
Stress Response and Translation Control in Rotavirus Infection


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Rotavirus
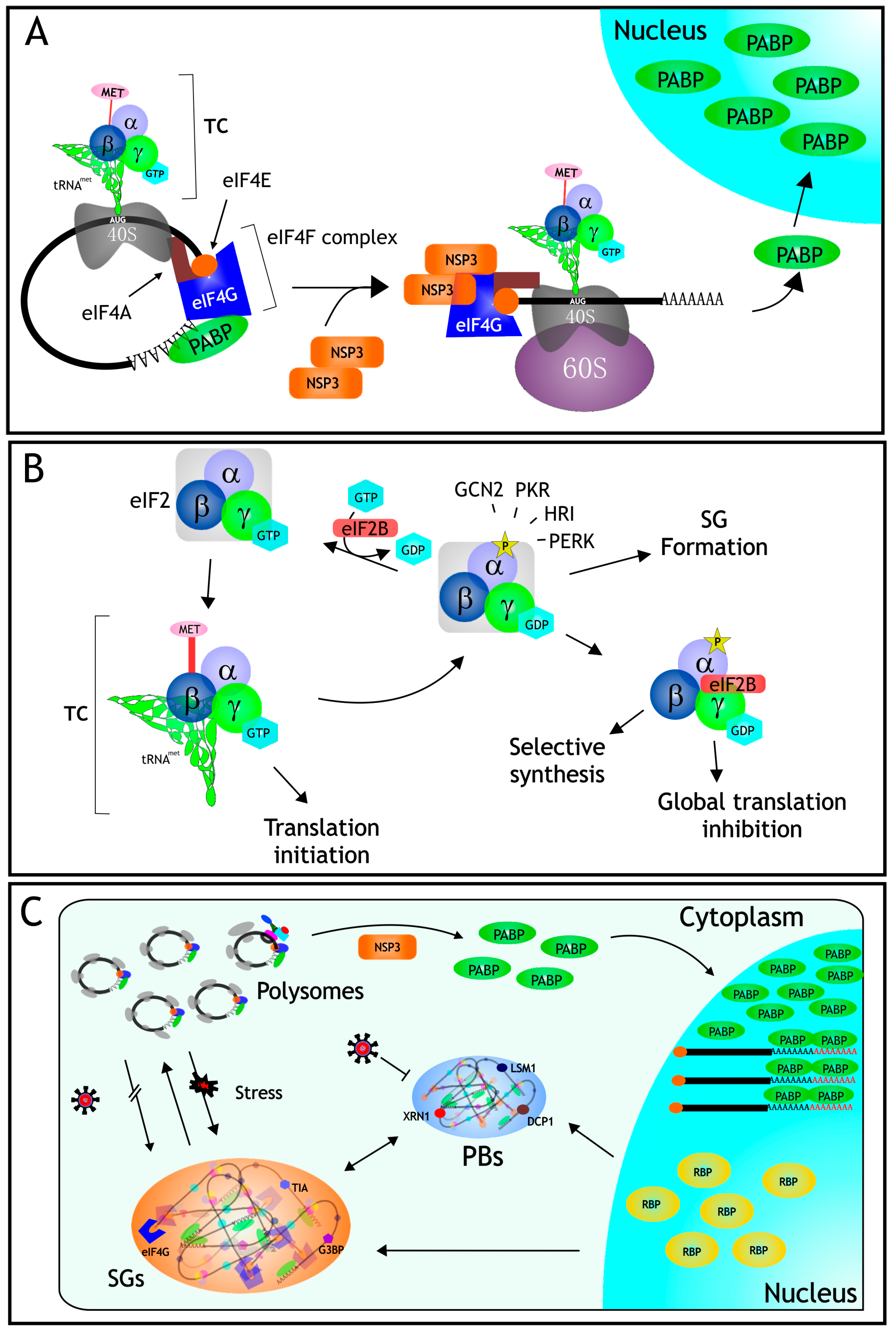
3. Protein Synthesis
4. mRNA Translation in Rotavirus Infected Cells
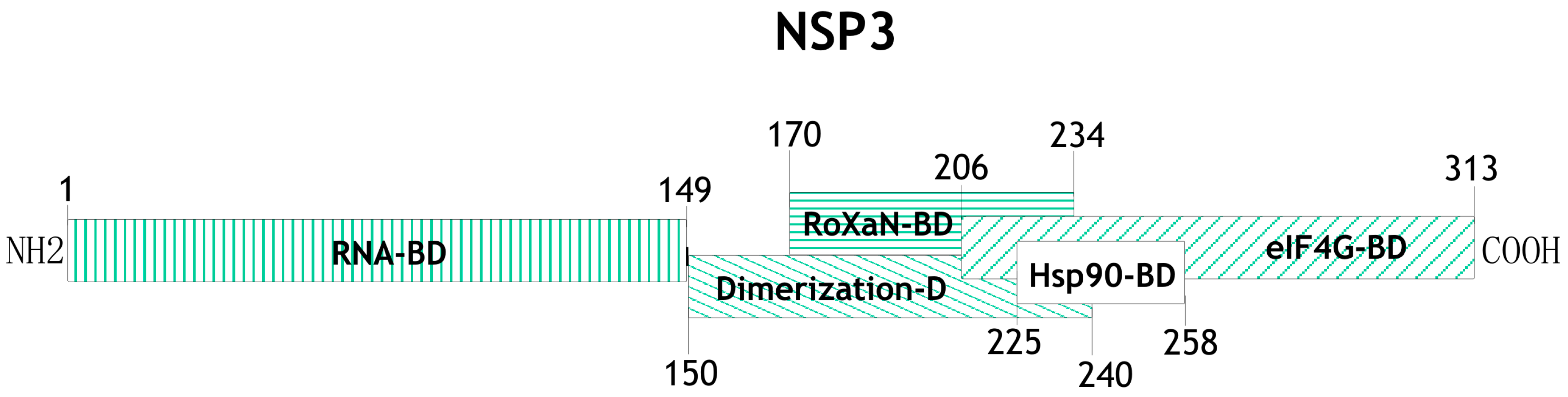
5. The Role of the Nonstructural Protein NSP3 during mRNA Translation
6. RNA Granules
6.1. Stress Granules
6.2. Processing Bodies
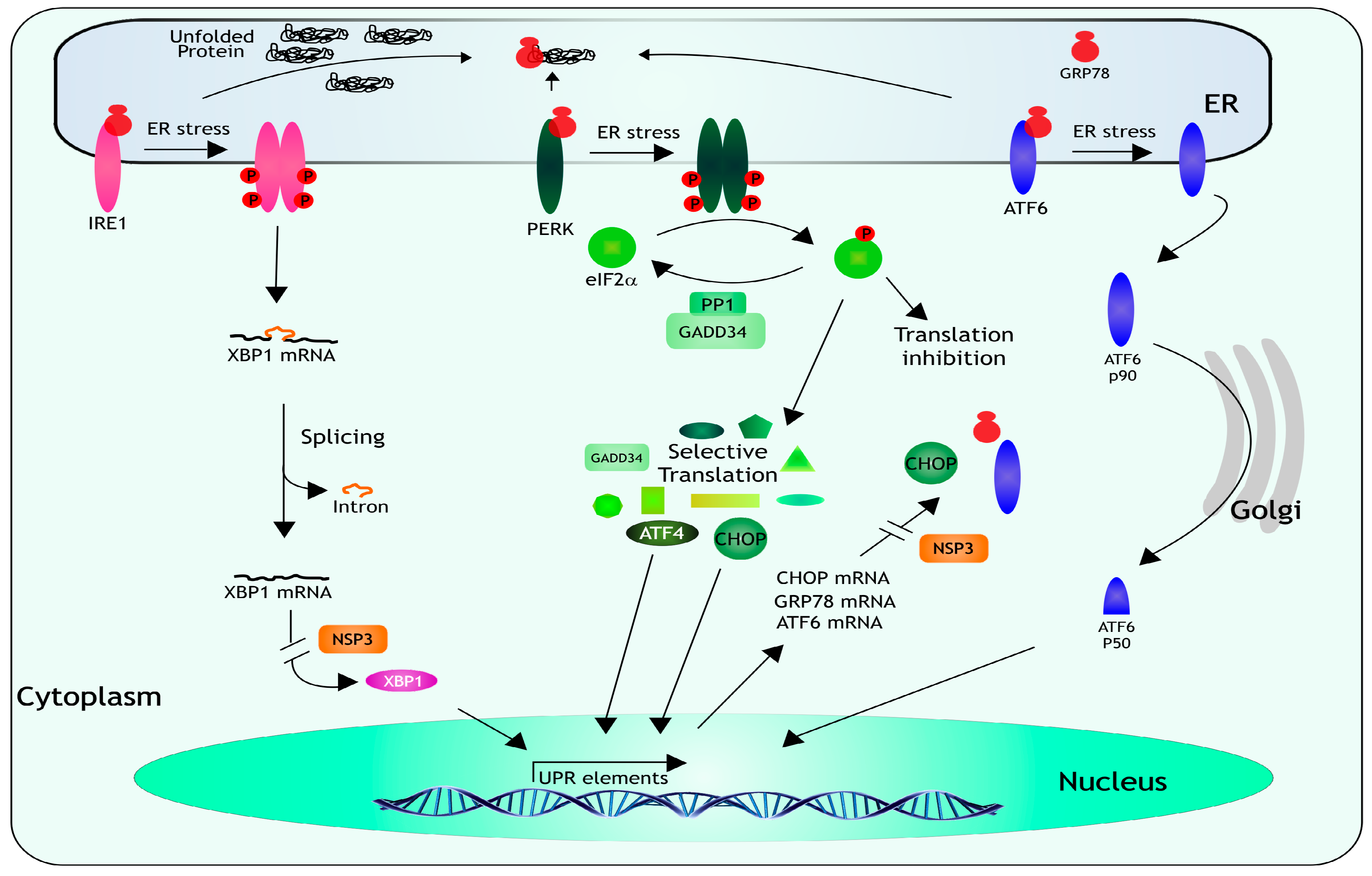
7. Unfolded Protein Response
8. Concluding Remarks
- How can viral and cellular mRNAs be efficiently translated although eIF2α is phosphorylated?
- Does NSP3 participate in the relocalization of PABP to the nucleus?
- Are there additional cellular proteins that interact with NSP3?
- Why does the knockdown of NSP3 result in an increased viral progeny?
- What is the cellular function of RoXaN?
- Which viral proteins are involved in preventing the formation of SGs and PBs?
- What is the function of the UPR proteins found in viroplasms?
- Which is the consequence of suppressing the UPR in rotavirus-infected cells?
Acknowledgments
Conflicts of Interest
Abbreviations
| ATF4 | Activating Transcription Factor 4 |
| ATF6 | Activating Transcription Factor 6 |
| CHOP | C/EBP Homologous Protein (CCAAT-Enhancer-Binding Protein Homologous Proteins) |
| DCP1 | Decapping Protein 1 |
| eIF2B | Eukaryotic Initiation Factor 2B |
| eIF2α | Eukaryotic Initiation Factor 2 Alpha Subunit |
| eIF4G | Eukaryotic Initiation Factor 4G |
| ER | Endoplasmic Reticulum |
| G3BP | GTPase Activating Protein (SH3 Domain) Binding Protein 1 |
| GADD34 | Growth Arrest and DNA Damage-Inducible Protein |
| GCN2 | General Control Nonderepressible 2 |
| GRP78 | 78 kDa Glucose-Regulated Protein |
| HRI | Heme-Regulated Kinase |
| IRE1 | Inositol-Requiring Enzyme-1 |
| LSM1 | U6 snRNA-Associated Sm-Like Protein |
| PABP | Poli(A)-Binding Protein |
| PBs | Processing Bodies |
| PERK | Protein Kinase-R-Like ER kinase |
| PKR | Protein Kinase-R |
| PP1 | Protein Phosphatase 1 |
| RBP | RNA Binding Protein |
| RoXaN | Rotavirus X Protein Associated with NSP3 |
| SGs | Stress Granules |
| TC | Ternary Complex |
| TIA | T-Cell intracellular Antigen 1 |
