
Development of an Oncolytic Adenovirus with Enhanced Spread Ability through Repeated UV Irradiation and Cancer Selection

 , ,
, , 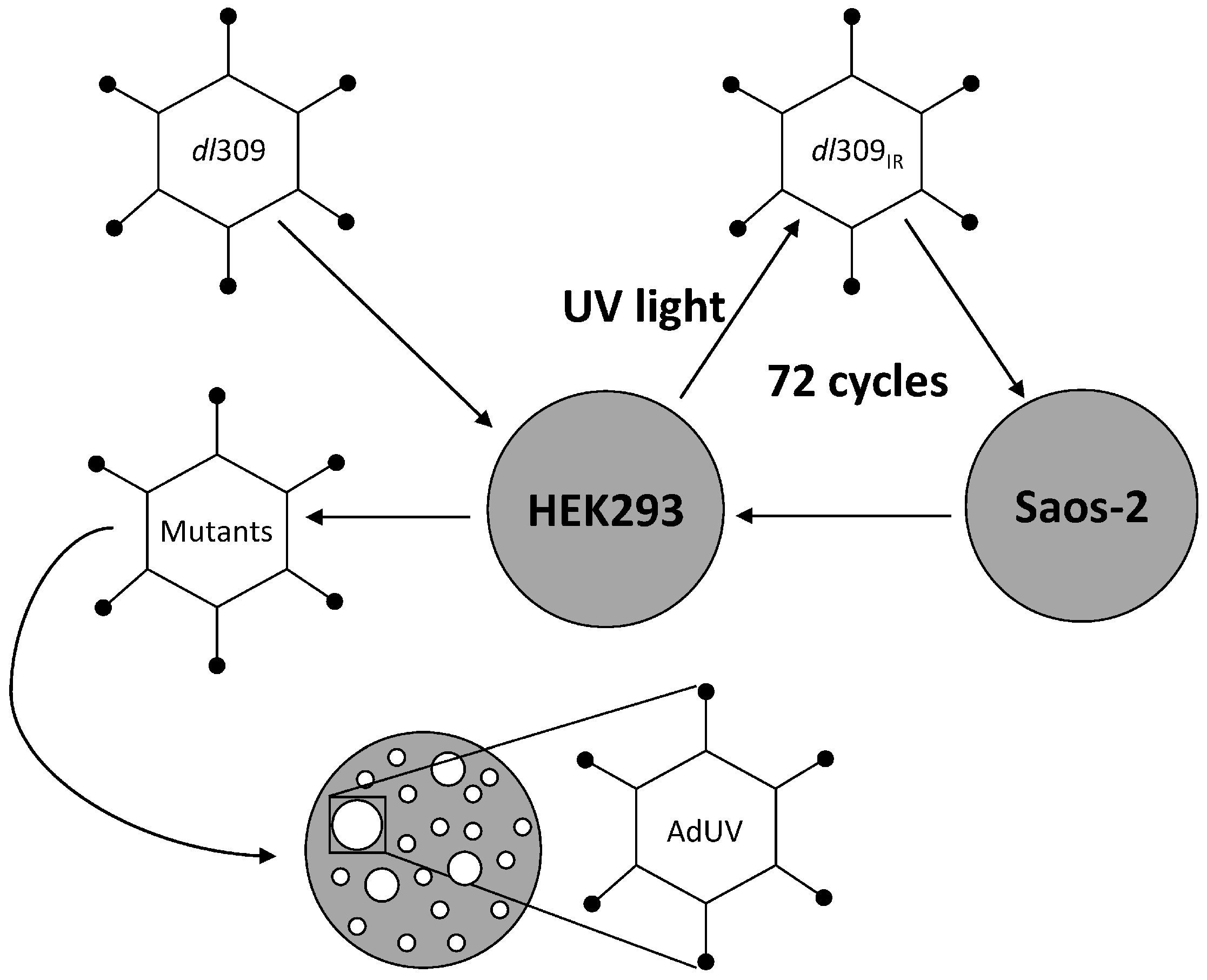{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines and Culture Conditions
2.2. Adenoviral Vectors
2.3. Virus Titration and Release
2.4. Western Blot Analysis
2.5. Plaque Formation Assay
2.6. Quantification of Band Intensity
2.7. Cytotoxicity Assay
2.8. Statistical Analysis
3. Results
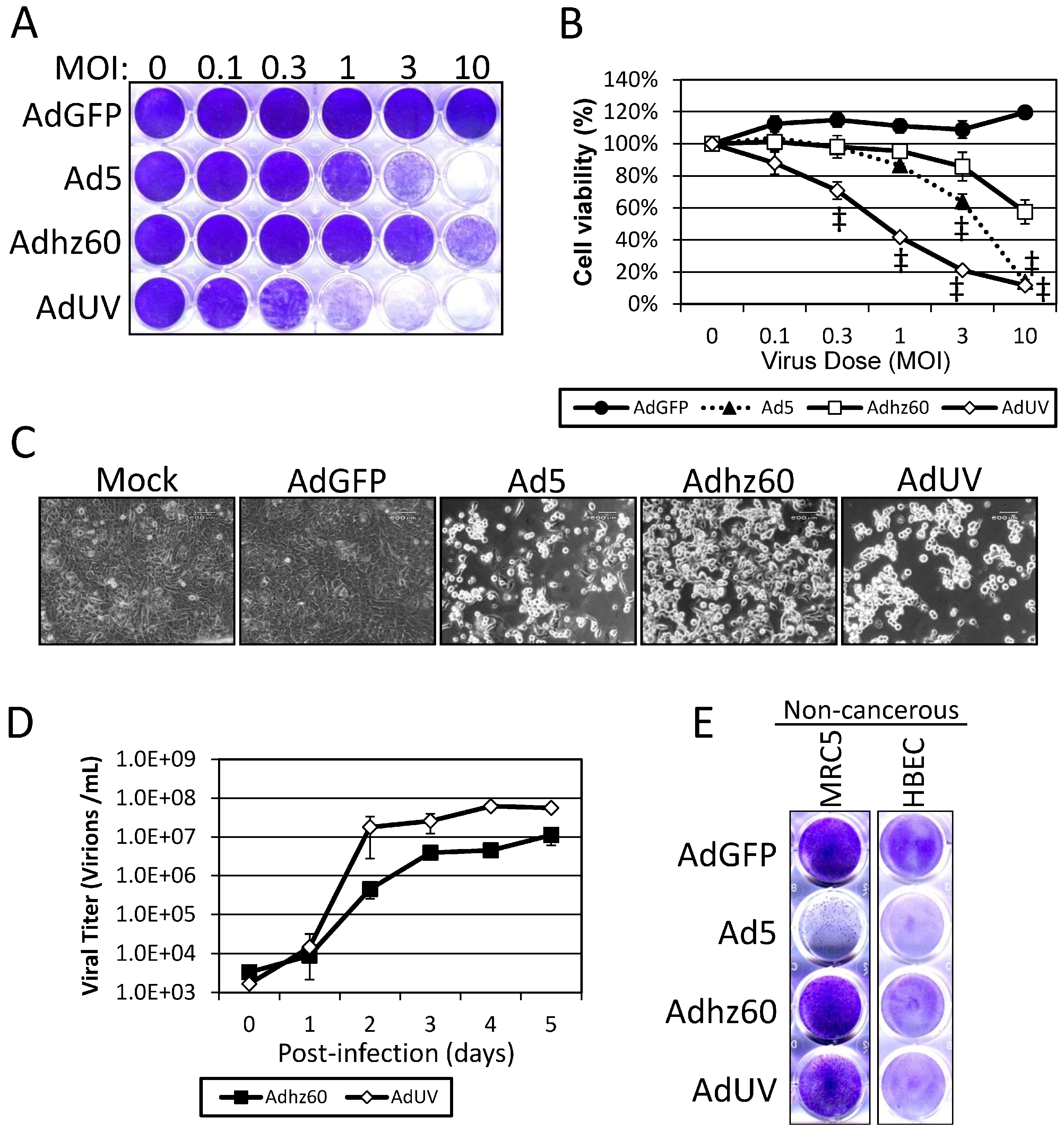
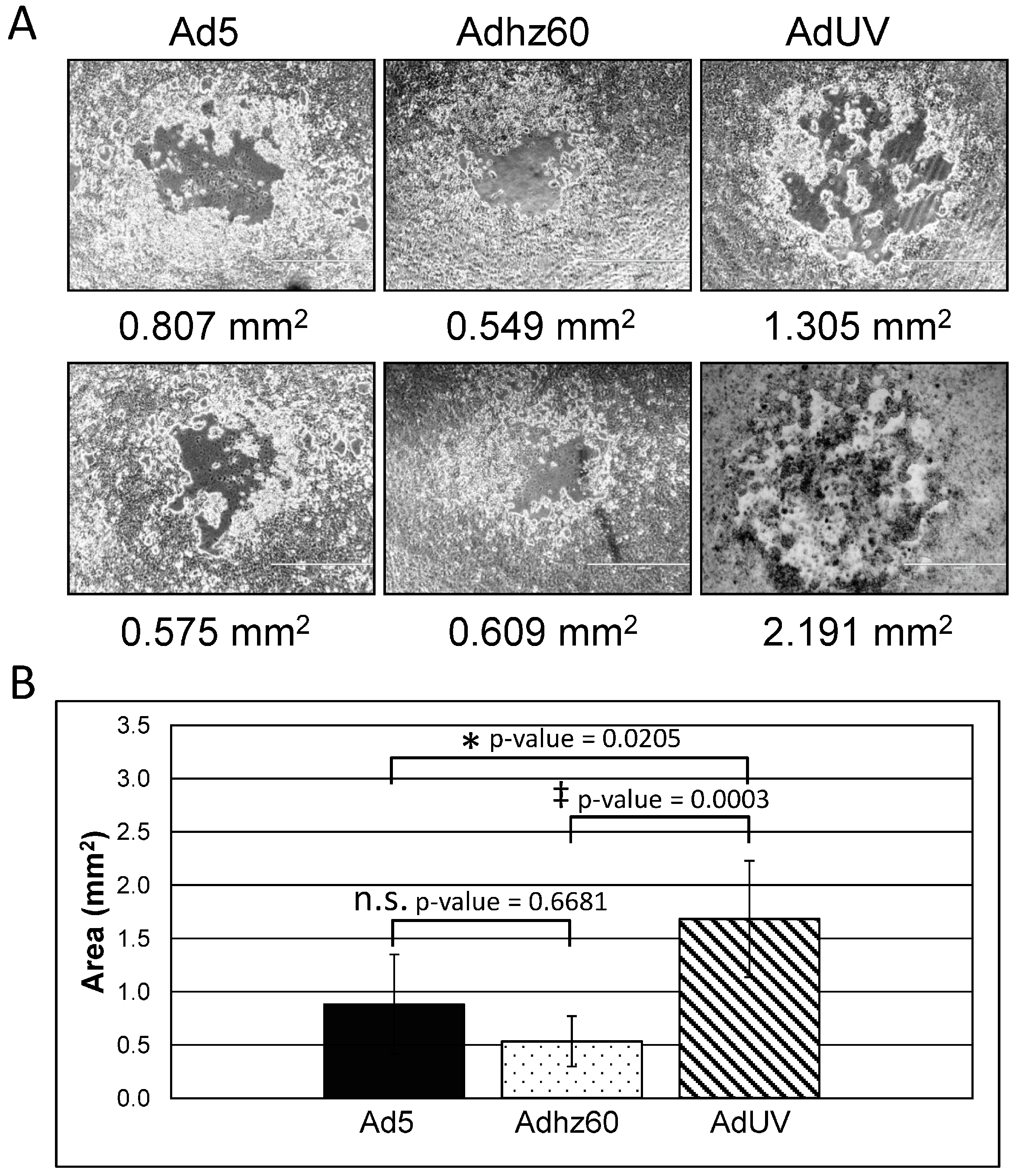
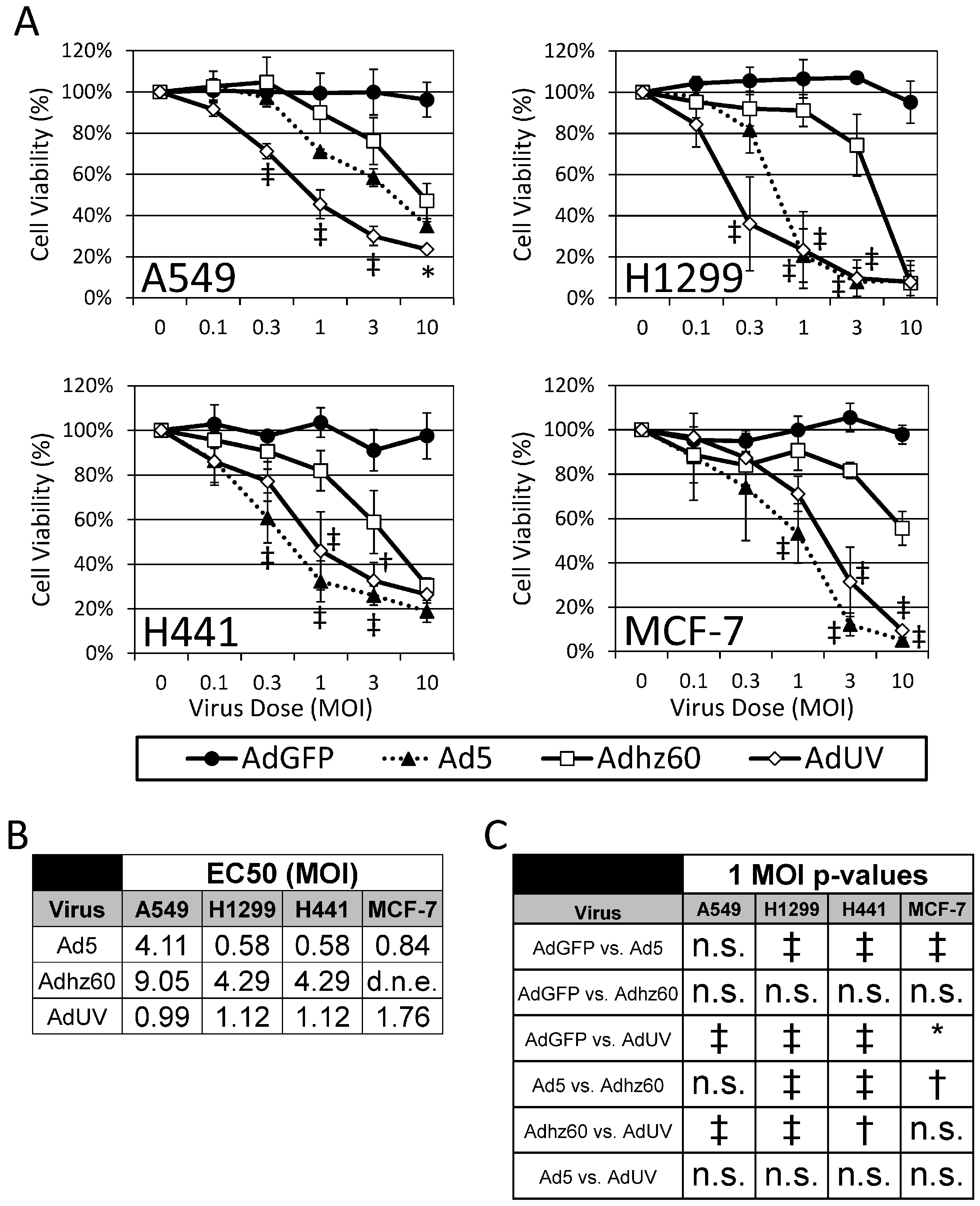
3.1. AdUV Displays More Efficient Release and Cancer Cell Lysis
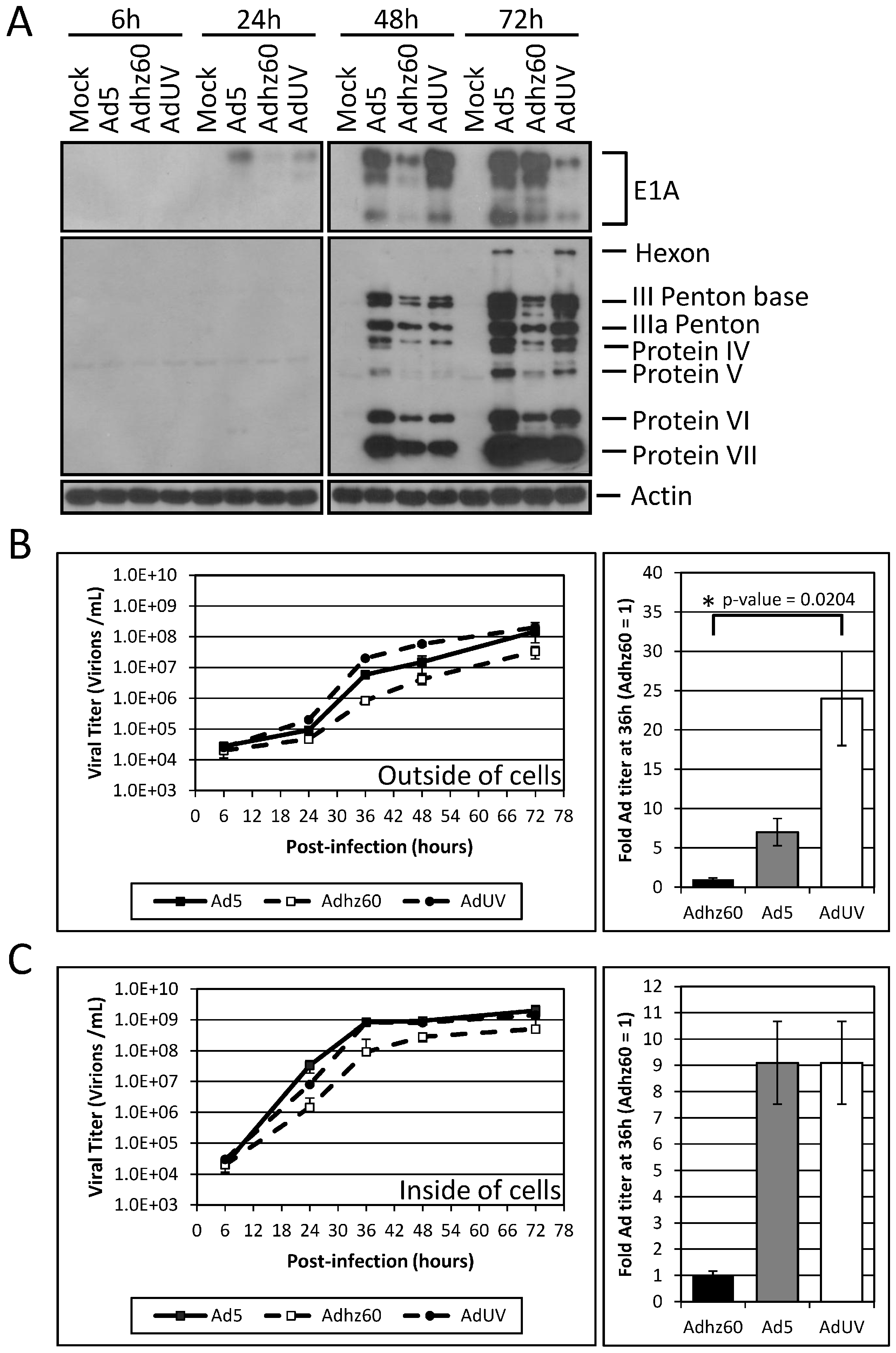
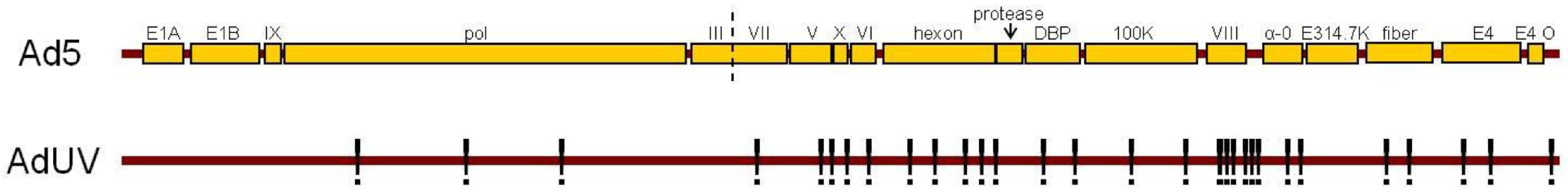
3.2. Improved AdUV Replication in Cancer Cells
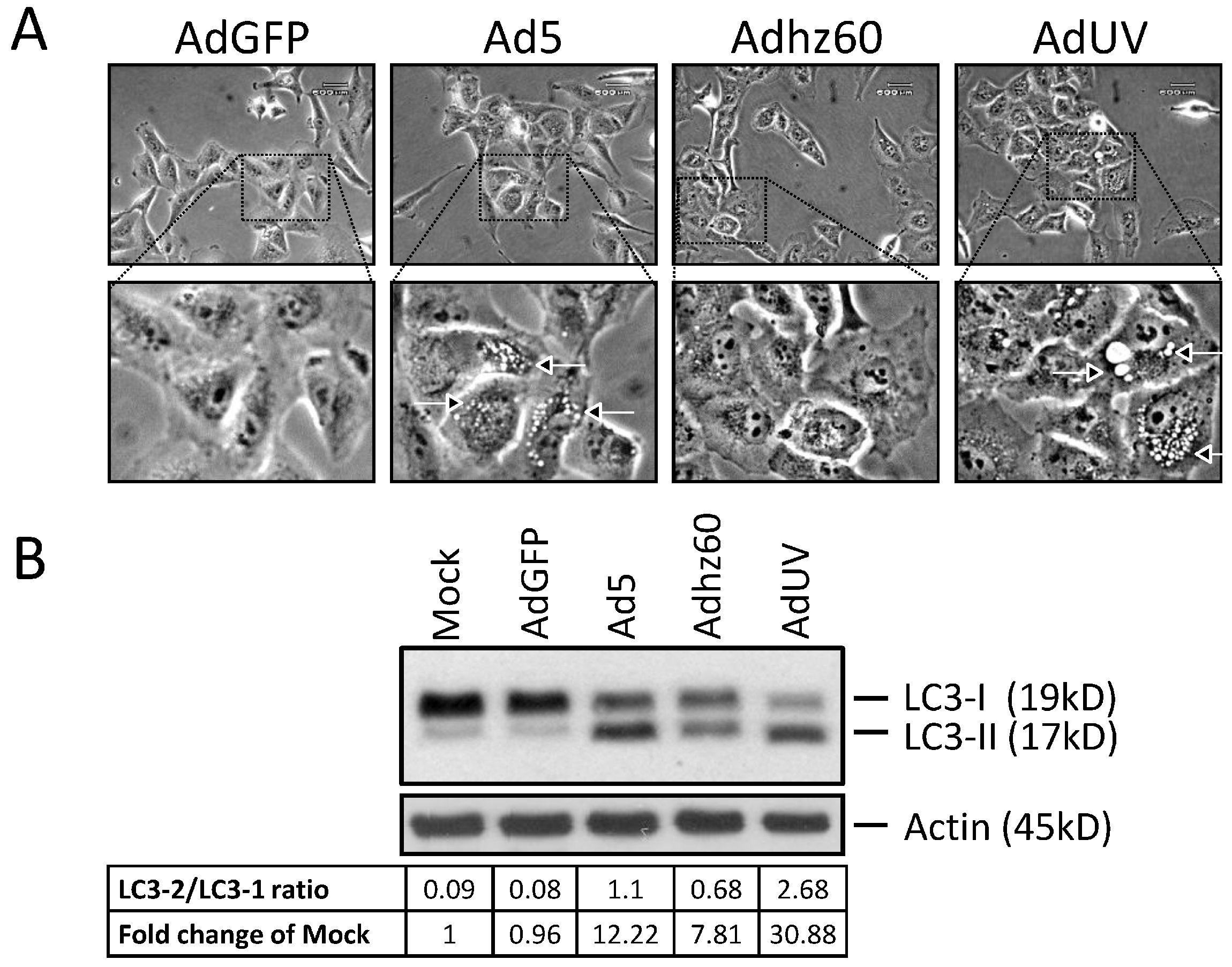
3.3. AdUV Induces Greater Autophagy
3.4. AdUV Kills a Variety of Cancer Cell Types
4. Discussion
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| Ad | Adenovirus |
| E1A | Early gene 1 A |
| E1B | Early gene 1 B |
| LC3 | Microtuble-associated protein light chain 3 |
| MOI | Multiplicity of infection |
References
- Mulvihill, S.; Warren, R.; Venook, A.; Adler, A.; Randlev, B.; Heise, C.; Kirn, D. Safety and feasibility of injection with an E1B-55 kDa gene-deleted, replication-selective adenovirus (ONYX-015) into primary carcinomas of the pancreas: A phase I trial. Gene Ther. 2001, 8, 308–315. [Google Scholar] [CrossRef] [PubMed]
- Berk, A.J. Adenoviridae: The Viruses and Their Replication. In HowleyDMKP (ed) Fields Virology; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2006; pp. 2354–2394. [Google Scholar]
- Parker, J.N.; Bauer, D.F.; Cody, J.J.; Markert, J.M. Oncolytic viral therapy of malignant glioma. Neurother. J. Am. Soc. Exp. NeuroTher. 2009, 6, 558–569. [Google Scholar] [CrossRef] [PubMed]
