
Regulation of Stress Responses and Translational Control by Coronavirus

{kind=link}
{kind=link}
Abstract
:1. ER Stress Responses Regulated by Coronavirus and Its Implication in Pathogenesis
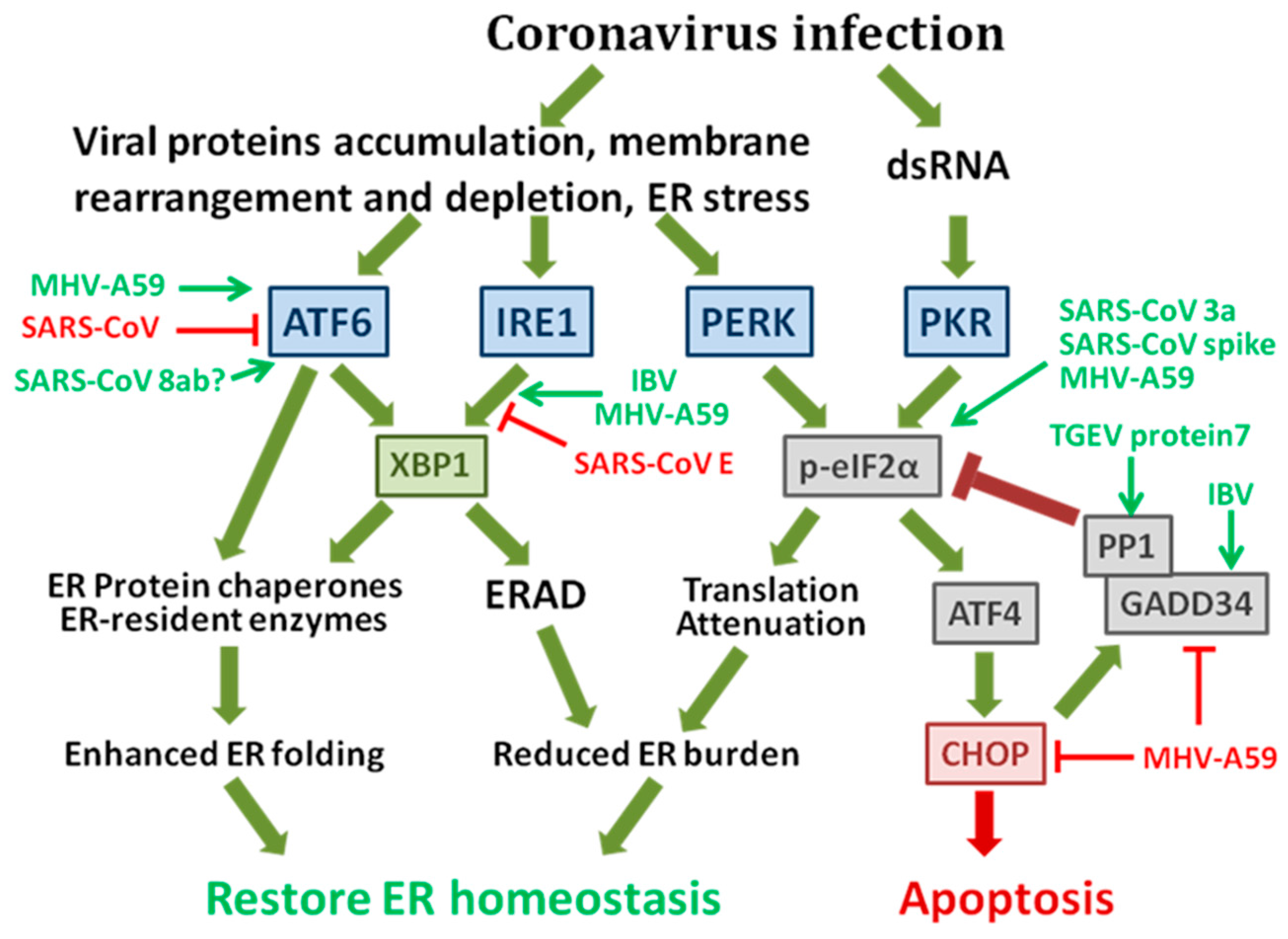
1.1. The Integrated Signaling Network of the Unfolded Protein Response (UPR)
1.2. The Possible Mechanisms of Coronavirus-Induced ER Stress Responses
1.3. Coronavirus-Induced UPR and Its Implication in Pathogenesis
2. Activation and subversion of p38 and JNK signaling pathways by coronavirus infection
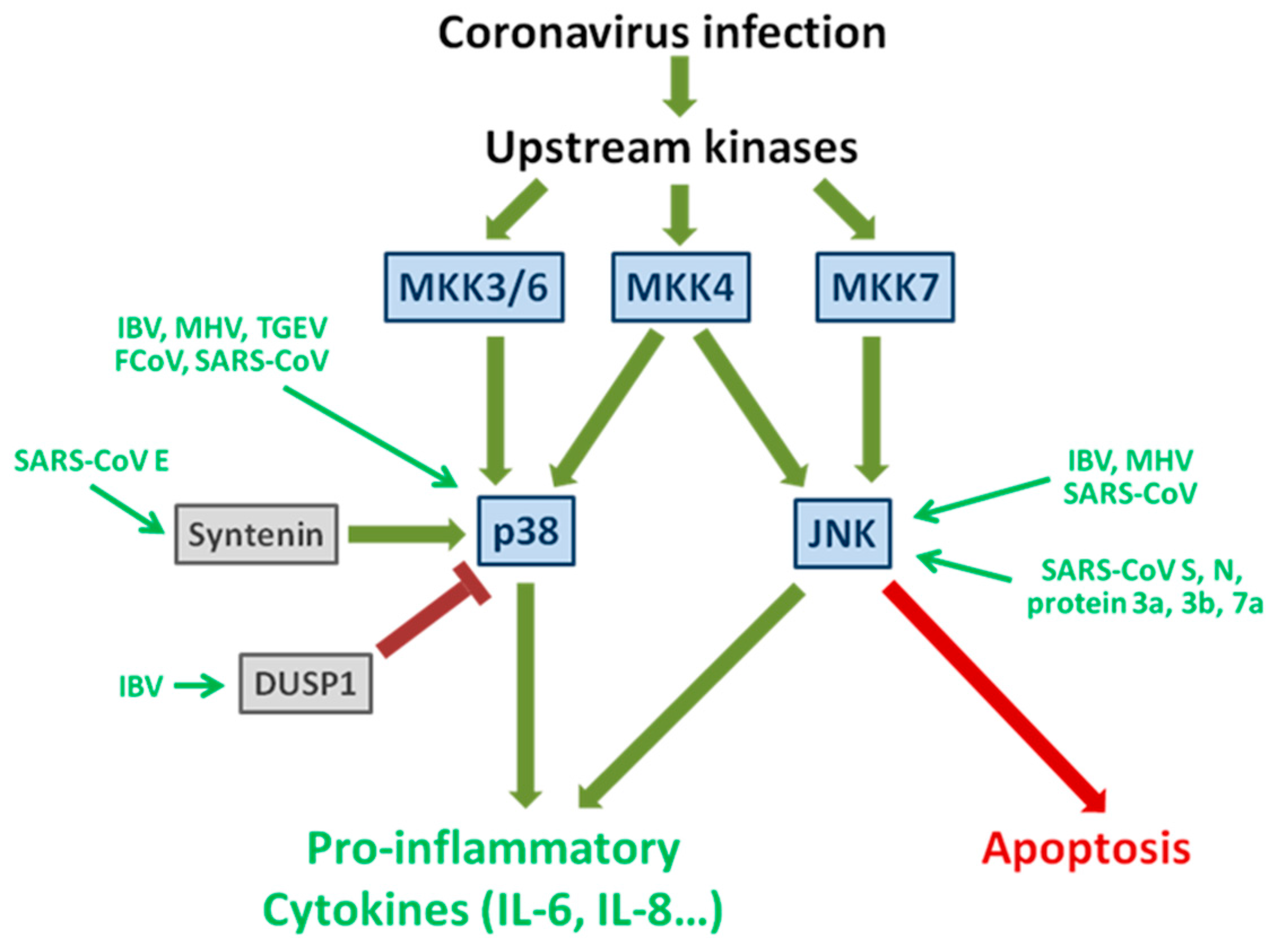
2.1. The Signaling Pathways of Stress-Activated Protein Kinase p38 and JNK
2.2. Induction and Subversion of the p38 Pathway by Coronavirus Infection
2.3. Activation of JNK during Coronavirus Infection
3. Translational Control by Coronavirus and Its Implication in Pathogenesis
3.1. Viral Protein Translation of Coronavirus
3.2. Regulation of Host Protein Synthesis via Targeting to eIF2α
3.3.Suppression of Host Protein Synthesis and IFN Response by Coronavirus nsp1
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Vattem, K.M.; Sood, R.; An, J.; Liang, J.; Stramm, L.; Wek, R.C. Identification and characterization of pancreatic eukaryotic initiation factor 2 α-subunit kinase, PEK, involved in translational control. Mol. Cell. Biol. 1998, 18, 7499–7509. [Google Scholar] [CrossRef] [PubMed]
- Harding, H.P.; Zhang, Y.; Ron, D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 1999, 397, 271–274. [Google Scholar] [PubMed]
- Harding, H.P.; Novoa, I.; Zhang, Y.; Zeng, H.; Wek, R.; Schapira, M.; Ron, D. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol. Cell 2000, 6, 1099–1108. [Google Scholar] [CrossRef]
- Brush, M.H.; Weiser, D.C.; Shenolikar, S. Growth arrest and DNA damage-inducible protein GADD34 targets protein phosphatase 1αto the endoplasmic reticulum and promotes dephosphorylation of the αsubunit of eukaryotic translation initiation factor 2. Mol. Cell. Biol. 2003, 23, 1292–1303. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Matsui, T.; Yamamoto, A.; Okada, T.; Mori, K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 2001, 107, 881–891. [Google Scholar] [CrossRef]
- Hollien, J.; Lin, J.H.; Li, H.; Stevens, N.; Walter, P.; Weissman, J.S. Regulated IRE1-dependent decay of messenger RNAs in mammalian cells. J. Cell Biol. 2009, 186, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Urano, F.; Wang, X.; Bertolotti, A.; Zhang, Y.; Chung, P.; Harding, H.P.; Ron, D. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 2000, 287, 664–666. [Google Scholar] [CrossRef] [PubMed]
- Yoneda, T.; Imaizumi, K.; Oono, K.; Yui, D.; Gomi, F.; Katayama, T.; Tohyama, M. Activation of caspase-12, an endoplastic reticulum (ER) resident caspase, through tumor necrosis factor receptor-associated factor 2-dependent mechanism in response to the ER stress. J. Biol. Chem. 2001, 276, 13935–13940. [Google Scholar] [PubMed]
