
PKR Activation Favors Infectious Pancreatic Necrosis Virus Replication in Infected Cells


Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Virus Propagation
2.3. Cloning, Prokaryotic Expression of Salmon PKR and Production of Rabbit Antiserum
2.4. Recombinant IFNα Treatment
2.5. Effect of IPNV Infection on PKR Expression
2.6. Western Blot
2.7. Quantitative Real-Time PCR Analysis
2.8. Pre-Treatment of Cells with PKR Inhibitor C16 on eIF2α Phosphorylation
2.9. Flow Cytometry
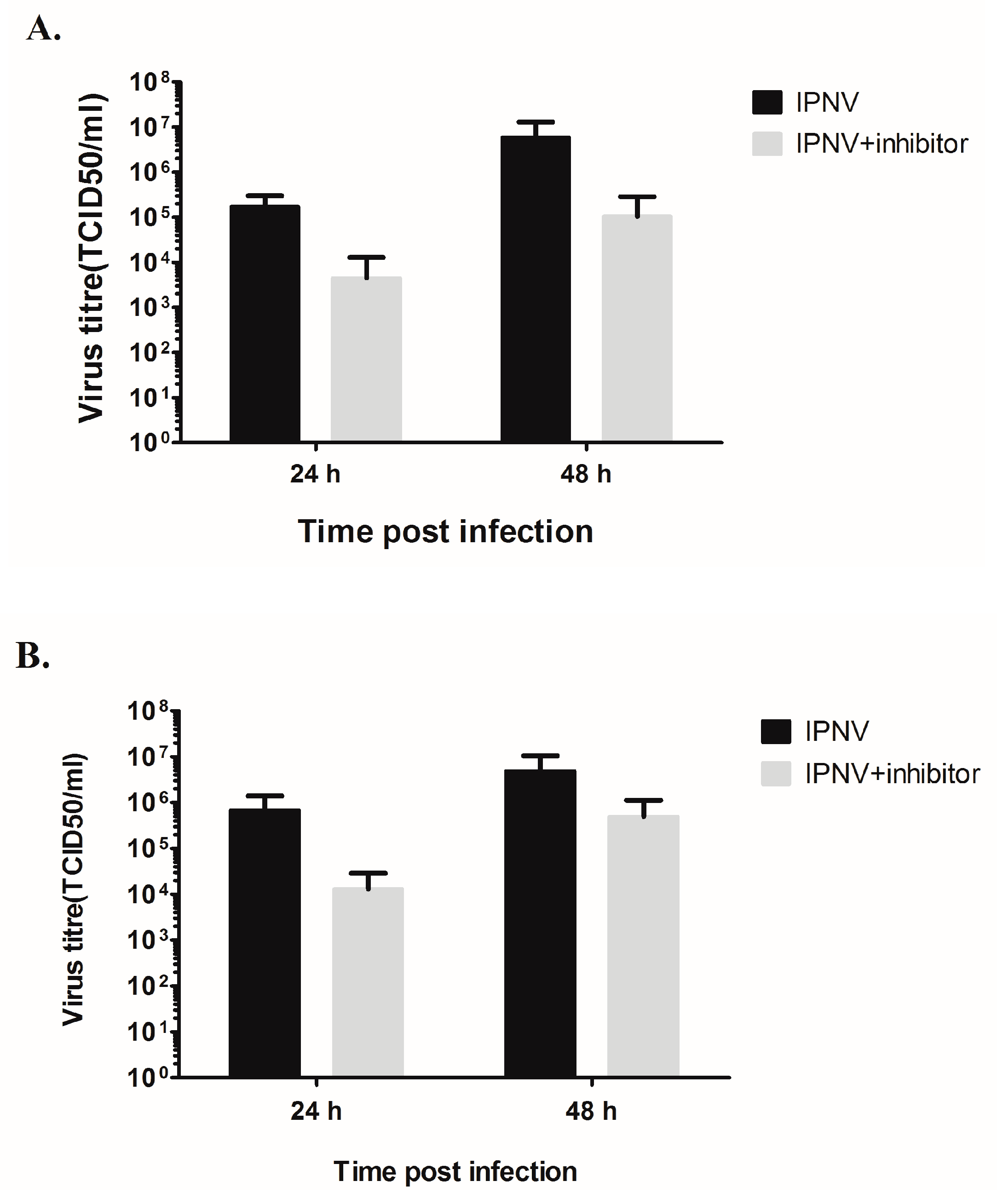
2.10. Effect of PKR Inhibition on Virus Loads in the Supernatant
2.11. Effect of PKR Inhibition on the Intracellular Virus Loads
2.12. Effect of Over-Expressing a Catalytic, Inactive Form of PKR
2.13. Statistics and Graphics
3. Results
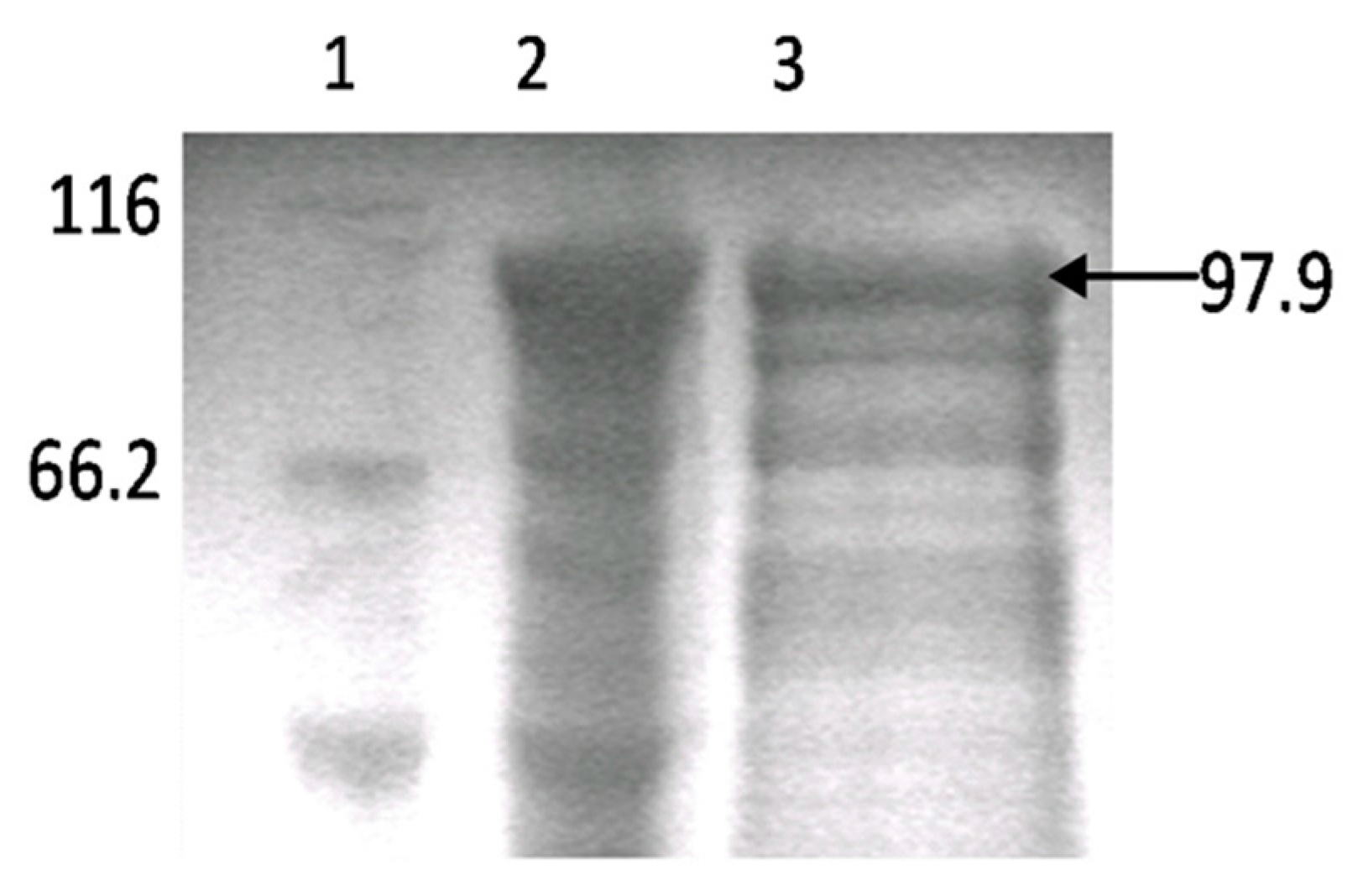
3.1. Production of Rabbit Antiserum against Atlantic Salmon PKR
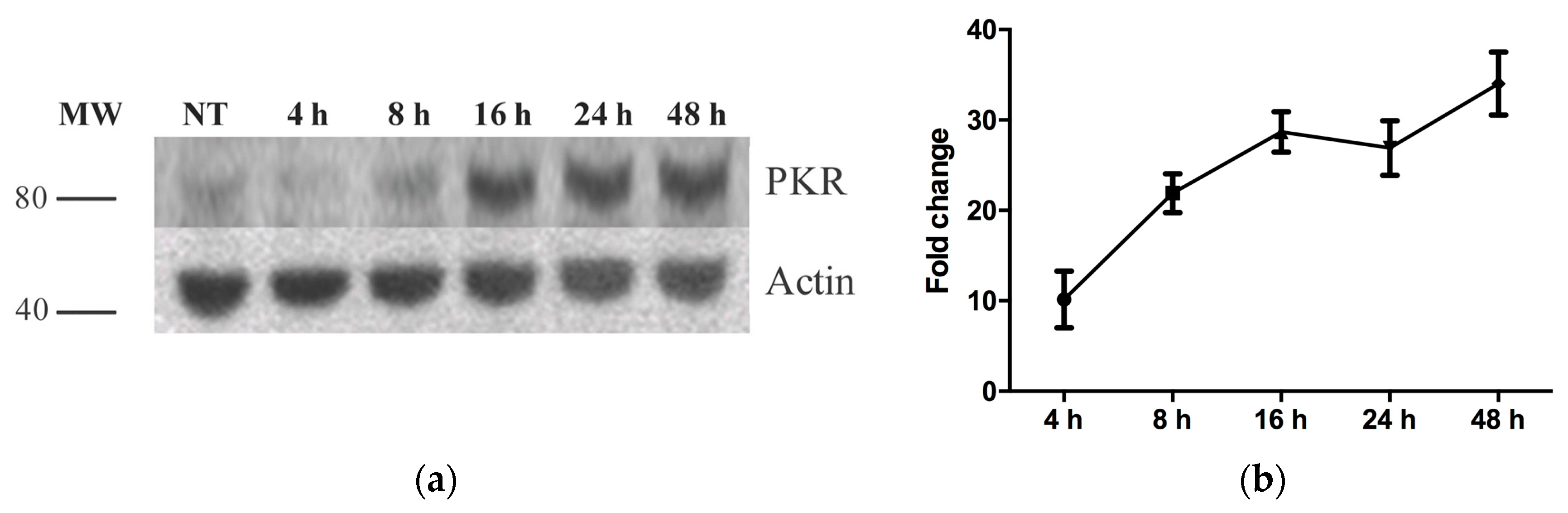
3.2. PKR Expression in CHSE-214 Cells Is Induced following IFNα Treatment
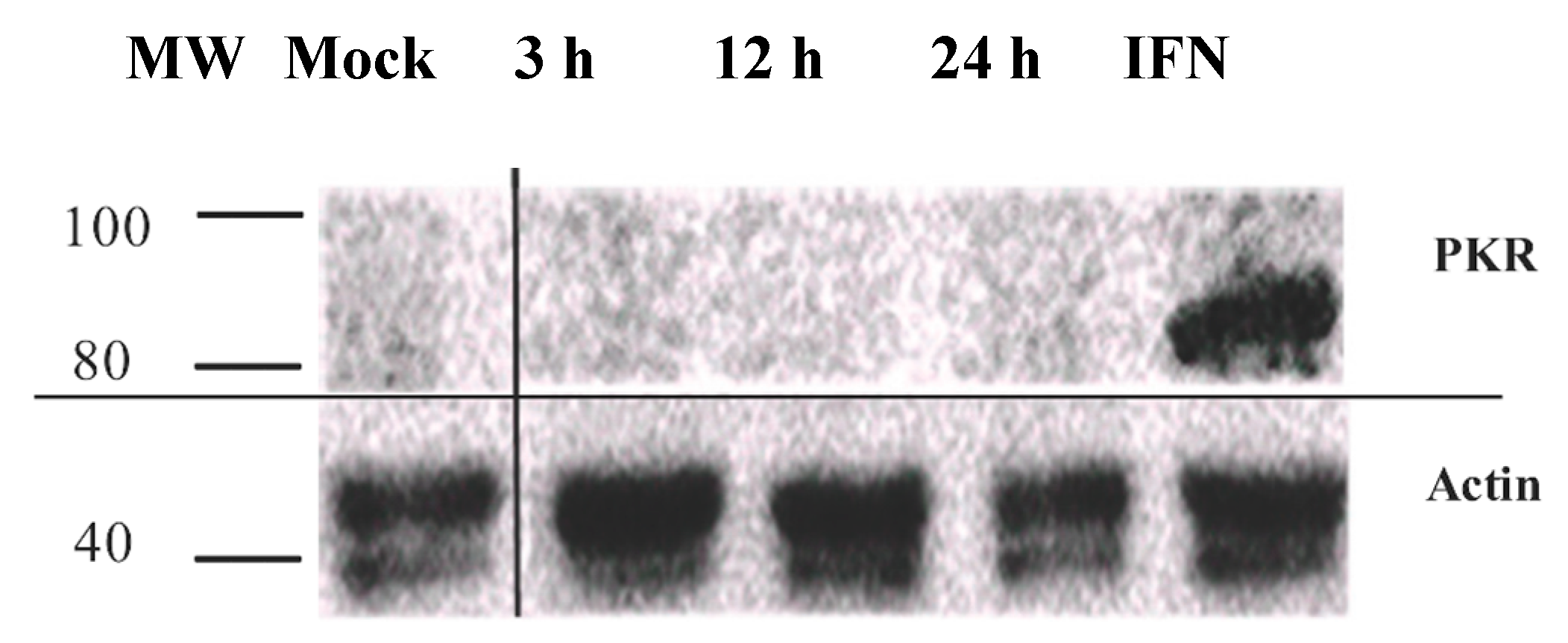
3.3. PKR Expression Is Not Induced in IPNV Infected Cells
3.4. Pretreatment of Cells with PKR Inhibitor Alters eIF2α Phosphorylation after IPNV Infection
3.5. Inhibition of PKR Reduces Virus Yield in the Supernatant
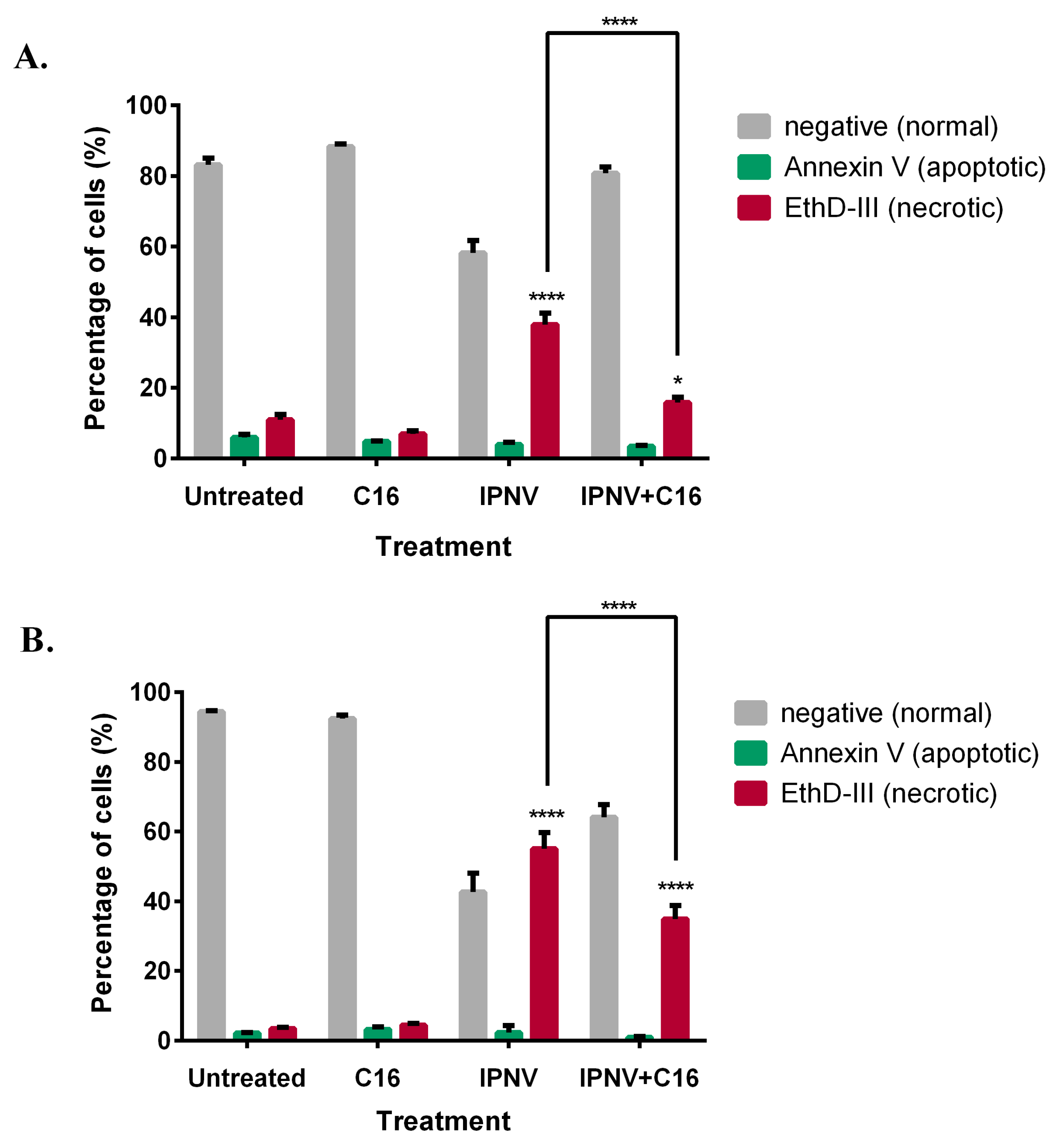
3.6. Inhibition of PKR Results in Decreased IPNV-Induced Membrane Damage
3.7. The Decrease in Membrane Damage and Virus Replication Results from Inhibition of Virus Replication
4. Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| IPNV | infectious pancreatic necrosis virus |
| PKR | phosphokinase R |
| eIF2a | eukaryotic initiation factor 2 alpha |
References
- Honda, K.; Yanai, H.; Takaoka, A.; Taniguchi, T. Regulation of the type I IFN induction: A current view. Int. Immunol. 2005, 17, 1367–1378. [Google Scholar] [CrossRef] [PubMed]
- Schoggins, J.W.; Rice, C.M. Interferon-stimulated genes and their antiviral effector functions. Curr. Opin. Virol. 2011, 1, 519–525. [Google Scholar] [CrossRef] [PubMed]