| UPR | Unfolded Protein Response |
| UTR | Untranslated Region |
| XBP1 | X-Box Binding Protein 1 |
| XRN1 | 5’–3’ Exoribonuclease 1 |
References
- Hetz, C.; Chevet, E.; Oakes, S.A. Proteostasis control by the unfolded protein response. Nat. Cell Biol. 2015, 17, 829–838. [Google Scholar] [CrossRef] [PubMed]
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Buchan, J.R. mRNP granules: Assembly, function, and connections with disease. RNA Biol. 2014, 11, 1019–1030. [Google Scholar] [CrossRef] [PubMed]
- Kedersha, N.; Ivanov, P.; Anderson, P. Stress granules and cell signaling: More than just a passing phase? Trends Biochem. Sci. 2013, 38, 494–506. [Google Scholar] [CrossRef] [PubMed]
- Lee, L.A.; Franzel, L.; Atwell, J.; Datta, S.D.; Friberg, I.K.; Goldie, S.J.; Reef, S.E.; Schwalbe, N.; Simons, E.; Strebel, P.M.; et al. The estimated mortality impact of vaccinations forecast to be administered during 2011–2020 in 73 countries supported by the GAVI alliance. Vaccine 2013, 31, B61–B72. [Google Scholar] [CrossRef] [PubMed]
- Tate, J.E.; Burton, A.H.; Boschi-Pinto, C.; Steele, A.D.; Duque, J.; Parashar, U.D.; Network, W.H.-C.G.R.S. 2008 Estimate of worldwide rotavirus-associated mortality in children younger than 5 years before the introduction of universal rotavirus vaccination programmes: A systematic review and meta-analysis. Lancet Infect. Dis. 2012, 12, 136–141. [Google Scholar] [CrossRef]
- Walker, C.L.; Rudan, I.; Liu, L.; Nair, H.; Theodoratou, E.; Bhutta, Z.A.; O’Brien, K.L.; Campbell, H.; Black, R.E. Global burden of childhood pneumonia and diarrhoea. Lancet 2013, 381, 1405–1416. [Google Scholar] [CrossRef]
- Armah, G.E.; Sow, S.O.; Breiman, R.F.; Dallas, M.J.; Tapia, M.D.; Feikin, D.R.; Binka, F.N.; Steele, A.D.; Laserson, K.F.; Ansah, N.A.; et al. Efficacy of pentavalent rotavirus vaccine against severe rotavirus gastroenteritis in infants in developing countries in sub-saharan africa: A randomised, double-blind, placebo-controlled trial. Lancet 2010, 376, 606–614. [Google Scholar] [CrossRef]
