
Engineering Hepadnaviruses as Reporter-Expressing Vectors: Recent Progress and Future Perspectives

Abstract
:1. Introduction
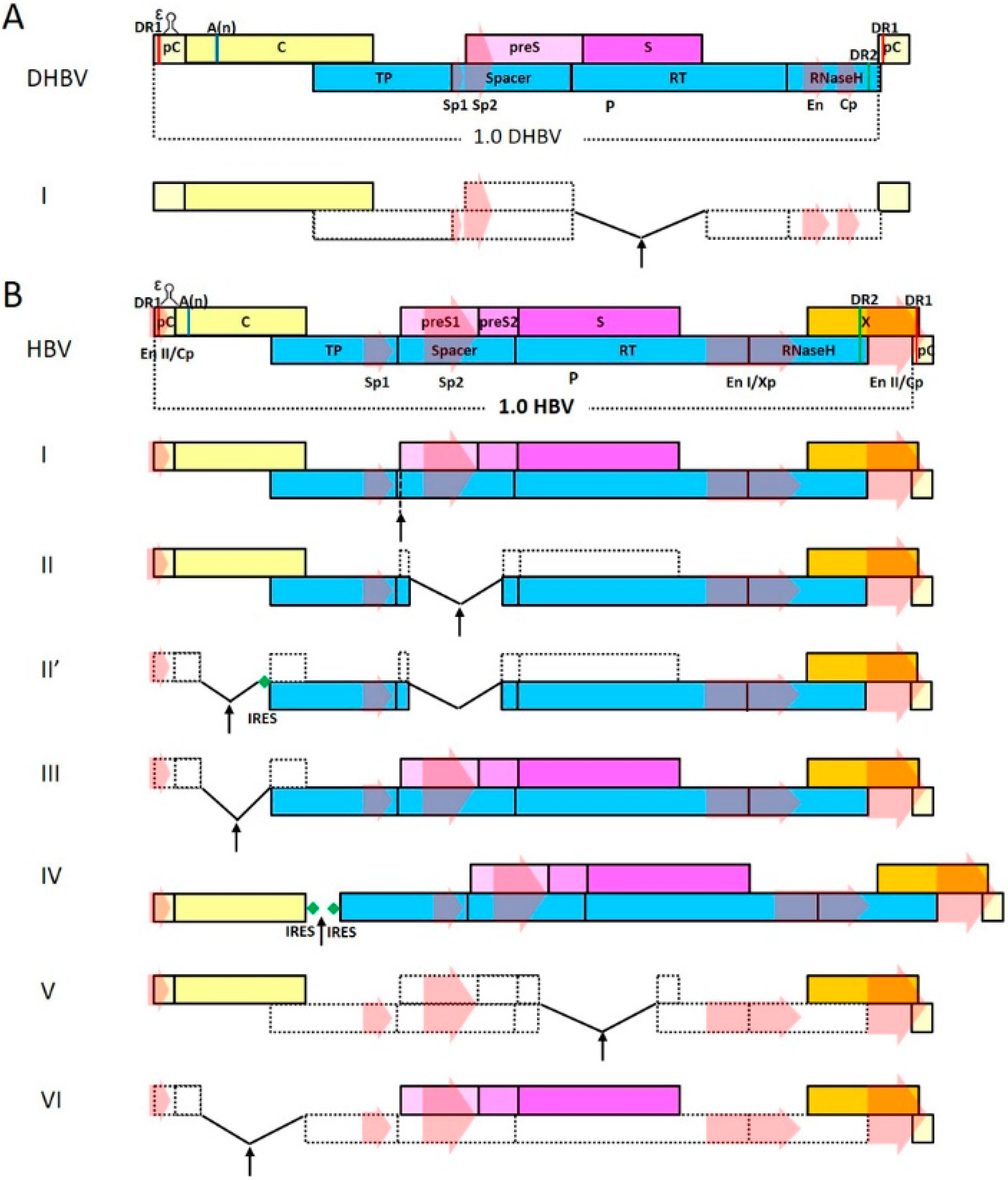
2. Genome Organization and Life Cycle of Hepadnaviruses
3. Hepadnavirus-Specific Difficulties for the Design and Development of Viral Vectors
4. Hepadnavirus-Derived Viral Vectors
4.1. Vectors that Encode Functional Polymerase
4.1.1. Vectors that Use Polymerase Spacer Region for Cargo Insertion
4.1.2. Vectors that Use Core Region for Cargo Insertion
4.2. Vectors that Do Not Encode Functional Polymerase
4.3. Comparison of Vector Designs from the Perspective of Potential Applications
5. Future Perspectives on Recombinant Hepadnavirus Vector Design
6. Future Perspectives on Applications of Recombinant Hepadnavirus Vector
Acknowledgments
Conflicts of Interest
References
- Liang, T.J. Hepatitis B: The virus and disease. Hepatology 2009, 49, S13–S21. [Google Scholar] [CrossRef] [PubMed]
- Seeger, C.; Mason, W.S. Hepatitis B virus biology. Microbiol. Mol. Biol. Rev. 2000, 64, 51–68. [Google Scholar] [CrossRef] [PubMed]
- Seeger, C.; Mason, W.S. Molecular biology of hepatitis B virus infection. Virology 2015, 479, 672–686. [Google Scholar] [CrossRef] [PubMed]
- Littlejohn, M.; Locarnini, S.; Yuen, L. Origins and evolution of hepatitis B virus and hepatitis D virus. Cold Spring Harb. Perspect. Med. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- MacLachlan, J.H.; Cowie, B.C. Hepatitis B virus epidemiology. Cold Spring Harb. Perspect. Med. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Drexler, J.F.; Geipel, A.; Konig, A.; Corman, V.M.; van Riel, D.; Leijten, L.M.; Bremer, C.M.; Rasche, A.; Cottontail, V.M.; Maganga, G.D.; et al. Bats carry pathogenic hepadnaviruses antigenically related to hepatitis B virus and capable of infecting human hepatocytes. Proc. Natl. Acad. Sci. USA 2013, 110, 16151–16156. [Google Scholar] [CrossRef] [PubMed]