- Rothenburg, S.; Deigendesch, N.; Dey, M.; Dever, T.E.; Tazi, L. Double-stranded RNA-activated protein kinase PKR of fishes and amphibians: Varying the number of double-stranded rna binding domains and lineage-specific duplications. BMC Biol. 2008, 6, 12. [Google Scholar] [CrossRef] [PubMed]
- Perelygin, A.A.; Lear, T.L.; Zharkikh, A.A.; Brinton, M.A. Comparative analysis of vertebrate eIF2AK2 (PKR) genes and assignment of the equine gene to ECA15q24-q25 and the bovine gene to BTA11q12-q15. Genet. Sel. Evol. 2006, 38, 551–563. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.S.; Li, W.; Li, D.M.; Liu, Y.; Fan, L.H.; Rao, Z.C.; Lin, G.; Hu, C.Y. Cloning, expression and functional analysis of PKR from grass carp (Ctenopharyngodon idellus). Fish Shellfish Immunol. 2013, 35, 1874–1881. [Google Scholar] [CrossRef] [PubMed]
- Bevilacqua, P.C.; George, C.X.; Samuel, C.E.; Cech, T.R. Binding of the protein kinase PKR to rnas with secondary structure defects: Role of the tandem A-G mismatch and noncontiguous helixes. Biochemistry 1998, 37, 6303–6316. [Google Scholar] [CrossRef] [PubMed]
- Dauber, B.; Martinez-Sobrido, L.; Schneider, J.; Hai, R.; Waibler, Z.; Kalinke, U.; Garcia-Sastre, A.; Wolff, T. Influenza B virus ribonucleoprotein is a potent activator of the antiviral kinase PKR. PLoS Pathog. 2009, 5, e1000473. [Google Scholar] [CrossRef] [PubMed]
- Manche, L.; Green, S.R.; Schmedt, C.; Mathews, M.B. Interactions between double-stranded-RNA regulators and the protein-kinase DAI. Mol. Cell. Biol. 1992, 12, 5238–5248. [Google Scholar] [CrossRef] [PubMed]
- Dey, M.; Cao, C.; Dar, A.C.; Tamura, T.; Ozato, K.; Sicheri, F.; Dever, T.E. Mechanistic link between PKR dimerization, autophosphorylation, and eLF2 alpha substrate recognition. Cell 2005, 122, 901–913. [Google Scholar] [CrossRef] [PubMed]
- Asano, K.; Clayton, J.; Shalev, A.; Hinnebusch, A.G. A multifactor complex of eukaryotic initiation factors, eIE1, eIF2, eIF3, eIF5, and initiator tRNA(Met) is an important translation initiation intermediate in vivo. Genes Dev. 2000, 14, 2534–2546. [Google Scholar] [CrossRef] [PubMed]
