
DNA Damage Reduces the Quality, but Not the Quantity of Human Papillomavirus 16 E1 and E2 DNA Replication

 ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Plasmids
2.3. Transient DNA Replication Assay
2.4. DNA Mutagenesis Analysis
2.5. Western Blots
2.6. Fluorescence-Activated Cell Sorting Analysis
2.7. Etoposide Treatment
2.8. Small interfering RNA (siRNA) and Metaphase Spread
2.9. Chromatin Immunoprecipitation
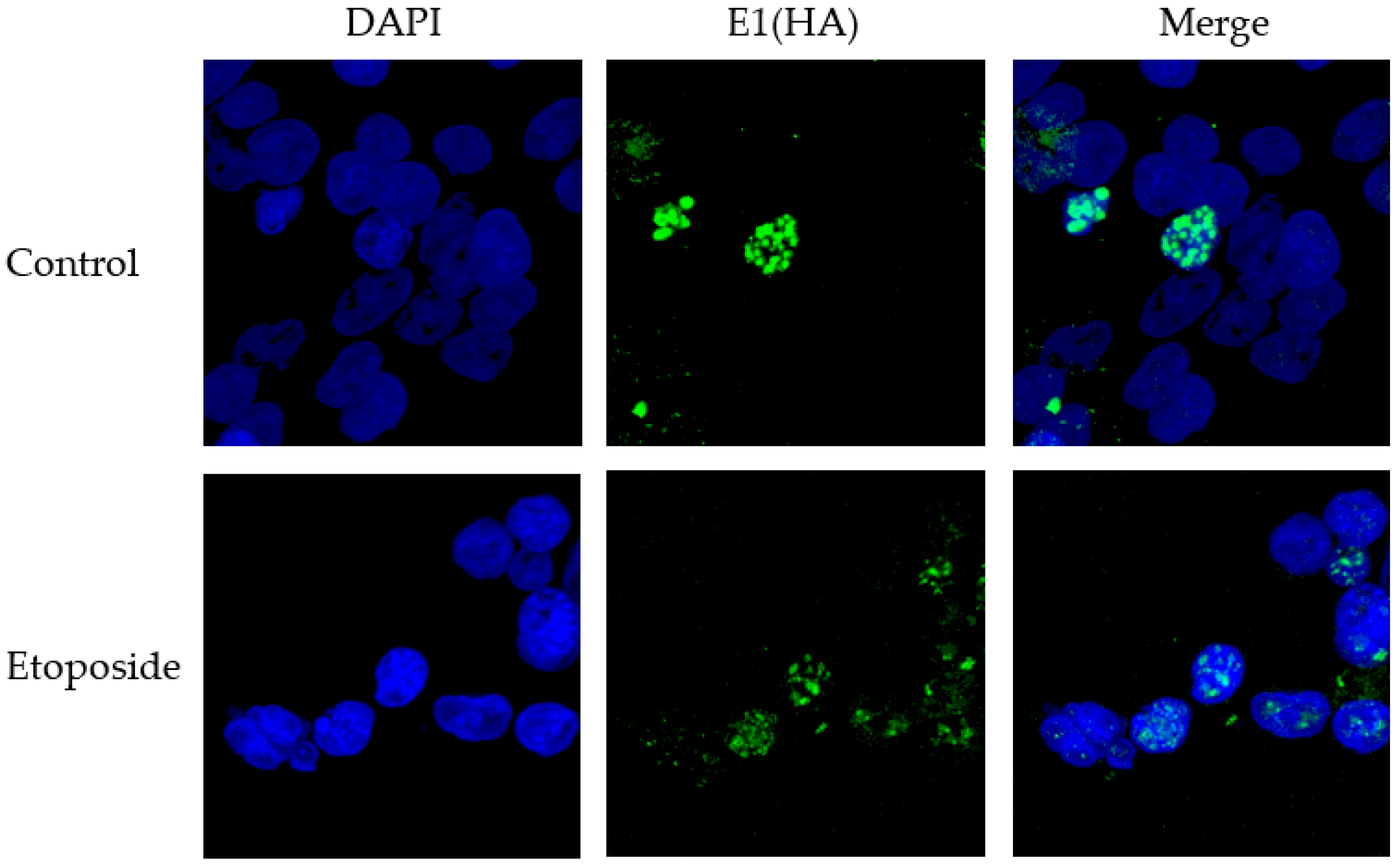
2.10. Immunofluorescence
3. Results
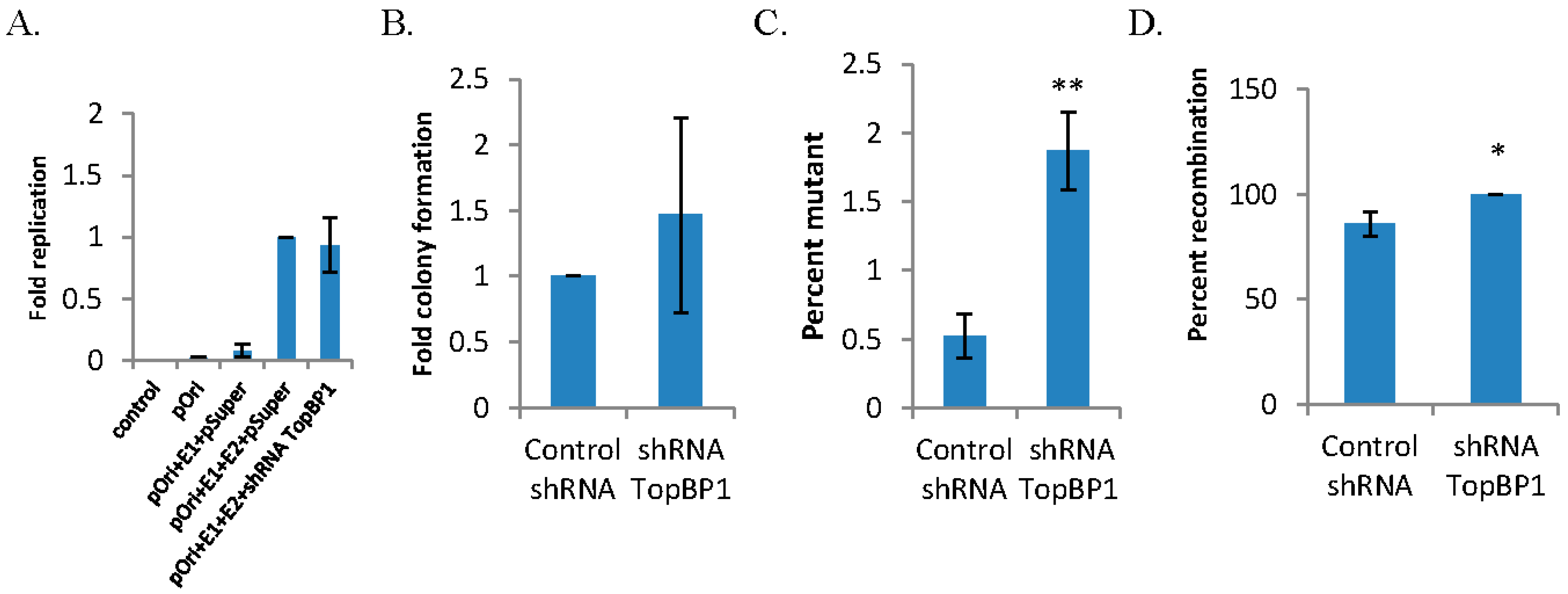
3.1. shRNA Knockdown of TopBP1 is Mutagenic for HPV16 E1–E2-Mediated DNA Replication
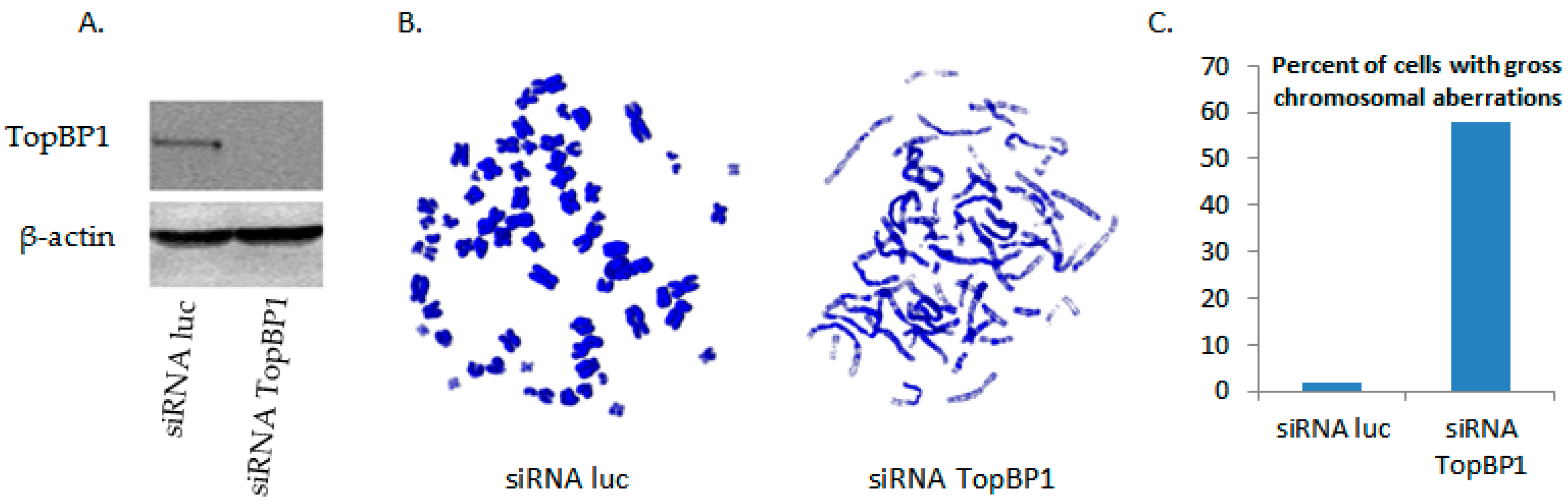
3.2. siRNA Knockout of TopBP1 Leads to Chromosome Dysfunction in Metaphase Cells
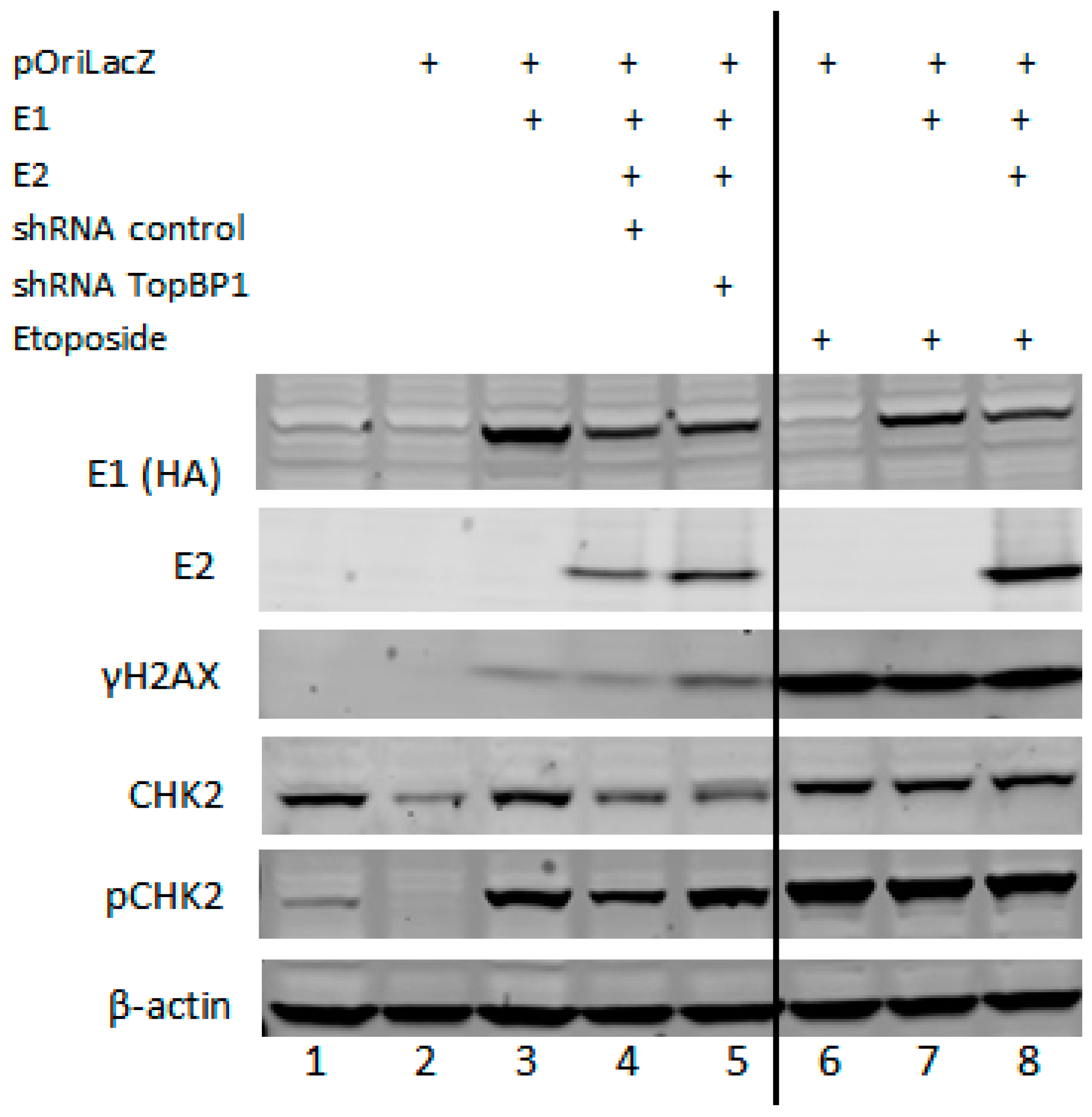
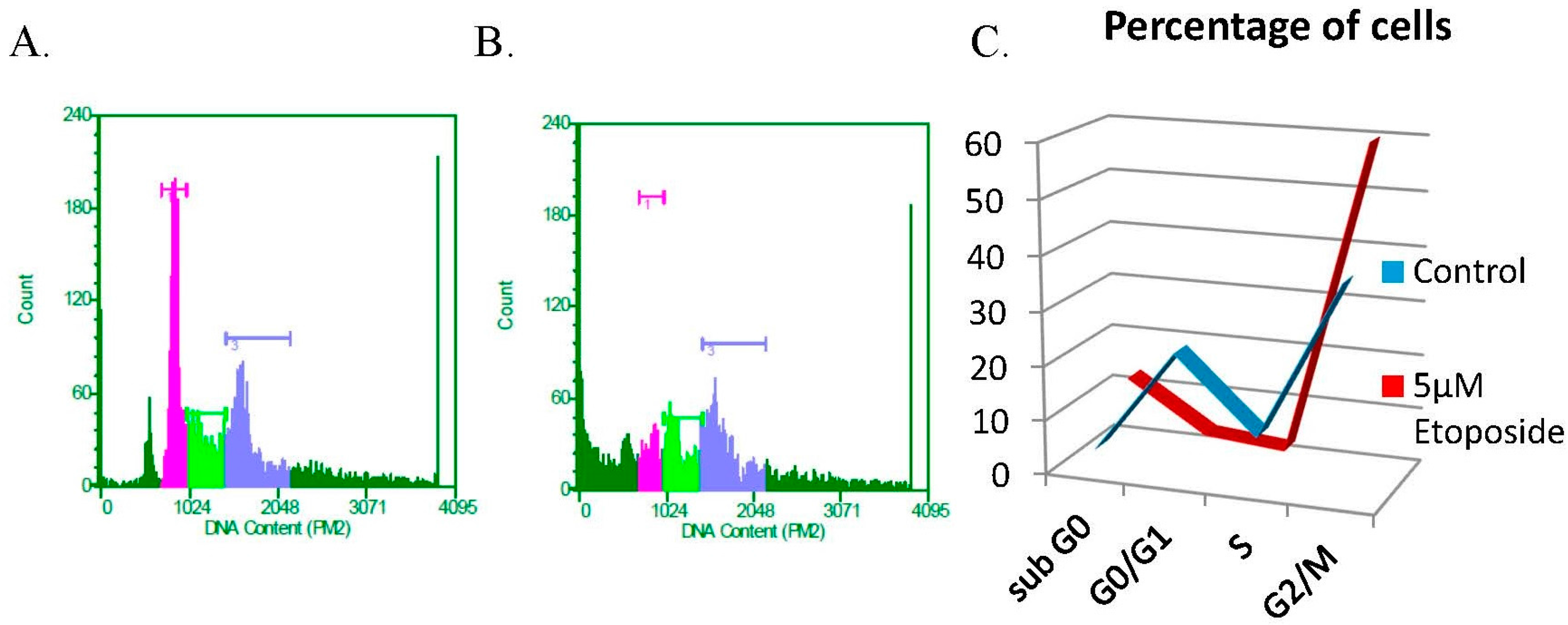
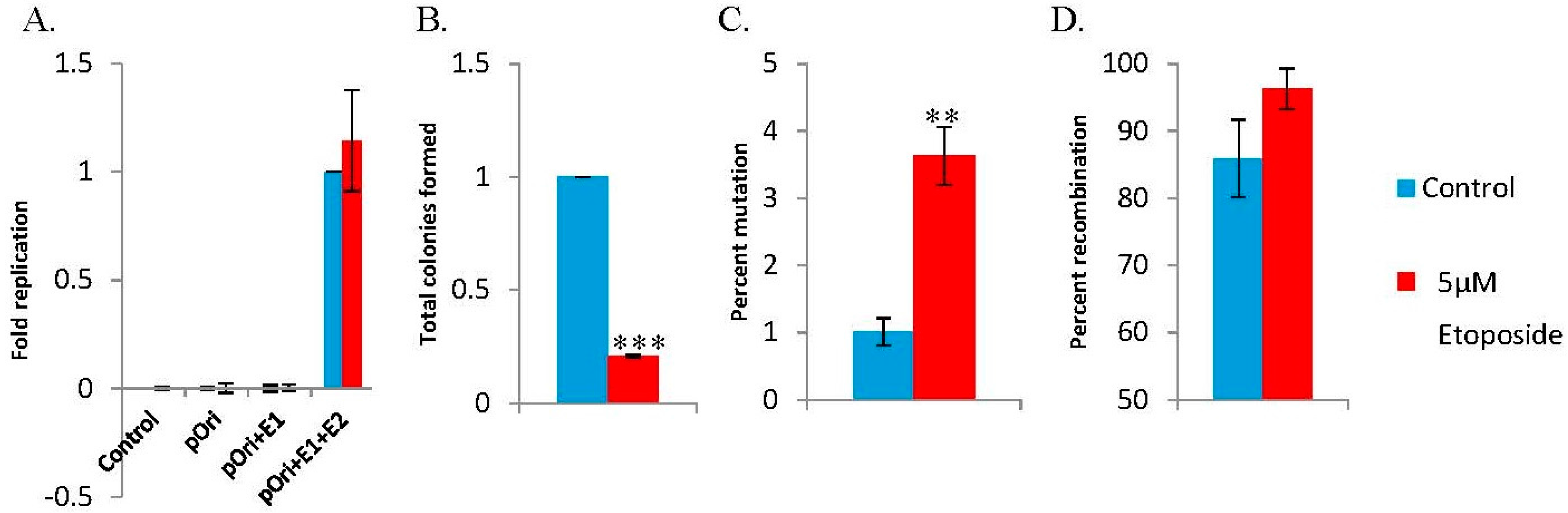
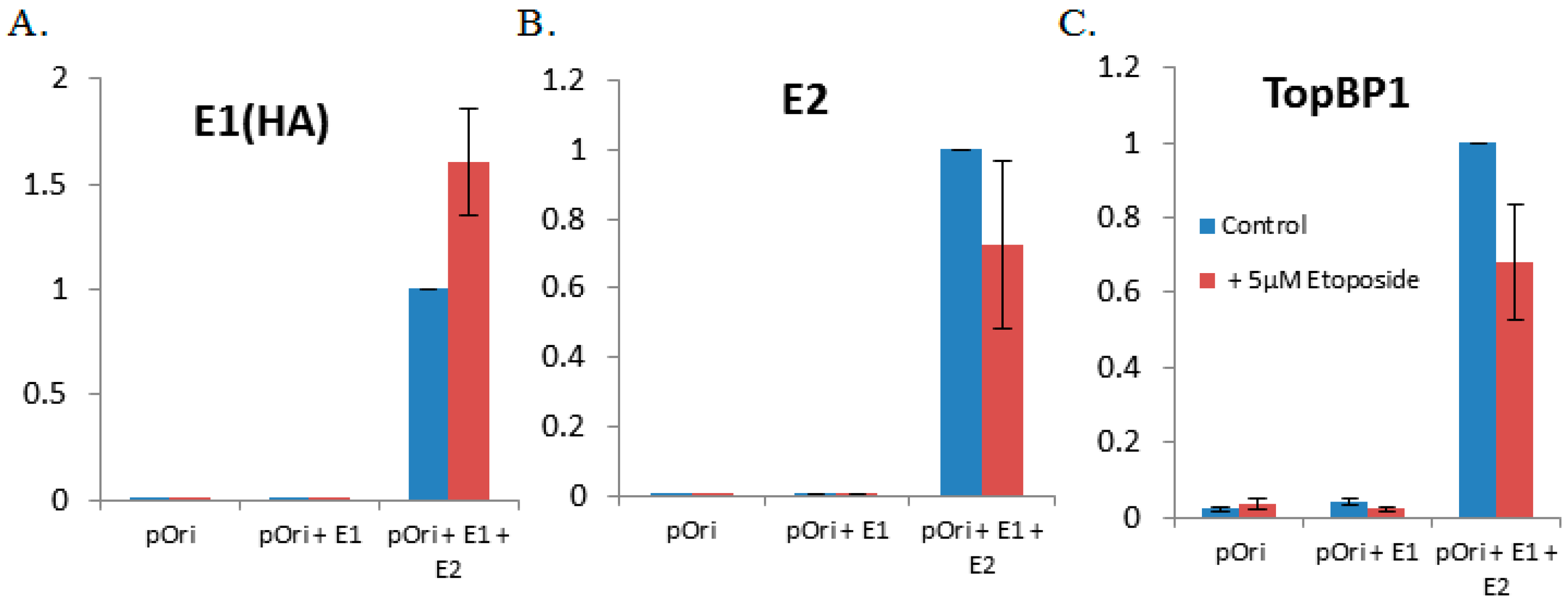
3.3. The DNA Damaging Agent Etoposide Promotes Low Fidelity E1–E2 DNA Replication
4. Discussion
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- IARC Working Group on the Evaluation of Carcinogenic Risk to Humans. IARC Monographs on the Evaluation of Carcinogenic Risks to Humans, Volume 90, Human Papillomavirus; Lyon, France, 2007; Volume 90. [Google Scholar]
- Psyrri, A.; DiMaio, D. Human Papillomavirus in Cervical and Head-and-Neck Cancer. Nat. Clin. Pract. Oncol. 2008, 5, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Dall, K.L.; Scarpini, C.G.; Roberts, I.; Winder, D.M.; Stanley, M.A.; Muralidhar, B.; Herdman, M.T.; Pett, M.R.; Coleman, N. Characterization of Naturally Occurring HPV16 Integration Sites Isolated from Cervical Keratinocytes Under Noncompetitive Conditions. Cancer Res. 2008, 68, 8249–8259. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Shen, C.; Zhao, L.; Wang, J.; McCrae, M.; Chen, X.; Lu, F. Dysregulation of Host Cellular Genes Targeted by Human Papillomavirus (HPV) Integration Contributes to HPV-Related Cervical Carcinogenesis. Int. J. Cancer 2016, 138, 1163–1174. [Google Scholar] [CrossRef] [PubMed]
- Fisher, C. Recent Insights into the Control of Human Papillomavirus (HPV) Genome Stability, Loss, and Degradation. J. Clin. Med. 2015, 4, 204–230. [Google Scholar] [CrossRef] [PubMed]
- Bodily, J.; Laimins, L.A. Persistence of Human Papillomavirus Infection: Keys to Malignant Progression. Trends Microbiol. 2011, 19, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Lehoux, M.; D’Abramo, C.M.; Archambault, J. Molecular Mechanisms of Human Papillomavirus-Induced Carcinogenesis. Public. Health. Genomics 2009, 12, 268–280. [Google Scholar] [CrossRef] [PubMed]
- Melendy, T.; Sedman, J.; Stenlund, A. Cellular Factors Required for Papillomavirus DNA Replication. J. Virol. 1995, 69, 7857–7867. [Google Scholar] [PubMed]
- Narahari, J.; Fisk, J.C.; Melendy, T.; Roman, A. Interactions of the Cellular CCAAT Displacement Protein and Human Papillomavirus E2 Protein with the Viral Origin of Replication can Regulate DNA Replication. Virology 2006, 350, 302–311. [Google Scholar] [CrossRef] [PubMed]
- Clower, R.V.; Hu, Y.; Melendy, T. Papillomavirus E2 Protein Interacts with and Stimulates Human Topoisomerase I. Virology 2006, 348, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Morin, G.; Fradet-Turcotte, A.; Di Lello, P.; Bergeron-Labrecque, F.; Omichinski, J.G.; Archambault, J. A Conserved Amphipathic Helix in the N-Terminal Regulatory Region of the Papillomavirus E1 Helicase is Required for Efficient Viral DNA Replication. J. Virol. 2011, 85, 5287–5300. [Google Scholar] [CrossRef] [PubMed]