- Ye, J.; Rawson, R.B.; Komuro, R.; Chen, X.; Dave, U.P.; Prywes, R.; Brown, M.S.; Goldstein, J.L. ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol. Cell 2000, 6, 1355–1364. [Google Scholar] [CrossRef]
- Adachi, Y.; Yamamoto, K.; Okada, T.; Yoshida, H.; Harada, A.; Mori, K. ATF6 is a transcription factor specializing in the regulation of quality control proteins in the endoplasmic reticulum. Cell Struct. Funct. 2008, 33, 75–89. [Google Scholar] [CrossRef] [PubMed]
- Calfon, M.; Zeng, H.; Urano, F.; Till, J.H.; Hubbard, S.R.; Harding, H.P.; Clark, S.G.; Ron, D. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 2002, 415, 92–96. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.G.; Tang, N.; Thompson, S.; Miller, J.; Katze, M.G. The 58,000-dalton cellular inhibitor of the interferon-induced double-stranded RNA-activated protein kinase (PKR) is a member of the tetratricopeptide repeat family of proteins. Mol. Cell. Biol. 1994, 14, 2331–2342. [Google Scholar] [CrossRef] [PubMed]
- Yan, W.; Frank, C.L.; Korth, M.J.; Sopher, B.L.; Novoa, I.; Ron, D.; Katze, M.G. Control of PERK eIF2α kinase activity by the endoplasmic reticulum stress-induced molecular chaperone P58IPK. Proc. Natl. Acad. Sci. USA 2002, 99, 15920–15925. [Google Scholar] [CrossRef] [PubMed]
- Eletto, D.; Dersh, D.; Gidalevitz, T.; Argon, Y. Protein disulfide isomerase A6 controls the decay of IRE1α signaling via disulfide-dependent association. Mol. Cell 2014, 53, 562–576. [Google Scholar] [CrossRef] [PubMed]
- Teske, B.F.; Wek, S.A.; Bunpo, P.; Cundiff, J.K.; McClintick, J.N.; Anthony, T.G.; Wek, R.C. The eIF2 kinase PERK and the integrated stress response facilitate activation of ATF6 during endoplasmic reticulum stress. Mol. Biol. Cell 2011, 22, 4390–4405. [Google Scholar] [CrossRef] [PubMed]
- Vekich, J.A.; Belmont, P.J.; Thuerauf, D.J.; Glembotski, C.C. Protein disulfide isomerase-associated 6 is an ATF6-inducible ER stress response protein that protects cardiac myocytes from ischemia/reperfusion-mediated cell death. J. Mol. Cell. Cardiol. 2012, 53, 259–267. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Fung, T.S.; Huang, M.; Fang, S.G.; Zhong, Y.; Liu, D.X. Upregulation of CHOP/GADD153 during coronavirus infectious bronchitis virus infection modulates apoptosis by restricting activation of the extracellular signal-regulated kinase pathway. J. Virol. 2013, 87, 8124–8134. [Google Scholar] [CrossRef] [PubMed]
- Tang, B.S.F.; Chan, K.-H.; Cheng, V.C.C.; Woo, P.C.Y.; Lau, S.K.P.; Lam, C.C.K.; Chan, T.-L.; Wu, A.K.L.; Hung, I.F.N.; Leung, S.-Y.; et al. Comparative host gene transcription by microarray analysis early after infection of the Huh7 cell line by severe acute respiratory syndrome coronavirus and human coronavirus 229E. J. Virol. 2005, 79, 6180–6193. [Google Scholar] [CrossRef] [PubMed]