- Gil, J.; Esteban, M. The interferon-induced protein kinase (PKR), triggers apoptosis through FADD-mediated activation of caspase 8 in a manner independent of Fas and TNF-alpha receptors. Oncogene 2000, 19, 3665–3674. [Google Scholar] [CrossRef] [PubMed]
- Lindcluist, M.E.; Mainou, B.A.; Dermody, T.S.; Crowe, J.E. Activation of protein kinase R is required for induction of stress granules by respiratory syncytial virus but dispensable for viral replication. Virology 2011, 413, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Thapa, R.J.; Nogusa, S.; Chen, P.; Maki, J.L.; Lerro, A.; Andrake, M.; Rall, G.F.; Degterev, A.; Balachandran, S. Interferon-induced RIP1/RIP3-mediated necrosis requires PKR and is licensed by FADD and caspases. Proc. Natl. Acad. Sci. USA 2013, 110, E3109–E3118. [Google Scholar] [CrossRef] [PubMed]
- Gil, J.; Garcia, M.A.; Esteban, M. Caspase 9 activation by the dsRNA-dependent protein kinase, PKR: Molecular mechanism and relevance. FEBS Lett. 2002, 529, 249–255. [Google Scholar] [CrossRef]
- Courtney, S.C.; Scherbik, S.V.; Stockman, B.M.; Brinton, M.A. West nile virus infections suppress early viral RNA synthesis and avoid inducing the cell stress granule response. J. Virol. 2012, 86, 3647–3657. [Google Scholar] [CrossRef] [PubMed]
- Dobos, P. The molecular biology of infectious pancreatic necrosis virus (IPNV). Annu. Rev. Fish Dis. 1995, 5, 25–54. [Google Scholar] [CrossRef]
- Duncan, R.; Mason, C.L.; Nagy, E.; Leong, J.A.; Dobos, P. Sequence-analysis of infectious pancreatic necrosis virus genome segment-B and its encoded VP1 protein—A putative RNA-dependent RNA-polymerase lacking the Gly-Asp-Asp motif. Virology 1991, 181, 541–552. [Google Scholar] [CrossRef]
- Duncan, R.; Nagy, E.; Krell, P.J.; Dobos, P. Synthesis of the infectious pancreatic necrosis virus polyprotein, detection of a virus-encoded protease, and fine-structure mapping of genome segment A coding regions. J. Virol. 1987, 61, 3655–3664. [Google Scholar] [PubMed]
- Macdonald, R.D.; Dobos, P. Identification of the proteins encoded by each genome segment of infectious pancreatic necrosis virus. Virology 1981, 114, 414–422. [Google Scholar] [CrossRef]