- Lehoux, M.; Fradet-Turcotte, A.; Archambault, J. Methods to Assess the Nucleocytoplasmic Shuttling of the HPV E1 Helicase and its Effects on Cellular Proliferation and Induction of a DNA Damage Response. Methods Mol. Biol. 2015, 1249, 67–80. [Google Scholar]
- McBride, A.A. The Papillomavirus E2 Proteins. Virology 2013, 445, 57–79. [Google Scholar] [CrossRef] [PubMed]
- Weitzman, M.D.; Lilley, C.E.; Chaurushiya, M.S. Genomes in Conflict: Maintaining Genome Integrity during Virus Infection. Annu. Rev. Microbiol. 2010, 64, 61–81. [Google Scholar] [CrossRef] [PubMed]
- Moody, C.A.; Laimins, L.A. Human Papillomaviruses Activate the ATM DNA Damage Pathway for Viral Genome Amplification upon Differentiation. PLoS Pathog. 2009, 5, e1000605. [Google Scholar] [CrossRef] [PubMed]
- Kadaja, M.; Isok-Paas, H.; Laos, T.; Ustav, E.; Ustav, M. Mechanism of Genomic Instability in Cells Infected with the High-Risk Human Papillomaviruses. PLoS Pathog. 2009, 5, e1000397. [Google Scholar] [CrossRef] [PubMed]
- Kadaja, M.; Sumerina, A.; Verst, T.; Ojarand, M.; Ustav, E.; Ustav, M. Genomic Instability of the Host Cell Induced by the Human Papillomavirus Replication Machinery. EMBO J. 2007, 26, 2180–2191. [Google Scholar] [CrossRef] [PubMed]
- Jang, M.K.; Shen, K.; McBride, A.A. Papillomavirus Genomes Associate with BRD4 to Replicate at Fragile Sites in the Host Genome. PLoS Pathog. 2014, 10, e1004117. [Google Scholar] [CrossRef] [PubMed]
- Cardenas-Mora, J.; Spindler, J.E.; Jang, M.K.; McBride, A.A. Dimerization of the Papillomavirus E2 Protein is Required for Efficient Mitotic Chromosome Association and Brd4 Binding. J. Virol. 2008, 82, 7298–7305. [Google Scholar] [CrossRef] [PubMed]