- Hahn, C.M.; Iwanowicz, L.R.; Cornman, R.S.; Conway, C.M.; Winton, J.R.; Blazer, V.S. Characterization of a novel hepadnavirus in the White Sucker (Catostomus commersonii) from the Great Lakes Region of the United States. J. Virol. 2015, 89, 11801–11811. [Google Scholar] [CrossRef] [PubMed]
- Suh, A.; Brosius, J.; Schmitz, J.; Kriegs, J.O. The genome of a Mesozoic paleovirus reveals the evolution of hepatitis B viruses. Nat. Commun. 2013, 4. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Pan, S.; Yang, H.; Bai, W.; Shen, Z.; Liu, J.; Xie, Y. The first full-length endogenous hepadnaviruses: Identification and analysis. J. Virol. 2012, 86, 9510–9513. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, C.; Feschotte, C. Genomic fossils calibrate the long-term evolution of hepadnaviruses. PLoS Biol. 2010, 8. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, C.; Meik, J.M.; Dashevsky, D.; Card, D.C.; Castoe, T.A.; Schaack, S. Endogenous hepadnaviruses, bornaviruses and circoviruses in snakes. Proc. R. Soc. London B Bio. Sci. 2014, 281. [Google Scholar] [CrossRef] [PubMed]
- Watashi, K.; Wakita, T. Hepatitis B virus and hepatitis D virus entry, species specificity, and tissue tropism. Cold Spring Harb. Perspect. Med. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Winer, B.Y.; Ploss, A. Determinants of hepatitis B and delta virus host tropism. Curr. Opin. Virol. 2015, 13, 109–116. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization Hepatitis B. World Health Organization Fact Sheet 204 (Revised July 2013). Available online: http://www.who.int/mediacentre/factsheets/fs204/en/ (accessed on 10 December 2013).