- Nettelbeck, D.M. Cellular genetic tools to control oncolytic adenoviruses for virotherapy of cancer. J. Mol. Med. 2008, 86, 363–377. [Google Scholar] [CrossRef] [PubMed]
- Seth, P.; Higginbotham, J. Advantages and disadvantages of multiple different methods of adenoviral vector construction. Methods Mol. Med. 2000, 45, 189–198. [Google Scholar] [PubMed]
- Ben-Israel, H.; Kleinberger, T. Adenovirus and cell cycle control. Front. Biosci. J. Virtual Libr. 2002, 7, d1369–d1395. [Google Scholar] [CrossRef]
- Barker, D.D.; Berk, A.J. Adenovirus proteins from both E1B reading frames are required for transformation of rodent cells by viral infection and DNA transfection. Virology 1987, 156, 107–121. [Google Scholar] [CrossRef]
- Stillman, B. Functions of the adenovirus E1B tumour antigens. Cancer Surv. 1986, 5, 389–404. [Google Scholar] [PubMed]
- Lowe, S.W.; Ruley, H.E. Stabilization of the p53 tumor suppressor is induced by adenovirus 5 E1A and accompanies apoptosis. Genes Dev. 1993, 7, 535–545. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Rao, X.M.; Snodgrass, C.L.; McMasters, K.M.; Zhou, H.S. Selective replication of E1B55K-deleted adenoviruses depends on enhanced E1A expression in cancer cells. Cancer Gene Ther. 2006, 13, 572–583. [Google Scholar] [CrossRef] [PubMed]
- Rao, X.M.; Zheng, X.; Waigel, S.; Zacharias, W.; McMasters, K.M.; Zhou, H.S. Gene expression profiles of normal human lung cells affected by adenoviral E1B. Virology 2006, 350, 418–428. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Rao, X.M.; Gomez-Gutierrez, J.G.; Hao, H.; McMasters, K.M.; Zhou, H.S. Adenovirus E1B55K region is required to enhance cyclin E expression for efficient viral DNA replication. J. Virol. 2008, 82, 3415–3427. [Google Scholar] [CrossRef] [PubMed]
- Cheng, P.H.; Wechman, S.L.; McMasters, K.M.; Zhou, H.S. Oncolytic Replication of E1b-Deleted Adenoviruses. Viruses 2015, 7, 5767–5779. [Google Scholar] [CrossRef] [PubMed]