- Petit, S.; Lejal, N.; Huet, J.C.; Delmas, B. Active residues and viral substrate cleavage sites of the protease of the birnavirus infectious pancreatic necrosis virus. J. Virol. 2000, 74, 2057–2066. [Google Scholar] [CrossRef] [PubMed]
- Havarstein, L.S.; Kalland, K.H.; Christie, K.E.; Endresen, C. Sequence of the large double-stranded-RNA segment of the N1 strain of infectious pancreatic necrosis virus—A comparison with other Birnaviridae. J. Gen. Virol. 1990, 71, 299–308. [Google Scholar] [CrossRef] [PubMed]
- Magyar, G.; Dobos, P. Evidence for the detection of the infectious pancreatic necrosis virus polyprotein and the 17-kDa polypeptide in infected-cells and of the NS protease in purified virus. Virology 1994, 204, 580–589. [Google Scholar] [CrossRef] [PubMed]
- Evensen, Ø.; Santi, N. Infectious pancreatic necrosis virus. In Encyclopedia of Virology, 3rd ed.; Mahy, W.J., van Regenmortel, M.H.V., Eds.; Academic Press: Oxford, UK, 2008; pp. 83–89. [Google Scholar]
- Hong, J.R.; Lin, T.L.; Hsu, Y.L.; Wu, J.L. Apoptosis precedes necrosis of fish cell line with infectious pancreatic necrosis virus infection. Virology 1998, 250, 76–84. [Google Scholar] [CrossRef] [PubMed]
- Gamil, A.A.A.; Mutoloki, S.; Evensen, O. A piscine birnavirus induces inhibition of protein synthesis in CHSE-214 cells primarily through the induction of eIF2 alpha phosphorylation. Viruses 2015, 7, 1987–2005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Espinoza, J.C.; Cortes-Gutierrez, M.; Kuznar, J. Necrosis of infectious pancreatic necrosis virus (IPNV) infected cells rarely is preceded by apoptosis. Virus Res. 2005, 109, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Lannan, C.N.; Winton, J.R.; Fryer, J.L. Fish cell lines: Establishment and characterization of nine cell lines from salmonids. In Vitro 1984, 20, 671–676. [Google Scholar] [CrossRef] [PubMed]
- Froystad, M.K.; Rode, M.; Berg, T.; Gjoen, T. A role for scavenger receptors in phagocytosis of protein-coated particles in rainbow trout head kidney macrophages. Dev. Comp. Immunol. 1998, 22, 533–549. [Google Scholar] [CrossRef]