- Mason, W.S. Animal models and the molecular biology of hepadnavirus infection. Cold Spring Harb. Perspect. Med. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.M.; Seeger, C. Hepadnavirus genome replication and persistence. Cold Spring Harb. Perspect. Med. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Protzer, U.; Nassal, M.; Chiang, P.W.; Kirschfink, M.; Schaller, H. Interferon gene transfer by a hepatitis B virus vector efficiently suppresses wild-type virus infection. Proc. Natl. Acad. Sci. USA 1999, 96, 10818–10823. [Google Scholar] [CrossRef] [PubMed]
- Ho, T.C.; Jeng, K.S.; Hu, C.P.; Chang, C. Effects of genomic length on translocation of hepatitis B virus polymerase-linked oligomer. J. Virol. 2000, 74, 9010–9018. [Google Scholar] [CrossRef] [PubMed]
- Bartenschlager, R.; Schaller, H. Hepadnaviral assembly is initiated by polymerase binding to the encapsidation signal in the viral RNA genome. EMBO J. 1992, 11, 3413–3420. [Google Scholar] [PubMed]
- Feitelson, M.A.; Bonamassa, B.; Arzumanyan, A. The roles of hepatitis B virus-encoded X protein in virus replication and the pathogenesis of chronic liver disease. Expert Opin. Ther. Targets 2014, 18, 293–306. [Google Scholar] [CrossRef] [PubMed]
- Belloni, L.; Pollicino, T.; de Nicola, F.; Guerrieri, F.; Raffa, G.; Fanciulli, M.; Raimondo, G.; Levrero, M. Nuclear HBx binds the HBV minichromosome and modifies the epigenetic regulation of cccDNA function. Proc. Natl. Acad. Sci. USA 2009, 106, 19975–19979. [Google Scholar] [CrossRef] [PubMed]
- Lucifora, J.; Arzberger, S.; Durantel, D.; Belloni, L.; Strubin, M.; Levrero, M.; Zoulim, F.; Hantz, O.; Protzer, U. Hepatitis B virus X protein is essential to initiate and maintain virus replication after infection. J. Hepatol. 2011, 55, 996–1003. [Google Scholar] [CrossRef] [PubMed]
- Tsuge, M.; Hiraga, N.; Akiyama, R.; Tanaka, S.; Matsushita, M.; Mitsui, F.; Abe, H.; Kitamura, S.; Hatakeyama, T.; Kimura, T.; et al. HBx protein is indispensable for development of viraemia in human hepatocyte chimeric mice. J. Gen. Virol. 2010, 91, 1854–1864. [Google Scholar] [CrossRef] [PubMed]
- Thung, S.N.; Gerber, M.A.; Purcell, R.H.; London, W.T.; Mihalik, K.B.; Popper, H. Animal model of human disease. Chimpanzee carriers of hepatitis B virus. Chimpanzee hepatitis B carriers. Am. J. Pathol. 1981, 105, 328–332. [Google Scholar] [PubMed]
- Dandri, M.; Burda, M.R.; Zuckerman, D.M.; Wursthorn, K.; Matschl, U.; Pollok, J.M.; Rogiers, X.; Gocht, A.; Kock, J.; Blum, H.E.; et al. Chronic infection with hepatitis B viruses and antiviral drug evaluation in uPA mice after liver repopulation with tupaia hepatocytes. J. Hepatol. 2005, 42, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Dandri, M.; Burda, M.R.; Torok, E.; Pollok, J.M.; Iwanska, A.; Sommer, G.; Rogiers, X.; Rogler, C.E.; Gupta, S.; Will, H.; et al. Repopulation of mouse liver with human hepatocytes and in vivo infection with hepatitis B virus. Hepatology 2001, 33, 981–988. [Google Scholar] [CrossRef] [PubMed]
- Kock, J.; Nassal, M.; MacNelly, S.; Baumert, T.F.; Blum, H.E.; von Weizsacker, F. Efficient infection of primary tupaia hepatocytes with purified human and woolly monkey hepatitis B virus. J. Virol. 2001, 75, 5084–5089. [Google Scholar] [CrossRef] [PubMed]