- Versteeg, G.A.; van de Nes, P.S.; Bredenbeek, P.J.; Spaan, W.J.M. The coronavirus spike protein induces endoplasmic reticulum stress and upregulation of intracellular chemokine mRNA concentrations. J. Virol. 2007, 81, 10981–10990. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.-P.; Siu, K.-L.; Chin, K.-T.; Yuen, K.-Y.; Zheng, B.; Jin, D.-Y. Modulation of the unfolded protein response by the severe acute respiratory syndrome coronavirus spike protein. J. Virol. 2006, 80, 9279–9287. [Google Scholar] [CrossRef] [PubMed]
- Yeung, Y.-S.; Yip, C.-W.; Hon, C.-C.; Chow, K.Y.C.; Ma, I.C.M.; Zeng, F.; Leung, F.C.C. Transcriptional profiling of Vero E6 cells over-expressing SARS-CoV S2 subunit: Insights on viral regulation of apoptosis and proliferation. Virology 2008, 371, 32–43. [Google Scholar] [CrossRef] [PubMed]
- Minakshi, R.; Padhan, K.; Rani, M.; Khan, N.; Ahmad, F.; Jameel, S. The SARS coronavirus 3a protein causes endoplasmic reticulum stress and induces ligand-independent downregulation of the type 1 interferon receptor. PLoS ONE 2009, 4, e8342. [Google Scholar] [CrossRef] [PubMed]
- Ye, Z.; Wong, C.K.; Li, P.; Xie, Y. A SARS-CoV protein, ORF-6, induces caspase-3 mediated, ER stress and JNK-dependent apoptosis. Biochim. Biophys. Acta 2008, 1780, 1383–1387. [Google Scholar] [CrossRef] [PubMed]
- Sung, S.-C.; Chao, C.-Y.; Jeng, K.-S.; Yang, J.-Y.; Lai, M.M.C. The 8ab protein of SARS-CoV is a luminal ER membrane-associated protein and induces the activation of ATF6. Virology 2009, 387, 402–413. [Google Scholar] [CrossRef] [PubMed]
- Fung, T.S.; Huang, M.; Liu, D.X. Coronavirus-induced ER stress response and its involvement in regulation of coronavirus-host interactions. Virus Res. 2014, 194, 110–123. [Google Scholar] [CrossRef] [PubMed]
- Fukushi, M.; Yoshinaka, Y.; Matsuoka, Y.; Hatakeyama, S.; Ishizaka, Y.; Kirikae, T.; Sasazuki, T.; Miyoshi-Akiyama, T. Monitoring of S protein maturation in the endoplasmic reticulum by calnexin is important for the infectivity of severe acute respiratory syndrome coronavirus. J. Virol. 2012, 86, 11745–11753. [Google Scholar] [CrossRef] [PubMed]
- David-Ferreira, J.F.; Manaker, R.A. An electron microscope study of the development of a mouse hepatitis virus in tissue culture cells. J. Cell Biol. 1965, 24, 57–78. [Google Scholar] [CrossRef] [PubMed]
- Maier, H.J.; Hawes, P.C.; Cottam, E.M.; Mantell, J.; Verkade, P.; Monaghan, P.; Wileman, T.; Britton, P. Infectious bronchitis virus generates spherules from zippered endoplasmic reticulum membranes. mBio 2013, 4. [Google Scholar] [CrossRef] [PubMed]
- Angelini, M.M.; Akhlaghpour, M.; Neuman, B.W.; Buchmeier, M.J. Severe acute respiratory syndrome coronavirus nonstructural proteins 3, 4, and 6 induce double-membrane vesicles. mBio 2013, 4. [Google Scholar] [CrossRef] [PubMed]