- Madhi, S.A.; Cunliffe, N.A.; Steele, D.; Witte, D.; Kirsten, M.; Louw, C.; Ngwira, B.; Victor, J.C.; Gillard, P.H.; Cheuvart, B.B.; et al. Effect of human rotavirus vaccine on severe diarrhea in african infants. N. Engl. J. Med. 2010, 362, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Zaman, K.; Dang, D.A.; Victor, J.C.; Shin, S.; Yunus, M.; Dallas, M.J.; Podder, G.; Vu, D.T.; Le, T.P.; Luby, S.P.; et al. Efficacy of pentavalent rotavirus vaccine against severe rotavirus gastroenteritis in infants in developing countries in asia: A randomised, double-blind, placebo-controlled trial. Lancet 2010, 376, 615–623. [Google Scholar] [CrossRef]
- Estes, M.K.; Greenberg, H.B. Rotaviruses and their replication. In Fields Virology, 6th ed.; Knipe, D.N., Howley, P.M., Eds.; Wolters Kluwer Health, Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; pp. 1347–1401. [Google Scholar]
- Sonenberg, N.; Hinnebusch, A.G. Regulation of translation initiation in eukaryotes: Mechanisms and biological targets. Cell 2009, 136, 731–745. [Google Scholar] [CrossRef] [PubMed]
- Jackson, R.J.; Hellen, C.U.; Pestova, T.V. The mechanism of eukaryotic translation initiation and principles of its regulation. Nat. Rev. Mol. Cell Biol. 2010, 11, 113–127. [Google Scholar] [CrossRef] [PubMed]
- Hinnebusch, A.G. The scanning mechanism of eukaryotic translation initiation. Annu. Rev. Biochem. 2014, 83, 779–812. [Google Scholar] [CrossRef] [PubMed]
- Pestova, T.V.; Kolupaeva, V.G.; Lomakin, I.B.; Pilipenko, E.V.; Shatsky, I.N.; Agol, V.I.; Hellen, C.U. Molecular mechanisms of translation initiation in eukaryotes. Proc. Natl. Acad. Sci. USA 2001, 98, 7029–7036. [Google Scholar] [CrossRef] [PubMed]
- Walsh, D.; Mohr, I. Viral subversion of the host protein synthesis machinery. Nat. Rev. Microbiol. 2011, 9, 860–875. [Google Scholar] [CrossRef] [PubMed]
- Poncet, D.; Laurent, S.; Cohen, J. Four nucleotides are the minimal requirement for RNA recognition by rotavirus non-structural protein NSP3. EMBO J. 1994, 13, 4165–4173. [Google Scholar] [PubMed]
- Chizhikov, V.; Patton, J.T. A four-nucleotide translation enhancer in the 3′-terminal consensus sequence of the nonpolyadenylated mRNAs of rotavirus. RNA 2000, 6, 814–825. [Google Scholar] [CrossRef] [PubMed]