- Gripon, P.; Rumin, S.; Urban, S.; le Seyec, J.; Glaise, D.; Cannie, I.; Guyomard, C.; Lucas, J.; Trepo, C.; Guguen-Guillouzo, C. Infection of a human hepatoma cell line by hepatitis B virus. Proc. Natl. Acad. Sci. USA 2002, 99, 15655–15660. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Zhong, G.; Xu, G.; He, W.; Jing, Z.; Gao, Z.; Huang, Y.; Qi, Y.; Peng, B.; Wang, H.; et al. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. eLife 2012, 1. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.J.; Hirsch, R.C.; Ganem, D.; Varmus, H.E. Effects of insertional and point mutations on the functions of the duck hepatitis B virus polymerase. J. Virol. 1990, 64, 5553–5558. [Google Scholar] [PubMed]
- Chaisomchit, S.; Tyrrell, D.L.; Chang, L.J. Development of replicative and nonreplicative hepatitis B virus vectors. Gene Ther. 1997, 4, 1330–1340. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Kaneko, S.; Honda, M.; Kobayashi, K. Approach to establishing a liver targeting gene therapeutic vector using naturally occurring defective hepatitis B viruses devoid of immunogenic T cell epitope. Virus Res. 2002, 85, 187–197. [Google Scholar] [CrossRef]
- Yoo, J.; Rho, J.; Lee, D.; Shin, S.; Jung, G. Hepatitis B virus vector carries a foreign gene into liver cells in vitro. Virus Genes 2002, 24, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Deng, Q.; Mancini-Bourgine, M.; Zhang, X.; Cumont, M.C.; Zhu, R.; Lone, Y.C.; Michel, M.L. Hepatitis B virus as a gene delivery vector activating foreign antigenic T cell response that abrogates viral expression in mouse models. Hepatology 2009, 50, 1380–1391. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhu, K.; Bai, W.; Jia, B.; Hu, H.; Zhou, D.; Zhang, X.; Zhang, X.; Xie, Y.; Bourgine, M.M.; et al. Adenoviral delivery of recombinant hepatitis B virus expressing foreign antigenic epitopes for immunotherapy of persistent viral infection. J. Virol. 2014, 88, 3004–3015. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wu, L.; Cheng, X.; Liu, S.; Li, B.; Li, H.; Kang, F.; Wang, J.; Xia, H.; Ping, C.; et al. Replication-competent infectious hepatitis B virus vectors carrying substantially sized transgenes by redesigned viral polymerase translation. PLoS ONE 2013, 8, e60306. [Google Scholar] [CrossRef] [PubMed]
- Hong, R.; Bai, W.; Zhai, J.; Liu, W.; Li, X.; Zhang, J.; Cui, X.; Zhao, X.; Ye, X.; Deng, Q.; et al. Novel recombinant hepatitis B virus vectors efficiently deliver protein and RNA encoding genes into primary hepatocytes. J. Virol. 2013, 87, 6615–6624. [Google Scholar] [CrossRef] [PubMed]
- Untergasser, A.; Protzer, U. Hepatitis B virus-based vectors allow the elimination of viral gene expression and the insertion of foreign promoters. Hum. Gene Ther. 2004, 15, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Cheng, X.; Guo, Z.; Wang, Z.; Li, D.; Kang, F.; Li, H.; Li, B.; Cao, Z.; Nassal, M.; et al. Truncated active human matrix metalloproteinase-8 delivered by a chimeric adenovirus-hepatitis B virus vector ameliorates rat liver cirrhosis. PLoS ONE 2013, 8, e53392. [Google Scholar] [CrossRef] [PubMed]