- Cheng, P.H.; Rao, X.M.; McMasters, K.M.; Zhou, H.S. Molecular basis for viral selective replication in cancer cells: Activation of CDK2 by adenovirus-induced cyclin E. PLoS ONE 2013, 8, e57340. [Google Scholar] [CrossRef] [PubMed]
- Cheng, P.H.; Rao, X.M.; Duan, X.; Li, X.F.; Egger, M.E.; McMasters, K.M.; Zhou, H.S. Virotherapy targeting cyclin E overexpression in tumors with adenovirus-enhanced cancer-selective promoter. J. Mol. Med. 2015, 93, 211–223. [Google Scholar] [CrossRef] [PubMed]
- Cheng, P.H.; Rao, X.M.; Wechman, S.L.; Li, X.F.; McMasters, K.M.; Zhou, H.S. Oncolytic adenovirus targeting cyclin E overexpression repressed tumor growth in syngeneic immunocompetent mice. BMC Cancer 2015, 15, 716. [Google Scholar] [CrossRef] [PubMed]
- Debbas, M.; White, E. Wild-type p53 mediates apoptosis by E1A, which is inhibited by E1B. Genes Dev. 1993, 7, 546–554. [Google Scholar] [CrossRef] [PubMed]
- Rao, L.; Debbas, M.; Sabbatini, P.; Hockenbery, D.; Korsmeyer, S.; White, E. The adenovirus E1A proteins induce apoptosis, which is inhibited by the E1B 19-kDa and Bcl-2 proteins. Proc. Natl. Acad. Sci. USA 1992, 89, 7742–7746. [Google Scholar] [CrossRef] [PubMed]
- White, E.; Sabbatini, P.; Debbas, M.; Wold, W.S.; Kusher, D.I.; Gooding, L.R. The 19-kilodalton adenovirus E1B transforming protein inhibits programmed cell death and prevents cytolysis by tumor necrosis factor alpha. Mol. Cell. Biol. 1992, 12, 2570–2580. [Google Scholar] [CrossRef] [PubMed]
- Cuconati, A.; Degenhardt, K.; Sundararajan, R.; Anschel, A.; White, E. Bak and Bax function to limit adenovirus replication through apoptosis induction. J. Virol. 2002, 76, 4547–4558. [Google Scholar] [CrossRef] [PubMed]
- Bischoff, J.R.; Kim, D.H.; Williams, A.; Heise, C.; Horn, S.; Muna, M.; Ng, L.; Nye, J.A.; SampsonJohannes, A.; Fattaey, A.; McCormick, F. An adenovirus mutant that replicates selectively in p53-deficient human tumor cells. Science 1996, 274, 373–376. [Google Scholar] [CrossRef] [PubMed]
