. Infectivity of Moloney murine leukemia virus defective in late assembly events is restored by late assembly domains of other retroviruses. J. Virol. 2000, 74, 7250–7260. [Google Scholar] [CrossRef] [PubMed]
- Yuan, B.; Li, X.; Goff, S.P. Mutations altering the moloney murine leukemia virus p12 Gag protein affect virion production and early events of the virus life cycle. EMBO J. 1999, 18, 4700–4710. [Google Scholar] [CrossRef] [PubMed]
- Zhai, Q.; Landesman, M.B.; Robinson, H.; Sundquist, W.I.; Hill, C.P. Identification and structural characterization of the ALIX-binding late domains of simian immunodeficiency virus SIVmac239 and SIVagmTan-1. J. Virol. 2011, 85, 632–637. [Google Scholar] [CrossRef] [PubMed]










© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Friedrich, M.; Setz, C.; Hahn, F.; Matthaei, A.; Fraedrich, K.; Rauch, P.; Henklein, P.; Traxdorf, M.; Fossen, T.; Schubert, U. Glutamic Acid Residues in HIV-1 p6 Regulate Virus Budding and Membrane Association of Gag. Viruses 2016, 8, 117. https://0-doi-org.brum.beds.ac.uk/10.3390/v8040117
Friedrich M, Setz C, Hahn F, Matthaei A, Fraedrich K, Rauch P, Henklein P, Traxdorf M, Fossen T, Schubert U. Glutamic Acid Residues in HIV-1 p6 Regulate Virus Budding and Membrane Association of Gag. Viruses. 2016; 8(4):117. https://0-doi-org.brum.beds.ac.uk/10.3390/v8040117
Chicago/Turabian StyleFriedrich, Melanie, Christian Setz, Friedrich Hahn, Alina Matthaei, Kirsten Fraedrich, Pia Rauch, Petra Henklein, Maximilian Traxdorf, Torgils Fossen, and Ulrich Schubert. 2016. "Glutamic Acid Residues in HIV-1 p6 Regulate Virus Budding and Membrane Association of Gag" Viruses 8, no. 4: 117. https://0-doi-org.brum.beds.ac.uk/10.3390/v8040117