- Knoops, K.; Kikkert, M.; van de Worm, S.H.E.; Zevenhoven-Dobbe, J.C.; van der Meer, Y.; Koster, A.J.; Mommaas, A.M.; Snijder, E.J. SARS-coronavirus replication is supported by a reticulovesicular network of modified endoplasmic reticulum. PLoS Biol. 2008, 6. [Google Scholar] [CrossRef] [PubMed]
- Reggiori, F.; Monastyrska, I.; Verheije, M.H.; Calì, T.; Ulasli, M.; Bianchi, S.; Bernasconi, R.; de Haan, C.A.M.; Molinari, M. Coronaviruses Hijack the LC3-I-positive EDEMosomes, ER-derived vesicles exporting short-lived ERAD regulators, for replication. Cell Host Microbe 2010, 7, 500–508. [Google Scholar] [CrossRef] [PubMed]
- Klumperman, J.; Locker, J.K.; Meijer, A.; Horzinek, M.C.; Geuze, H.J.; Rottier, P.J. Coronavirus M proteins accumulate in the Golgi complex beyond the site of virion budding. J. Virol. 1994, 68, 6523–6534. [Google Scholar] [PubMed]
- Stertz, S.; Reichelt, M.; Spiegel, M.; Kuri, T.; Martínez-Sobrido, L.; García-Sastre, A.; Weber, F.; Kochs, G. The intracellular sites of early replication and budding of SARS-coronavirus. Virology 2007, 361, 304–315. [Google Scholar] [CrossRef] [PubMed]
- Testerink, N.; van de Sanden, M.H.M.; Houweling, M.; Helms, J.B.; Vaandrager, A.B. Depletion of phosphatidylcholine affects endoplasmic reticulum morphology and protein traffic at the Golgi complex. J. Lipid Res. 2009, 50, 2182–2192. [Google Scholar] [CrossRef] [PubMed]
- Bechill, J.; Chen, Z.; Brewer, J.W.; Baker, S.C. Coronavirus infection modulates the unfolded protein response and mediates sustained translational repression. J. Virol. 2008, 82, 4492–4501. [Google Scholar] [CrossRef] [PubMed]
- Fung, T.S.; Liao, Y.; Liu, D.X. The endoplasmic reticulum stress sensor IRE1α protects cells from apoptosis induced by the coronavirus infectious bronchitis virus. J. Virol. 2014, 88, 12752–12764. [Google Scholar] [CrossRef] [PubMed]
- DeDiego, M.L.; Nieto-Torres, J.L.; Jiménez-Guardeño, J.M.; Regla-Nava, J.A.; Álvarez, E.; Oliveros, J.C.; Zhao, J.; Fett, C.; Perlman, S.; Enjuanes, L. Severe acute respiratory syndrome coronavirus envelope protein regulates cell stress response and apoptosis. PLoS Pathog. 2011, 7, e1002315. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.A.; Lee, A.-H.; Platzer, B.; Cross, B.C.S.; Gardner, B.M.; de Luca, H.; Luong, P.; Harding, H.P.; Glimcher, L.H.; Walter, P.; et al. The unfolded protein response element IRE1α senses bacterial proteins invading the ER to activate RIG-I and innate immune signaling. Cell Host Microbe 2013, 13, 558–569. [Google Scholar] [CrossRef] [PubMed]
- Tam, A.B.; Mercado, E.L.; Hoffmann, A.; Niwa, M. ER stress activates NF-κB by integrating functions of basal IKK activity, IRE1 and PERK. PLoS ONE 2012, 7, e45078. [Google Scholar] [CrossRef] [PubMed]