- Kirn, D. Oncolytic virotherapy for cancer with the adenovirus dl1520 (Onyx-015): Results of phase I and II trials. Expert Opin. Biol. Ther. 2001, 1, 525–538. [Google Scholar] [CrossRef] [PubMed]
- Kirn, D. Clinical research results with dl1520 (Onyx-015), a replication-selective adenovirus for the treatment of cancer: What have we learned? Gene Ther. 2001, 8, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Fang, H. Clinical trials with oncolytic adenovirus in China. Curr. Cancer Drug Targets 2007, 7, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Cafferata, E.G.; Maccio, D.R.; Lopez, M.V.; Viale, D.L.; Carbone, C.; Mazzolini, G.; Podhajcer, O.L. A novel A33 promoter-based conditionally replicative adenovirus suppresses tumor growth and eradicates hepatic metastases in human colon cancer models. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2009, 15, 3037–3049. [Google Scholar] [CrossRef] [PubMed]
- Cheng, P.H.; Lian, S.; Zhao, R.; Rao, X.M.; McMasters, K.M.; Zhou, H.S. Combination of autophagy inducer rapamycin and oncolytic adenovirus improves antitumor effect in cancer cells. Virol. J. 2013, 10, 293. [Google Scholar] [CrossRef] [PubMed]
- Malumbres, M.; Barbacid, M. To cycle or not to cycle: A critical decision in cancer. Nat. Rev. Cancer 2001, 1, 222–231. [Google Scholar] [CrossRef] [PubMed]
- Nemunaitis, J.; Tong, A.W.; Nemunaitis, M.; Senzer, N.; Phadke, A.P.; Bedell, C.; Adams, N.; Zhang, Y.A.; Maples, P.B.; Chen, S.; et al. A phase I study of telomerase-specific replication competent oncolytic adenovirus (telomelysin) for various solid tumors. Mol. Ther. J. Am. Soc. Gene Ther. 2010, 18, 429–434. [Google Scholar] [CrossRef] [PubMed]