- Munang’andu, H.M.; Fredriksen, B.N.; Mutoloki, S.; Brudeseth, B.; Kuo, T.Y.; Marjara, I.S.; Dalmo, R.A.; Evensen, O. Comparison of vaccine efficacy for different antigen delivery systems for infectious pancreatic necrosis virus vaccines in atlantic salmon (Salmo salar L.) in a cohabitation challenge model. Vaccine 2012, 30, 4007–4016. [Google Scholar] [CrossRef] [PubMed]
- Song, H.C.; Santi, N.; Evensen, O.; Vakharia, V.N. Molecular determinants of infectious pancreatic necrosis virus virulence and cell culture adaptation. J. Virol. 2005, 79, 10289–10299. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Guo, T.C.; Mutoloki, S.; Haugland, O.; Marjara, I.S.; Evensen, O. Alpha interferon and not gamma interferon inhibits salmonid alphavirus subtype 3 replication in vitro. J. Virol. 2010, 84, 8903–8912. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Kärber, G. Beitrag zur kollektiven Behandlung pharmakologischer Reihenversuche. Arch. Exp. Path. Pharmak 1931, 162, 480–487. (In Germany) [Google Scholar] [CrossRef]
- Liu, T.K.; Zhang, Y.B.; Liu, Y.; Sun, F.; Gui, J.F. Cooperative roles of fish protein kinase containing Z-DNA binding domains and double-stranded RNA-dependent protein kinase in interferon-mediated antiviral response. J. Virol. 2011, 85, 12769–12780. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.L.; Wu, J.L.; Chen, M.H.; Hong, J.R. Aquatic birnavirus-induced ER stress-mediated death signaling contribute to downregulation of Bcl-2 family proteins in salmon embryo cells. PLoS ONE 2011, 6, e22935. [Google Scholar] [CrossRef] [PubMed]
- Garner, J.N.; Joshi, B.; Jagus, R. Characterization of rainbow trout and zebrafish eukaryotic initiation factor 2 alpha and its response to endoplasmic reticulum stress and ipnv infection. Dev. Comp. Immunol. 2003, 27, 217–231. [Google Scholar] [CrossRef]
- Arnaud, N.; Dabo, S.; Maillard, P.; Budkowska, A.; Kalliampakou, K.I.; Mavromara, P.; Garcin, D.; Hugon, J.; Gatignol, A.; Akazawa, D.; et al. Hepatitis c virus controls interferon production through PKR activation. PLoS ONE 2010, 5, e10575. [Google Scholar] [CrossRef] [PubMed]