- Hu, F.; Yu, X.; Wang, H.; Zuo, D.; Guo, C.; Yi, H.; Tirosh, B.; Subjeck, J.R.; Qiu, X.; Wang, X.-Y. ER stress and its regulator X-box-binding protein-1 enhance polyIC-induced innate immune response in dendritic cells. Eur. J. Immunol. 2011, 41, 1086–1097. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F.; Chen, X.; Lee, A.-H.; Glimcher, L.H. TLR activation of the transcription factor XBP1 regulates innate immune responses in macrophages. Nat. Immunol. 2010, 11, 411–418. [Google Scholar] [CrossRef] [PubMed]
- Zeng, L.; Liu, Y.-P.; Sha, H.; Chen, H.; Qi, L.; Smith, J.A. XBP-1 couples endoplasmic reticulum stress to augmented IFN-β induction via a cis-acting enhancer in macrophages. J. Immunol. 2010, 185, 2324–2330. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Liao, Y.; Yap, P.L.; Png, K.J.; Tam, J.P.; Liu, D.X. Inhibition of protein kinase R activation and upregulation of GADD34 expression play a synergistic role in facilitating coronavirus replication by maintaining de novo protein synthesis in virus-infected cells. J. Virol. 2009, 83, 12462–12472. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Hauns, K.; Langland, J.O.; Jacobs, B.L.; Hogue, B.G. Mouse hepatitis coronavirus A59 nucleocapsid protein is a type I interferon antagonist. J. Virol. 2007, 81, 2554–2563. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.P.; Siu, K.L.; Chin, K.T.; Yuen, K.Y.; Zheng, B.; Jin, D.Y. Modulation of the unfolded protein response by the severe acute respiratory syndrome coronavirus spike protein. J. Virol. 2006, 80, 9279–9287. [Google Scholar] [CrossRef] [PubMed]
- Krahling, V.; Stein, D.A.; Spiegel, M.; Weber, F.; Muhlberger, E. Severe acute respiratory syndrome coronavirus triggers apoptosis via protein kinase R but is resistant to its antiviral activity. J. Virol. 2009, 83, 2298–2309. [Google Scholar] [CrossRef] [PubMed]
- Keshet, Y.; Seger, R. The MAP kinase signaling cascades: A system of hundreds of components regulates a diverse array of physiological functions. Methods Mol. Biol. 2010, 661, 3–38. [Google Scholar] [PubMed]
- Kyriakis, J.M.; Avruch, J. Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol. Rev. 2001, 81, 807–869. [Google Scholar] [PubMed]
- Nishimoto, S.; Nishida, E. MAPK signalling: ERK5 versus ERK1/2. EMBO Rep. 2006, 7, 782–786. [Google Scholar] [CrossRef] [PubMed]
- Davis, R.J. Signal transduction by the JNK group of MAP kinases. Cell 2000, 103, 239–252. [Google Scholar] [CrossRef]
- Derijard, B.; Raingeaud, J.; Barrett, T.; Wu, I.H.; Han, J.; Ulevitch, R.J.; Davis, R.J. Independent human MAP-kinase signal transduction pathways defined by MEK and MKK isoforms. Science 1995, 267, 682–685. [Google Scholar] [CrossRef] [PubMed]