- DeWeese, T.L.; van der Poel, H.; Li, S.; Mikhak, B.; Drew, R.; Goemann, M.; Hamper, U.; DeJong, R.; Detorie, N.; Rodriguez, R.; et al. A phase I trial of CV706, a replication-competent, PSA selective oncolytic adenovirus, for the treatment of locally recurrent prostate cancer following radiation therapy. Cancer Res. 2001, 61, 7464–7472. [Google Scholar] [PubMed]
- Yamamoto, M.; Curiel, D.T. Current issues and future directions of oncolytic adenoviruses. Mol. Ther. J. Am. Soc. Gene Ther. 2010, 18, 243–250. [Google Scholar] [CrossRef] [PubMed]
- McCormick, F. Cancer-specific viruses and the development of ONYX-015. Cancer Biol. Ther. 2003, 2 (Suppl. S1), S157–S160. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.; Rao, X.M.; Xie, X.; Li, L.; Thompson, T.C.; McMasters, K.M.; Zhou, H.S. Adenovirus with insertion-mutated E1A selectively propagates in liver cancer cells and destroys tumors in vivo. Cancer Res. 2003, 63, 3073–3078. [Google Scholar] [PubMed]
- Russell, S.J.; Peng, K.W.; Bell, J.C. Oncolytic virotherapy. Nat. Biotechnol. 2012, 30, 658–670. [Google Scholar] [CrossRef] [PubMed]
- Jones, N.; Shenk, T. Isolation of adenovirus type 5 host range deletion mutants defective for transformation of rat embryo cells. Cell 1979, 17, 683–689. [Google Scholar] [CrossRef]
- Rao, X.M.; Tseng, M.T.; Zheng, X.; Dong, Y.; Jamshidi-Parsian, A.; Thompson, T.C.; Brenner, M.K.; McMasters, K.M.; Zhou, H.S. E1A-induced apoptosis does not prevent replication of adenoviruses with deletion of E1b in majority of infected cancer cells. Cancer Gene Ther. 2004, 11, 585–593. [Google Scholar] [CrossRef] [PubMed]
- Ishiyama, M.; Tominaga, H.; Shiga, M.; Sasamoto, K.; Ohkura, Y.; Ueno, K. A combined assay of cell viability and in vitro cytotoxicity with a highly water-soluble tetrazolium salt, neutral red and crystal violet. Biol. Pharm. Bull. 1996, 19, 1518–1520. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Rocha, H.; Gomez-Gutierrez, J.G.; Garcia-Garcia, A.; Rao, X.M.; Chen, L.; McMasters, K.M.; Zhou, H.S. Adenoviruses induce autophagy to promote virus replication and oncolysis. Virology 2011, 416, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Taegtmeyer, H. Too much or not enough of a good thing—The Janus faces of autophagy in cardiac fuel and protein homeostasis. J. Mol. Cell. Cardiol. 2015, 84, 223–226. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.H.; Hu, D.H.; Zhang, Z.F.; Bai, X.Z.; Wang, H.T.; Zhu, X.X.; Su, Y.J.; Tang, C.W. Reduced expression of microtubule-associated protein 1 light chain 3 in hypertrophic scars. Arch. Dermatol. Res. 2012, 304, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Kabeya, Y.; Mizushima, N.; Ueno, T.; Yamamoto, A.; Kirisako, T.; Noda, T.; Kominami, E.; Ohsumi, Y.; Yoshimori, T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000, 19, 5720–5728. [Google Scholar] [CrossRef] [PubMed]
- Barth, S.; Glick, D.; Macleod, K.F. Autophagy: Assays and artifacts. J. Pathol. 2010, 221, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Opyrchal, M.; Aderca, I.; Galanis, E. Phase I clinical trial of locoregional administration of the oncolytic adenovirus ONYX-015 in combination with mitomycin-C, doxorubicin, and cisplatin chemotherapy in patients with advanced sarcomas. Methods Mol. Biol. 2009, 542, 705–717. [Google Scholar] [PubMed]
- Lu, W.; Zheng, S.; Li, X.F.; Huang, J.J.; Zheng, X.; Li, Z. Intra-tumor injection of H101, a recombinant adenovirus, in combination with chemotherapy in patients with advanced cancers: A pilot phase II clinical trial. World J. Gastroenterol. 2004, 10, 3634–3638. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.H.; Yuan, Z.Y.; Guan, Z.Z.; Cao, Y.; Wang, H.Q.; Hu, X.H.; Feng, J.F.; Zhang, Y.; Li, F.; Chen, Z.T.; et al. [Phase II clinical study of intratumoral H101, an E1B deleted adenovirus, in combination with chemotherapy in patients with cancer]. Chin. J. Cancer 2003, 22, 1307–1310. [Google Scholar]