- McPhillips, M.G.; Oliveira, J.G.; Spindler, J.E.; Mitra, R.; McBride, A.A. Brd4 is Required for e2-Mediated Transcriptional Activation but Not Genome Partitioning of all Papillomaviruses. J. Virol. 2006, 80, 9530–9543. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Laimins, L.A. Regulation of the Life Cycle of HPVs by Differentiation and the DNA Damage Response. Future Microbiol. 2013, 8, 1547–1557. [Google Scholar] [CrossRef] [PubMed]
- Gillespie, K.A.; Mehta, K.P.; Laimins, L.A.; Moody, C.A. Human Papillomaviruses Recruit Cellular DNA Repair and Homologous Recombination Factors to Viral Replication Centers. J. Virol. 2012, 86, 9520–9526. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Zhu, D.; Wang, W.; Li, W.; Jia, W.; Zeng, X.; Ding, W.; Yu, L.; Wang, X.; Wang, L.; et al. Genome-Wide Profiling of HPV Integration in Cervical Cancer Identifies Clustered Genomic Hot Spots and a Potential Microhomology-Mediated Integration Mechanism. Nat. Genet. 2015, 47, 158–163. [Google Scholar] [CrossRef] [PubMed]
- Das, P.; Thomas, A.; Kannan, S.; Deodhar, K.; Shrivastava, S.K.; Mahantshetty, U.; Mulherkar, R. Human Papillomavirus (HPV) Genome Status & Cervical Cancer Outcome—A Retrospective Study. Indian J. Med. Res. 2015, 142, 525–532. [Google Scholar] [PubMed]
- Shin, H.J.; Joo, J.; Yoon, J.H.; Yoo, C.W.; Kim, J.Y. Physical Status of Human Papillomavirus Integration in Cervical Cancer is Associated with Treatment Outcome of the Patients Treated with Radiotherapy. PLoS One 2014, 9, e78995. [Google Scholar] [CrossRef] [PubMed]
- Vega-Pena, A.; Illades-Aguiar, B.; Flores-Alfaro, E.; Lopez-Bayghen, E.; Leyva-Vazquez, M.A.; Castaneda-Saucedo, E.; Alarcon-Romero Ldel, C. Risk of Progression of Early Cervical Lesions is Associated with Integration and Persistence of HPV-16 and Expression of E6, Ki-67, and Telomerase. J. Cytol. 2013, 30, 226–232. [Google Scholar] [PubMed]