- Raingeaud, J.; Whitmarsh, A.J.; Barrett, T.; Dérijard, B.; Davis, R.J. MKK3- and MKK6-regulated gene expression is mediated by the p38 mitogen-activated protein kinase signal transduction pathway. Mol. Cell. Biol. 1996, 16, 1247–1255. [Google Scholar] [CrossRef] [PubMed]
- Tournier, C.; Dong, C.; Turner, T.K.; Jones, S.N.; Flavell, R.A.; Davis, R.J. MKK7 is an essential component of the JNK signal transduction pathway activated by proinflammatory cytokines. Genes Dev. 2001, 15, 1419–1426. [Google Scholar] [CrossRef] [PubMed]
- Zarubin, T.; Han, J. Activation and signaling of the p38 MAP kinase pathway. Cell Res. 2005, 15, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Bulavin, D.V.; Saito, S.; Hollander, M.C.; Sakaguchi, K.; Anderson, C.W.; Appella, E.; Fornace, A.J. Phosphorylation of human p53 by p38 kinase coordinates N-terminal phosphorylation and apoptosis in response to UV radiation. EMBO J. 1999, 18, 6845–6854. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Ron, D. Stress-induced phosphorylation and activation of the transcription factor CHOP (GADD153) by p38 MAP kinase. Science 1996, 272, 1347–1349. [Google Scholar] [CrossRef] [PubMed]
- Smeal, T.; Binetruy, B.; Mercola, D.A.; Birrer, M.; Karin, M. Oncogenic and transcriptional cooperation with Ha-Ras requires phosphorylation of c-Jun on serines 63 and 73. Nature 1991, 354, 494–496. [Google Scholar] [CrossRef] [PubMed]
- Hess, J.; Angel, P.; Schorpp-Kistner, M. AP-1 subunits: Quarrel and harmony among siblings. J. Cell Sci. 2004, 117, 5965–5973. [Google Scholar] [CrossRef] [PubMed]
- McGilvray, I.D.; Lu, Z.; Wei, A.C.; Dackiw, A.P.; Marshall, J.C.; Kapus, A.; Levy, G.; Rotstein, O.D. Murine hepatitis virus strain 3 induces the macrophage prothrombinase fgl-2 through p38 mitogen-activated protein kinase activation. J. Biol. Chem. 1998, 273, 32222–32229. [Google Scholar] [CrossRef] [PubMed]
- Mizutani, T.; Fukushi, S.; Saijo, M.; Kurane, I.; Morikawa, S. Phosphorylation of p38 MAPK and its downstream targets in SARS coronavirus-infected cells. Biochem. Biophys. Res. Commun. 2004, 319, 1228–1234. [Google Scholar] [CrossRef] [PubMed]
- Regan, A.D.; Cohen, R.D.; Whittaker, G.R. Activation of p38 MAPK by feline infectious peritonitis virus regulates pro-inflammatory cytokine production in primary blood-derived feline mononuclear cells. Virology 2009, 384, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Wang, X.; Huang, M.; Tam, J.P.; Liu, D.X. Regulation of the p38 mitogen-activated protein kinase and dual-specificity phosphatase 1 feedback loop modulates the induction of interleukin 6 and 8 in cells infected with coronavirus infectious bronchitis virus. Virology 2011, 420, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Ding, L.; Li, Z.; Dai, M.; Zhao, X.; Li, W.; Du, Q.; Xu, X.; Tong, D. Transmissible gastroenteritis virus infection induces cell apoptosis via activation of p53 signalling. J. Gen. Virol. 2013, 94, 1807–1817. [Google Scholar] [CrossRef] [PubMed]