- Chung, K.T.; McCrae, M.A. Regulation of gene expression by the NSP1 and NSP3 non-structural proteins of rotavirus. Arch. Virol. 2011, 156, 2197–2203. [Google Scholar] [CrossRef] [PubMed]
- Gratia, M.; Sarot, E.; Vende, P.; Charpilienne, A.; Baron, C.H.; Duarte, M.; Pyronnet, S.; Poncet, D. Rotavirus NSP3 is a translational surrogate of the poly(A) binding protein-poly(A) complex. J. Virol. 2015, 89, 8773–8782. [Google Scholar] [CrossRef] [PubMed]
- Gratia, M.; Vende, P.; Charpilienne, A.; Baron, H.C.; Laroche, C.; Sarot, E.; Pyronnet, S.; Duarte, M.; Poncet, D. Challenging the roles of NSP3 and untranslated regions in rotavirus mRNA translation. PLoS ONE 2016, 11, e0145998. [Google Scholar] [CrossRef] [PubMed]
- Rubio, R.M.; Mora, S.I.; Romero, P.; Arias, C.F.; Lopez, S. Rotavirus prevents the expression of host responses by blocking the nucleocytoplasmic transport of polyadenylated mRNAs. J. Virol. 2013, 87, 6336–6345. [Google Scholar] [CrossRef] [PubMed]
- Vende, P.; Piron, M.; Castagne, N.; Poncet, D. Efficient translation of rotavirus mRNA requires simultaneous interaction of NSP3 with the eukaryotic translation initiation factor eIF4G and the mRNA 3′ end. J. Virol. 2000, 74, 7064–7071. [Google Scholar] [CrossRef] [PubMed]
- Deo, R.C.; Groft, C.M.; Rajashankar, K.R.; Burley, S.K. Recognition of the rotavirus mRNA 3′ consensus by an asymmetric NSP3 homodimer. Cell 2002, 108, 71–81. [Google Scholar] [CrossRef]
- Piron, M.; Delaunay, T.; Grosclaude, J.; Poncet, D. Identification of the RNA-binding, dimerization, and eIF4GI-binding domains of rotavirus nonstructural protein NSP3. J. Virol. 1999, 73, 5411–5421. [Google Scholar] [PubMed]
- Piron, M.; Vende, P.; Cohen, J.; Poncet, D. Rotavirus RNA-binding protein NSP3 interacts with eIF4GI and evicts the poly(A) binding protein from eIF4F. EMBO J. 1998, 17, 5811–5821. [Google Scholar] [CrossRef] [PubMed]
- Poncet, D.; Aponte, C.; Cohen, J. Rotavirus protein NSP3 (ns34) is bound to the 3′ end consensus sequence of viral mRNAs in infected cells. J. Virol. 1993, 67, 3159–3165. [Google Scholar] [PubMed]
- Groft, C.M.; Burley, S.K. Recognition of eIF4G by rotavirus NSP3 reveals a basis for mRNA circularization. Mol. Cell 2002, 9, 1273–1283. [Google Scholar] [CrossRef]