- Yoshida, T.; Sano, T.; Oyama, T.; Kanuma, T.; Fukuda, T. Prevalence, Viral Load, and Physical Status of HPV 16 and 18 in Cervical Adenosquamous Carcinoma. Virchows Arch. 2009, 455, 253–259. [Google Scholar] [CrossRef] [PubMed]
- Pett, M.; Coleman, N. Integration of High-Risk Human Papillomavirus: A Key Event in Cervical Carcinogenesis? J. Pathol. 2007, 212, 356–367. [Google Scholar] [CrossRef] [PubMed]
- Veldman, T.; Horikawa, I.; Barrett, J.C.; Schlegel, R. Transcriptional Activation of the Telomerase hTERT Gene by Human Papillomavirus Type 16 E6 Oncoprotein. J. Virol. 2001, 75, 4467–4472. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. The Hallmarks of Cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- King, L.E.; Fisk, J.C.; Dornan, E.S.; Donaldson, M.M.; Melendy, T.; Morgan, I.M. Human Papillomavirus E1 and E2 Mediated DNA Replication is Not Arrested by DNA Damage Signalling. Virology 2010, 406, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Gauson, E.J.; Donaldson, M.M.; Dornan, E.S.; Wang, X.; Bristol, M.; Bodily, J.M.; Morgan, I.M. Evidence Supporting a Role for TopBP1 and Brd4 in the Initiation but Not Continuation of Human Papillomavirus 16 E1/E2-Mediated DNA Replication. J. Virol. 2015, 89, 4980–4991. [Google Scholar] [CrossRef] [PubMed]
- Taylor, E.R.; Dornan, E.S.; Boner, W.; Connolly, J.A.; McNair, S.; Kannouche, P.; Lehmann, A.R.; Morgan, I.M. The Fidelity of HPV16 E1/E2-Mediated DNA Replication. J. Biol. Chem. 2003, 278, 52223–52230. [Google Scholar] [CrossRef] [PubMed]