- Makower, D.; Rozenblit, A.; Kaufman, H.; Edelman, M.; Lane, M.E.; Zwiebel, J.; Haynes, H.; Wadler, S. Phase II clinical trial of intralesional administration of the oncolytic adenovirus ONYX-015 in patients with hepatobiliary tumors with correlative p53 studies. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2003, 9, 693–702. [Google Scholar]
- Parato, K.A.; Senger, D.; Forsyth, P.A.; Bell, J.C. Recent progress in the battle between oncolytic viruses and tumours. Nat. Rev. Cancer 2005, 5, 965–976. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.; Breznik, J.; Lichty, B.D. Strategies to enhance viral penetration of solid tumors. Hum. Gene Ther. 2011, 22, 1053–1060. [Google Scholar] [CrossRef] [PubMed]
- Galanis, E.; Okuno, S.H.; Nascimento, A.G.; Lewis, B.D.; Lee, R.A.; Oliveira, A.M.; Sloan, J.A.; Atherton, P.; Edmonson, J.H.; Erlichman, C.; et al. Phase I-II trial of ONYX-015 in combination with MAP chemotherapy in patients with advanced sarcomas. Gene Ther. 2005, 12, 437–445. [Google Scholar] [CrossRef] [PubMed]
- Beier, R.; Hermiston, T.; Mumberg, D. Isolation of more potent oncolytic paramyxovirus by bioselection. Gene Ther. 2013, 20, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Gros, A.; Martinez-Quintanilla, J.; Puig, C.; Guedan, S.; Mollevi, D.G.; Alemany, R.; Cascallo, M. Bioselection of a gain of function mutation that enhances adenovirus 5 release and improves its antitumoral potency. Cancer Res. 2008, 68, 8928–8937. [Google Scholar] [CrossRef] [PubMed]
- Yan, W.; Kitzes, G.; Dormishian, F.; Hawkins, L.; Sampson-Johannes, A.; Watanabe, J.; Holt, J.; Lee, V.; Dubensky, T.; Fattaey, A.; et al. Developing novel oncolytic adenoviruses through bioselection. J. Virol. 2003, 77, 2640–2650. [Google Scholar] [CrossRef] [PubMed]
- Ohsumi, Y. Molecular dissection of autophagy: Two ubiquitin-like systems. Nat. Rev. Mol. Cell Biol. 2001, 2, 211–216. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Klionsky, D.J. Development by self-digestion: Molecular mechanisms and biological functions of autophagy. Dev. Cell 2004, 6, 463–477. [Google Scholar] [CrossRef]