- Vitour, D.; Lindenbaum, P.; Vende, P.; Becker, M.M.; Poncet, D. RoXaN, a novel cellular protein containing TPR, LD, and zinc finger motifs, forms a ternary complex with eukaryotic initiation factor 4G and rotavirus NSP3. J. Virol. 2004, 78, 3851–3862. [Google Scholar] [CrossRef] [PubMed]
- Dutta, D.; Chattopadhyay, S.; Bagchi, P.; Halder, U.C.; Nandi, S.; Mukherjee, A.; Kobayashi, N.; Taniguchi, K.; Chawla-Sarkar, M. Active participation of cellular chaperone Hsp90 in regulating the function of rotavirus nonstructural protein 3 (NSP3). J. Biol. Chem. 2011, 286, 20065–20077. [Google Scholar] [CrossRef] [PubMed]
- Padilla-Noriega, L.; Paniagua, O.; Guzman-Leon, S. Rotavirus protein NSP3 shuts off host cell protein synthesis. Virology 2002, 298, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Michel, Y.M.; Poncet, D.; Piron, M.; Kean, K.M.; Borman, A.M. Cap-poly(A) synergy in mammalian cell-free extracts. Investigation of the requirements for poly(A)-mediated stimulation of translation initiation. J. Biol. Chem. 2000, 275, 32268–32276. [Google Scholar] [CrossRef] [PubMed]
- Montero, H.; Arias, C.F.; Lopez, S. Rotavirus nonstructural protein NSP3 is not required for viral protein synthesis. J. Virol. 2006, 80, 9031–9038. [Google Scholar] [CrossRef] [PubMed]
- Arnold, M.M.; Brownback, C.S.; Taraporewala, Z.F.; Patton, J.T. Rotavirus variant replicates efficiently although encoding an aberrant NSP3 that fails to induce nuclear localization of poly(A)-binding protein. J. Gen. Virol. 2012, 93, 1483–1494. [Google Scholar] [CrossRef] [PubMed]
- Harb, M.; Becker, M.M.; Vitour, D.; Baron, C.H.; Vende, P.; Brown, S.C.; Bolte, S.; Arold, S.T.; Poncet, D. Nuclear localization of cytoplasmic poly(A)-binding protein upon rotavirus infection involves the interaction of NSP3 with eIF4G and RoXaN. J. Virol. 2008, 82, 11283–11293. [Google Scholar] [CrossRef] [PubMed]