- Taylor, E.R.; Morgan, I.M. A Novel Technique with Enhanced Detection and Quantitation of HPV-16 E1- and E2-Mediated DNA Replication. Virology 2003, 315, 103–109. [Google Scholar] [CrossRef]
- Bouvard, V.; Storey, A.; Pim, D.; Banks, L. Characterization of the Human Papillomavirus E2 Protein: Evidence of Trans-Activation and Trans-Repression in Cervical Keratinocytes. EMBO J. 1994, 13, 5451–5459. [Google Scholar] [PubMed]
- Kingston, R.E.; Chen, C.A.; Rose, J.K. Calcium Phosphate Transfection. Curr. Protoc. Mol. Biol. 2003. [Google Scholar] [CrossRef]
- Boner, W.; Taylor, E.R.; Tsirimonaki, E.; Yamane, K.; Campo, M.S.; Morgan, I.M. A Functional Interaction between the Human Papillomavirus 16 transcription/replication Factor E2 and the DNA Damage Response Protein TopBP1. J. Biol. Chem. 2002, 277, 22297–22303. [Google Scholar] [CrossRef] [PubMed]
- Donaldson, M.M.; Mackintosh, L.J.; Bodily, J.M.; Dornan, E.S.; Laimins, L.A.; Morgan, I.M. An Interaction between Human Papillomavirus 16 E2 and TopBP1 is Required for Optimum Viral DNA Replication and Episomal Genome Establishment. J. Virol. 2012, 86, 12806–12815. [Google Scholar] [CrossRef] [PubMed]
- Donaldson, M.M.; Boner, W.; Morgan, I.M. TopBP1 Regulates Human Papillomavirus Type 16 E2 Interaction with Chromatin. J. Virol. 2007, 81, 4338–4342. [Google Scholar] [CrossRef] [PubMed]
- Sakakibara, N.; Mitra, R.; McBride, A.A. The Papillomavirus E1 Helicase Activates a Cellular DNA Damage Response in Viral Replication Foci. J. Virol. 2011, 85, 8981–8995. [Google Scholar] [CrossRef] [PubMed]
- Edwards, T.G.; Vidmar, T.J.; Koeller, K.; Bashkin, J.K.; Fisher, C. DNA Damage Repair Genes Controlling Human Papillomavirus (HPV) Episome Levels Under Conditions of Stability and Extreme Instability. PLoS One 2013, 8, e75406. [Google Scholar] [CrossRef] [PubMed]