- Nishitsuji, H.; Ujino, S.; Shimizu, Y.; Harada, K.; Zhang, J.; Sugiyama, M.; Mizokami, M.; Shimotohno, K. Novel reporter system to monitor early stages of the hepatitis B virus life cycle. Cancer Sci. 2015, 106, 1616–1624. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.; Kao, Y.F.; Brown, C.M. Translation of the first upstream ORF in the hepatitis B virus pregenomic RNA modulates translation at the core and polymerase initiation codons. Nucleic Acids Res. 2005, 33, 1169–1181. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Publication | Virus Base | Required trans-Complementation 1 | Cargo Insertion Site and Insertion Strategy | Tested Cargo(s) (Length) | Replication Evidence | Replication Efficiency | Virion Formation Evidence 2 | Virion Formation Efficiency | Virion Infectivity Evidence | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Recombinant vectors that express functional polymerase | ||||||||||||||||||
| Chang et al., 1990 [30] | DHBV | preS/S | Cargo ORF inserted in-frame in Pol spacer between preS and S with own ATG but no stop codon | Protein A (369) | EPA, Southern blot | Comparable | N.D. | N.D. | N.D. | |||||||||
| Chaisomchit et al., 1997 [31] | HBV | None | Cargo ORF inserted in-frame in Pol spacer between Sp1 and preS1 start codon with own start but no stop codon | HIV Tat (267) | EPA | Severely reduced | Capture by S antibodies followed by Southern blot | Severely reduced | N.D. | |||||||||
| Wang et al., 2002 [32] | HBV | C | Cargo ORF replaced C ORF between ε/A(n) and Pol start codon, fused to remaining N-terminal of C | Flag (48) | Southern blot | Severely reduced | Density gradient ultracentrifuge followed by Southern blot and PCR | N.D. | PHH | |||||||||
| Yoo et al., 2002 [33] | HBV | C | Cargo ORF replaced C ORF between ε/A(n) and Pol start codon | GFP | EPA, Southern blot | Severely reduced | Density gradient ultracentrifuge and capture by S antibodies followed by Southern blot | N.D. | PHH, HepG2 3 | |||||||||
| Deng et al., 2009 [34] Wang et al., 2014 [35] | HBV | C | Cargo ORF replaced C ORF between ε/A(n) and Pol start codon, fused to remaining N-terminal of C. Kozak sequences of Pol were optimized. | Peptide (180) | Southern blot | Increased | Density gradient ultracentrifuge followed by dot blot | Reduced | PTH | |||||||||
| Wang et al., 2013 [36] | HBV | None | Cargo ORF inserted between separated C and Pol ORF with intervening short IRES | BsdR (399) GFP (720) | EPA, Southern blot | Comparable to severely reduced depending on cargo | Density gradient ultracentrifuge followed by Southern blot | Comparable to severely reduced depending on cargo | HepaRG | |||||||||
| Hong et al., 2013 [37] | HBV | preS1/preS2/S | Cargo sequences replaced 384 bp of Pol spacer (preS1/preS2) in-frame with preS1, without terminating Pol ORF. Start codon of preS1 mutated. | HIV Tat (207) DsRed (678) shRNA cassette (294) | Southern blot | Comparable to reduced depending on cargo | Capture by preS1 mAb followed by Southern blot | Comparable to reduced depending on cargo | PTH | |||||||||
| Bai et al., (submitted) | HBV | C preS1/preS2/S | Cargo sequences replaced C ORF between ε/A(n) and Pol start codon. Short IRES placed before Pol start codon and 384 bp of Pol spacer (preS1/preS2), same as above, deleted. C ORF prematurely terminated. | ZeoR (375) NanoLuc (522/606) 4 DsRed (678) GFP (747) shRNA cassette (294) | Southern blot | Comparable to reduced depending on cargo | Capture by preS1 mAb followed by Southern blot | Comparable to reduced depending on cargo | PTH | |||||||||