- Montero, H.; Rojas, M.; Arias, C.F.; Lopez, S. Rotavirus infection induces the phosphorylation of eIF2alpha but prevents the formation of stress granules. J. Virol. 2008, 82, 1496–1504. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, N.; Gorman, A.M.; Gupta, S.; Samali, A. The eIF2alpha kinases: Their structures and functions. Cell. Mol. Life Sci. 2013, 70, 3493–3511. [Google Scholar] [CrossRef] [PubMed]
- Proud, C.G. eIF2 and the control of cell physiology. Semin. Cell Dev. Biol. 2005, 16, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Rojas, M.; Arias, C.F.; Lopez, S. Protein kinase R is responsible for the phosphorylation of eIF2alpha in rotavirus infection. J. Virol. 2010, 84, 10457–10466. [Google Scholar] [CrossRef] [PubMed]
- Anderson, P.; Kedersha, N. Stress granules: The tao of RNA triage. Trends Biochem. Sci. 2008, 33, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Reineke, L.C.; Kedersha, N.; Langereis, M.A.; van Kuppeveld, F.J.; Lloyd, R.E. Stress granules regulate double-stranded RNA-dependent protein kinase activation through a complex containing G3BP1 and Caprin1. mBio 2015, 6, e02486. [Google Scholar] [CrossRef] [PubMed]
- Tsai, W.C.; Lloyd, R.E. Cytoplasmic RNA granules and viral infection. Annu. Rev. Virol. 2014, 1, 147–170. [Google Scholar] [CrossRef] [PubMed]
- Panas, M.D.; Kedersha, N.; McInerney, G.M. Methods for the characterization of stress granules in virus infected cells. Methods 2015, 90, 57–64. [Google Scholar] [CrossRef] [PubMed]
- White, J.P.; Lloyd, R.E. Regulation of stress granules in virus systems. Trends Microbiol. 2012, 20, 175–183. [Google Scholar] [CrossRef] [PubMed]
- Decker, C.J.; Parker, R. P-bodies and stress granules: Possible roles in the control of translation and mRNA degradation. Cold Spring Harb. Perspect. Biol. 2012, 4, a012286. [Google Scholar] [CrossRef] [PubMed]
- Eulalio, A.; Behm-Ansmant, I.; Izaurralde, E. P bodies: At the crossroads of post-transcriptional pathways. Nat. Rev. Mol. Cell Biol. 2007, 8, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, R.E. How do viruses interact with stress-associated RNA granules? PLoS Pathog. 2012, 8, e1002741. [Google Scholar] [CrossRef] [PubMed]
- Kedersha, N.; Stoecklin, G.; Ayodele, M.; Yacono, P.; Lykke-Andersen, J.; Fritzler, M.J.; Scheuner, D.; Kaufman, R.J.; Golan, D.E.; Anderson, P. Stress granules and processing bodies are dynamically linked sites of mRNP remodeling. J. Cell Biol. 2005, 169, 871–884. [Google Scholar] [CrossRef] [PubMed]