- Fradet-Turcotte, A.; Bergeron-Labrecque, F.; Moody, C.A.; Lehoux, M.; Laimins, L.A.; Archambault, J. Nuclear Accumulation of the Papillomavirus E1 Helicase Blocks S-Phase Progression and Triggers an ATM-Dependent DNA Damage Response. J. Virol. 2011, 85, 8996–9012. [Google Scholar] [CrossRef] [PubMed]
- Wardlaw, C.P.; Carr, A.M.; Oliver, A.W. TopBP1: A BRCT-Scaffold Protein Functioning in Multiple Cellular Pathways. DNA Repair (Amst) 2014, 22, 165–174. [Google Scholar] [CrossRef] [PubMed]
- McKinney, C.C.; Hussmann, K.L.; McBride, A.A. The Role of the DNA Damage Response Throughout the Papillomavirus Life Cycle. Viruses 2015, 7, 2450–2469. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.P.; Bartek, J. The DNA-Damage Response in Human Biology and Disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- Jeon, Y.; Ko, E.; Lee, K.Y.; Ko, M.J.; Park, S.Y.; Kang, J.; Jeon, C.H.; Lee, H.; Hwang, D.S. TopBP1 Deficiency Causes an Early Embryonic Lethality and Induces Cellular Senescence in Primary Cells. J. Biol. Chem. 2011, 286, 5414–5422. [Google Scholar] [CrossRef] [PubMed]
- Johansson, C.; Graham, S.V.; Dornan, E.S.; Morgan, I.M. The Human Papillomavirus 16 E2 Protein is Stabilised in S Phase. Virology 2009, 394, 194–199. [Google Scholar] [CrossRef] [PubMed]
- Leonhardt, H.; Rahn, H.P.; Weinzierl, P.; Sporbert, A.; Cremer, T.; Zink, D.; Cardoso, M.C. Dynamics of DNA Replication Factories in Living Cells. J. Cell Biol. 2000, 149, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Sakakibara, N.; Chen, D.; Jang, M.K.; Kang, D.W.; Luecke, H.F.; Wu, S.Y.; Chiang, C.M.; McBride, A.A. Brd4 is Displaced from HPV Replication Factories as they Expand and Amplify Viral DNA. PLoS Pathog. 2013, 9, e1003777. [Google Scholar] [CrossRef] [PubMed]