- Gros, A.; Puig, C.; Guedan, S.; Rojas, J.J.; Alemany, R.; Cascallo, M. Verapamil enhances the antitumoral efficacy of oncolytic adenoviruses. Mol. Ther. J. Am. Soc. Gene Ther. 2010, 18, 903–911. [Google Scholar] [CrossRef] [PubMed]
- Abou El Hassan, M.A.; van der Meulen-Muileman, I.; Abbas, S.; Kruyt, F.A. Conditionally replicating adenoviruses kill tumor cells via a basic apoptotic machinery-independent mechanism that resembles necrosis-like programmed cell death. J. Virol. 2004, 78, 12243–12251. [Google Scholar] [CrossRef] [PubMed]
- Baird, S.K.; Aerts, J.L.; Eddaoudi, A.; Lockley, M.; Lemoine, N.R.; McNeish, I.A. Oncolytic adenoviral mutants induce a novel mode of programmed cell death in ovarian cancer. Oncogene 2008, 27, 3081–3090. [Google Scholar] [CrossRef] [PubMed]







© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wechman, S.L.; Rao, X.-M.; Cheng, P.-H.; Gomez-Gutierrez, J.G.; McMasters, K.M.; Zhou, H.S. Development of an Oncolytic Adenovirus with Enhanced Spread Ability through Repeated UV Irradiation and Cancer Selection. Viruses 2016, 8, 167. https://0-doi-org.brum.beds.ac.uk/10.3390/v8060167
Wechman SL, Rao X-M, Cheng P-H, Gomez-Gutierrez JG, McMasters KM, Zhou HS. Development of an Oncolytic Adenovirus with Enhanced Spread Ability through Repeated UV Irradiation and Cancer Selection. Viruses. 2016; 8(6):167. https://0-doi-org.brum.beds.ac.uk/10.3390/v8060167
Chicago/Turabian StyleWechman, Stephen L., Xiao-Mei Rao, Pei-Hsin Cheng, Jorge G. Gomez-Gutierrez, Kelly M. McMasters, and H. Sam Zhou. 2016. "Development of an Oncolytic Adenovirus with Enhanced Spread Ability through Repeated UV Irradiation and Cancer Selection" Viruses 8, no. 6: 167. https://0-doi-org.brum.beds.ac.uk/10.3390/v8060167