- Bhowmick, R.; Mukherjee, A.; Patra, U.; Chawla-Sarkar, M. Rotavirus disrupts cytoplasmic P bodies during infection. Virus Res. 2015, 210, 344–354. [Google Scholar] [CrossRef] [PubMed]
- Charley, P.A.; Wilusz, J. Sponging of cellular proteins by viral RNAs. Curr. Opin. Virol. 2014, 9, 14–18. [Google Scholar] [CrossRef] [PubMed]
- Bernales, S.; Papa, F.R.; Walter, P. Intracellular signaling by the unfolded protein response. Annu. Rev. Cell Dev. Biol. 2006, 22, 487–508. [Google Scholar] [CrossRef] [PubMed]
- Schroder, M.; Kaufman, R.J. ER stress and the unfolded protein response. Mutat. Res. 2005, 569, 29–63. [Google Scholar] [CrossRef] [PubMed]
- Walter, P.; Ron, D. The unfolded protein response: From stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef] [PubMed]
- Trujillo-Alonso, V.; Maruri-Avidal, L.; Arias, C.F.; Lopez, S. Rotavirus infection induces the unfolded protein response of the cell and controls it through the nonstructural protein NSP3. J. Virol. 2011, 85, 12594–12604. [Google Scholar] [CrossRef] [PubMed]
- Zambrano, J.L.; Ettayebi, K.; Maaty, W.S.; Faunce, N.R.; Bothner, B.; Hardy, M.E. Rotavirus infection activates the UPR but modulates its activity. Virol. J. 2011, 8. [Google Scholar] [CrossRef] [PubMed]
- Rutkowski, D.T.; Kaufman, R.J. A trip to the ER: Coping with stress. Trends Cell Biol. 2004, 14, 20–28. [Google Scholar] [CrossRef] [PubMed]



© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
López, S.; Oceguera, A.; Sandoval-Jaime, C. Stress Response and Translation Control in Rotavirus Infection. Viruses 2016, 8, 162. https://0-doi-org.brum.beds.ac.uk/10.3390/v8060162
López S, Oceguera A, Sandoval-Jaime C. Stress Response and Translation Control in Rotavirus Infection. Viruses. 2016; 8(6):162. https://0-doi-org.brum.beds.ac.uk/10.3390/v8060162
Chicago/Turabian StyleLópez, Susana, Alfonso Oceguera, and Carlos Sandoval-Jaime. 2016. "Stress Response and Translation Control in Rotavirus Infection" Viruses 8, no. 6: 162. https://0-doi-org.brum.beds.ac.uk/10.3390/v8060162