- Padhan, K.; Minakshi, R.; Towheed, M.A.B.; Jameel, S. Severe acute respiratory syndrome coronavirus 3a protein activates the mitochondrial death pathway through p38 MAP kinase activation. J. Gen. Virol. 2008, 89, 1960–1969. [Google Scholar] [CrossRef] [PubMed]
- Kopecky-Bromberg, S.A.; Martinez-Sobrido, L.; Palese, P. 7a protein of severe acute respiratory syndrome coronavirus inhibits cellular protein synthesis and activates p38 mitogen-activated protein kinase. J. Virol. 2006, 80, 785–793. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Narayanan, K.; Mizutani, T.; Makino, S. Murine coronavirus replication-induced p38 mitogen-activated protein kinase activation promotes interleukin-6 production and virus replication in cultured cells. J. Virol. 2002, 76, 5937–5948. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.-L.; Chen, J.; Yang, F.; Li, G.-Q.; Zheng, L.-X.; Wu, Y.-Z. C5a/C5aR pathway is essential for the pathogenesis of murine viral fulminant hepatitis by way of potentiating Fgl2/fibroleukin expression. Hepatology 2014, 60, 114–124. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Guardeño, J.M.; Nieto-Torres, J.L.; DeDiego, M.L.; Regla-Nava, J.A.; Fernandez-Delgado, R.; Castaño-Rodriguez, C.; Enjuanes, L. The PDZ-binding motif of severe acute respiratory syndrome coronavirus envelope protein is a determinant of viral pathogenesis. PLoS Pathog. 2014, 10, e1004320. [Google Scholar] [CrossRef] [PubMed]
- Surjit, M.; Liu, B.; Kumar, P.; Chow, V.T.K.; Lal, S.K. The nucleocapsid protein of the SARS coronavirus is capable of self-association through a C-terminal 209 amino acid interaction domain. Biochem. Biophys. Res. Commun. 2004, 317, 1030–1036. [Google Scholar] [CrossRef] [PubMed]
- Varshney, B.; Lal, S.K. SARS-CoV accessory protein 3b induces AP-1 transcriptional activity through activation of JNK and ERK pathways. Biochemistry 2011, 50, 5419–5425. [Google Scholar] [CrossRef] [PubMed]
- Kanzawa, N.; Nishigaki, K.; Hayashi, T.; Ishii, Y.; Furukawa, S.; Niiro, A.; Yasui, F.; Kohara, M.; Morita, K.; Matsushima, K.; et al. Augmentation of chemokine production by severe acute respiratory syndrome coronavirus 3a/X1 and 7a/X4 proteins through NF-κB activation. FEBS Lett. 2006, 580, 6807–6812. [Google Scholar] [CrossRef] [PubMed]
- Mizutani, T.; Fukushi, S.; Saijo, M.; Kurane, I.; Morikawa, S. JNK and PI3k/Akt signaling pathways are required for establishing persistent SARS-CoV infection in Vero E6 cells. Biochim. Biophys. Acta 2005, 1741, 4–10. [Google Scholar] [CrossRef] [PubMed]