- Garcia, M.A.; Gil, J.; Ventoso, I.; Guerra, S.; Domingo, E.; Rivas, C.; Esteban, M. Impact of protein kinase pkr in cell biology: From antiviral to antiproliferative action. Microbiol. Mol. Biol. Rev. 2006, 70, 1032–1060. [Google Scholar] [CrossRef] [PubMed]
- Rosenberger, C.M.; Clark, A.E.; Treuting, P.M.; Johnson, C.D.; Aderem, A. ATF3 regulates MCMV infection in mice by modulating IFN-gamma expression in natural killer cells. Proc. Natl. Acad. Sci. USA 2008, 105, 2544–2549. [Google Scholar] [CrossRef] [PubMed]
- Hiscott, J.; Kwon, H.; Genin, P. Hostile takeovers: Viral appropriation of the NF-kappaB pathway. J. Clin. Invest. 2001, 107, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Andreev, D.E.; O’Connor, P.B.F.; Fahey, C.; Kenny, E.M.; Terenin, I.M.; Dmitriev, S.E.; Cormican, P.; Morris, D.W.; Shatsky, I.N.; Baranov, P.V. Translation of 5’ leaders is pervasive in genes resistant to eIF2 repression. eLife 2015, 4. [Google Scholar] [CrossRef] [PubMed]
- Shabman, R.S.; Hoenen, T.; Groseth, A.; Jabado, O.; Binning, J.M.; Amarasinghe, G.K.; Feldmann, H.; Basler, C.F. An upstream open reading frame modulates ebola virus polymerase translation and virus replication. PLoS. Pathog. 2013, 9, e1003147. [Google Scholar] [CrossRef] [PubMed]
- Weber, S.; Fichtner, D.; Mettenleiter, T.C.; Mundt, E. Expression of VP5 of infectious pancreatic necrosis virus strain VR299 is initiated at the second in-frame start codon. J. Gen. Virol. 2001, 82, 805–812. [Google Scholar] [CrossRef] [PubMed]
- Santi, N.; Song, H.; Vakharia, V.N.; Evensen, O. Infectious pancreatic necrosis virus VP5 is dispensable for virulence and persistence. J. Virol. 2005, 79, 9206–9216. [Google Scholar] [CrossRef] [PubMed]
- Galloux, M.; Libersou, S.; Morellet, N.; Bouaziz, S.; da Costa, B.; Ouldali, M.; Lepault, J.; Delmas, B. Infectious bursal disease virus, a non-enveloped virus, possesses a capsid-associated peptide that deforms and perforates biological membranes. J. Biol. Chem. 2007, 282, 20774–20784. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.L.; Hong, J.R.; Lin, G.H.; Liu, W.; Gong, H.Y.; Lu, M.W.; Lin, C.C.; Wu, J.L. Stage-specific expression of TNFalpha regulates Bad/Bid-mediated apoptosis and RIP1/ROS-mediated secondary necrosis in birnavirus-infected fish cells. PLoS ONE 2011, 6, e16740. [Google Scholar]