| Recombinant vectors that do not express functional polymerase | ||||||||||||||||||
| Chaisomchit et al., 1997 [31] | HBV | Pol | Cargo ORF inserted in-frame in Pol spacer between Sp1 and preS1 start codon with own start and stop codon | ZeoR (372) | EPA | Severely reduced | N.D. | N.D. | N.D. | |||||||||
| Protzer et al., 1999 [17] | DHBV | Pol preS/S | Cargo ORF replaced 558 bp of S ORF in-frame | GFP (733) Duck IFN (591) | N.D. | N.D. | Density gradient ultracentrifuge followed by dot blot | N.D. | PDH | |||||||||
| Ibid. | HBV | Pol preS1/preS2/S | Cargo ORF replaced 939 bp of S ORF in-frame | GFP (733) | N.D. | N.D. | Ibid. | N.D. | PHH | |||||||||
| Untergasser et al., 2004 [38] | HBV | All HBV ORFs | Cargo ORF replaced 939 nt of S ORF in-frame. All other HBV ORFs are prematurely terminated by mutation. Some constructs replaced ~311 nt of SP2 with exogenous promoters 366 nt or 575 nt long. | GFP (733) RLuc (942) | Southern blot | N.D. | Ibid. | Comparable | PHH | |||||||||
| Liu et al., 2013 [39] | HBV | All HBV ORFs | Cargo ORF replaced S ORF in-frame. All other HBV ORFs are prematurely terminated or nulled by mutation. | GFP RFP | Southern blot | Reduced | N.D. | N.D. | HepaRG | |||||||||
| Nishitsuji et al., 2015 [40] | HBV | Pol C | Cargo ORF with own start and stop codons replaced 562 nt of C ORF downstream of ε/A(n) and the N-terminal of P ORF | NanoLuc (513) | N.D. | N.D. | Density gradient ultracentrifuge followed by Southern blot | N.D. | PXB NTCP cell lines | |||||||||
| Representative Publication(s) | Obliterated ORF(s) | cccDNA Pool Expansion 1 | Progeny Virus Production 2 |
|---|---|---|---|
| Chaisomchit et al., 1997 [31] | None | Self-sufficient | Self-sufficient |
| Wang et al., 2013 [36] | |||
| Hong et al., 2013 [37] | S | Self-sufficient | Requires help |
| Wang et al., 2002 [32] | C | Requires help | Requires help |
| Yoo et al., 2002 [33] | |||
| Deng et al., 2009 [34] | |||
| Wang et al., 2014 [35] | |||
| Bai et al. (submitted) | C/S | Requires help | Requires help |
| Chaisomchit et al., 1997 [31] | P | Requires help | Requires help |
| Protzer et al., 1999 [17] | P/S | Requires help | Requires help |
| Chang et al., 1990 [30] | |||
| Nishitsuji et al., 2015 [40] | P/C | Requires help | Requires help |
| Untergasser et al., 2004 [38] | All | Requires help | Requires help |
| Liu et al., 2013 [39] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bai, W.; Cui, X.; Xie, Y.; Liu, J. Engineering Hepadnaviruses as Reporter-Expressing Vectors: Recent Progress and Future Perspectives. Viruses 2016, 8, 125. https://0-doi-org.brum.beds.ac.uk/10.3390/v8050125
Bai W, Cui X, Xie Y, Liu J. Engineering Hepadnaviruses as Reporter-Expressing Vectors: Recent Progress and Future Perspectives. Viruses. 2016; 8(5):125. https://0-doi-org.brum.beds.ac.uk/10.3390/v8050125
Chicago/Turabian StyleBai, Weiya, Xiaoxian Cui, Youhua Xie, and Jing Liu. 2016. "Engineering Hepadnaviruses as Reporter-Expressing Vectors: Recent Progress and Future Perspectives" Viruses 8, no. 5: 125. https://0-doi-org.brum.beds.ac.uk/10.3390/v8050125