- van Veelen, L.R.; Cervelli, T.; van de Rakt, M.W.; Theil, A.F.; Essers, J.; Kanaar, R. Analysis of Ionizing Radiation-Induced Foci of DNA Damage Repair Proteins. Mutat. Res. 2005, 574, 22–33. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| shRNA control | shRNA TopBP1 | |
|---|---|---|
| Totala | 11,210 | 17,428 |
| White | 59 | 326 |
| MF (×10-5)b | 526 | 1871 |
| Rearrangedc | 46/54 | 54/54 |
| Lane | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 |
|---|---|---|---|---|---|---|---|---|
| E1 | 0 | 0 | 2.7 | 1 | 1 | 0 | 1.43 | 0.9 |
| E2 | 0 | 0 | 0 | 1 | 1.5 | 0 | 0 | 2.9 |
| H2AX | 0 | 0 | 1 | 0.9 | 2.1 | 11.4 | 7.65 | 10 |
| Chk2 | 1 | 0.3 | 1 | 0.5 | 0.6 | 0.95 | 0.81 | 0.8 |
| pChk2 | 1 | 0 | 4.8 | 2.7 | 4.3 | 10 | 9.97 | 9.1 |
| Control | Etoposide | |
|---|---|---|
| Totala | 6431 | 1389 |
| White | 68 | 54 |
| MF (×10-5)b | 1057 | 3887 |
| Rearrangedc | 46/54 | 52/54 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bristol, M.L.; Wang, X.; Smith, N.W.; Son, M.P.; Evans, M.R.; Morgan, I.M. DNA Damage Reduces the Quality, but Not the Quantity of Human Papillomavirus 16 E1 and E2 DNA Replication. Viruses 2016, 8, 175. https://0-doi-org.brum.beds.ac.uk/10.3390/v8060175
Bristol ML, Wang X, Smith NW, Son MP, Evans MR, Morgan IM. DNA Damage Reduces the Quality, but Not the Quantity of Human Papillomavirus 16 E1 and E2 DNA Replication. Viruses. 2016; 8(6):175. https://0-doi-org.brum.beds.ac.uk/10.3390/v8060175
Chicago/Turabian StyleBristol, Molly L., Xu Wang, Nathan W. Smith, Minkyeong P. Son, Michael R. Evans, and Iain M. Morgan. 2016. "DNA Damage Reduces the Quality, but Not the Quantity of Human Papillomavirus 16 E1 and E2 DNA Replication" Viruses 8, no. 6: 175. https://0-doi-org.brum.beds.ac.uk/10.3390/v8060175