- Gainey, M.D.; Dillon, P.J.; Clark, K.M.; Manuse, M.J.; Parks, G.D. Paramyxovirus-induced shutoff of host and viral protein synthesis: Role of the P and V proteins in limiting PKR activation. J. Virol. 2008, 82, 828–839. [Google Scholar] [CrossRef] [PubMed]
- Busnadiego, I.; Maestre, A.M.; Rodriguez, D.; Rodriguez, J.F. The infectious bursal disease virus RNA-binding VP3 polypeptide inhibits PKR-mediated apoptosis. PLoS ONE 2012, 7, e46768. [Google Scholar] [CrossRef] [PubMed]
- Barry, G.; Breakwell, L.; Fragkoudis, R.; Attarzadeh-Yazdi, G.; Rodriguez-Andres, J.; Kohl, A.; Fazakerley, J.K. PKR acts early in infection to suppress semliki forest virus production and strongly enhances the type I interferon response. J. Gen. Virol. 2009, 90, 1382–1391. [Google Scholar] [CrossRef] [PubMed]
- Dauber, B.; Wolff, T. Activation of the antiviral kinase PKR and viral countermeasures. Viruses 2009, 1, 523–544. [Google Scholar] [CrossRef] [PubMed]
- Schneider, R.J.; Mohr, I. Translation initiation and viral tricks. Trends Biochem. Sci. 2003, 28, 130–136. [Google Scholar] [CrossRef]
- Taddeo, B.; Luo, T.R.; Zhang, W.R.; Roizman, B. Activation of NF-kappaB in cells productively infected with HSV-1 depends on activated protein kinase R and plays no apparent role in blocking apoptosis. Proc. Natl. Acad. Sci. USA 2003, 100, 12408–12413. [Google Scholar] [CrossRef] [PubMed]
- Santoro, M.G.; Rossi, A.; Amici, C. NF-kappaB and virus infection: Who controls whom. EMBO J. 2003, 22, 2552–2560. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.R.; Guan, B.J.; Her, G.M.; Evensen, O.; Santi, N.; Wu, J.L. Aquatic birnavirus infection activates the transcription factor NF-kappaB via tyrosine kinase signalling leading to cell death. J. Fish Dis. 2008, 31, 451–460. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Kim, C.; Yel, L.; Gollapudi, S. A role of Fas-associated death domain (FADD) in increased apoptosis in aged humans. J. Clin. Immunol. 2004, 24, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Thorburn, J.; Bender, L.M.; Morgan, M.J.; Thorburn, A. Caspase- and serine protease-dependent apoptosis by the death domain of fadd in normal epithelial cells. Mol. Biol. Cell 2003, 14, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y. RIP1-mediated signaling pathways in cell survival and death control. In Necrotic Cell Death; Shen, H.-M., Vandenabeele, P., Eds.; Springer: New York, NY, USA, 2014; pp. 23–43. [Google Scholar]
- Ingrand, S.; Barrier, L.; Lafay-Chebassier, C.; Fauconneau, B.; Page, G.; Hugon, J. The oxindole/imidazole derivative c16 reduces in vivo brain PKR activation. FEBS Lett. 2007, 581, 4473–4478. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Primer Sequence | Use | Genbank Accession No. |
|---|---|---|---|
| B actin-F | CCAGTCCTGCTCACTGAGGC | qPCR | AF012125 |
| B actin-R | GGTCTCAAACATGATCTGGGTCA | ||
| IPNV-F | CAACAGGGTTCGACAAACCATAC | qPCR | |
| IPNV-R | TTGACGATGTCGGCGTTTC | ||
| PKR-F1 | TCAACGCATTCTACTGCAC | pGEMT cloning | EF523422.1 |
| PKR-R1 | GAAACCCAGCCTAAAACCC | ||
| pET32c-PKR-F | GCGGAATTCGAGATTCCACAAATT | pET32c cloning | |
| pET32c-PKR-R | GCGAAGCTTTTAGATTGTTCTGTTG | ||
| PKR-F2 | ATGAACACAGCCAGAAGAAC | qPCR | EF523422.1 |
| PKR-R2 | TTCTCACCTACCACATACCTAC |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gamil, A.A.A.; Xu, C.; Mutoloki, S.; Evensen, Ø. PKR Activation Favors Infectious Pancreatic Necrosis Virus Replication in Infected Cells. Viruses 2016, 8, 173. https://0-doi-org.brum.beds.ac.uk/10.3390/v8060173
Gamil AAA, Xu C, Mutoloki S, Evensen Ø. PKR Activation Favors Infectious Pancreatic Necrosis Virus Replication in Infected Cells. Viruses. 2016; 8(6):173. https://0-doi-org.brum.beds.ac.uk/10.3390/v8060173
Chicago/Turabian StyleGamil, Amr A.A., Cheng Xu, Stephen Mutoloki, and Øystein Evensen. 2016. "PKR Activation Favors Infectious Pancreatic Necrosis Virus Replication in Infected Cells" Viruses 8, no. 6: 173. https://0-doi-org.brum.beds.ac.uk/10.3390/v8060173