- Mizutani, T.; Fukushi, S.; Ishii, K.; Sasaki, Y.; Kenri, T.; Saijo, M.; Kanaji, Y.; Shirota, K.; Kurane, I.; Morikawa, S. Mechanisms of establishment of persistent SARS-CoV-infected cells. Biochem. Biophys. Res. Commun. 2006, 347, 261–265. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.; Zhu, H.; Liu, Y.; Cao, J.; Zhang, X. Regulation of proinflammatory cytokine expression in primary mouse astrocytes by coronavirus infection. J. Virol. 2009, 83, 12204–12214. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.-J.; Liu, C.Y.Y.; Chiang, B.-L.; Chao, Y.-C.; Chen, C.-C. Induction of IL-8 release in lung cells via activator protein-1 by recombinant baculovirus displaying severe acute respiratory syndrome-coronavirus spike proteins: Identification of two functional regions. J. Immunol. 2004, 173, 7602–7614. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Yang, Y.; Gu, C.; Yue, Y.; Wu, K.K.; Wu, J.; Zhu, Y. Spike protein of SARS-CoV stimulates cyclooxygenase-2 expression via both calcium-dependent and calcium-independent protein kinase C pathways. FASEB J. 2007, 21, 1586–1596. [Google Scholar] [CrossRef] [PubMed]
- Pasternak, A.O.; Spaan, W.J.; Snijder, E.J. Nidovirus transcription: How to make sense...? J. Gen. Virol. 2006, 87, 1403–1421. [Google Scholar] [CrossRef] [PubMed]
- Tahara, S.M.; Dietlin, T.A.; Bergmann, C.C.; Nelson, G.W.; Kyuwa, S.; Anthony, R.P.; Stohlman, S.A. Coronavirus translational regulation: Leader affects mRNA efficiency. Virology 1994, 202, 621–630. [Google Scholar] [CrossRef] [PubMed]
- Tahara, S.M.; Dietlin, T.A.; Nelson, G.W.; Stohlman, S.A.; Manno, D.J. Mouse hepatitis virus nucleocapsid protein as a translational effector of viral mRNAs. Adv. Exp. Med. Biol. 1998, 440, 313–318. [Google Scholar] [PubMed]
- Hofmann, M.A.; Chang, R.Y.; Ku, S.; Brian, D.A. Leader-mRNA junction sequences are unique for each subgenomic mRNA species in the bovine coronavirus and remain so throughout persistent infection. Virology 1993, 196, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, M.A.; Sethna, P.B.; Brian, D.A. Bovine coronavirus mRNA replication continues throughout persistent infection in cell culture. J. Virol. 1990, 64, 4108–4114. [Google Scholar] [PubMed]
- Senanayake, S.D.; Brian, D.A. Translation from the 5′ untranslated region (UTR) of mRNA 1 is repressed, but that from the 5′ UTR of mRNA 7 is stimulated in coronavirus-infected cells. J. Virol. 1999, 73, 8003–8009. [Google Scholar] [PubMed]
- Kamitani, W.; Narayanan, K.; Huang, C.; Lokugamage, K.; Ikegami, T.; Ito, N.; Kubo, H.; Makino, S. Severe acute respiratory syndrome coronavirus nsp1 protein suppresses host gene expression by promoting host mRNA degradation. Proc. Natl. Acad. Sci. USA 2006, 103, 12885–12890. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, K.; Huang, C.; Lokugamage, K.; Kamitani, W.; Ikegami, T.; Tseng, C.T.; Makino, S. Severe acute respiratory syndrome coronavirus nsp1 suppresses host gene expression, including that of type I interferon, in infected cells. J. Virol. 2008, 82, 4471–4479. [Google Scholar] [CrossRef] [PubMed]
- Lokugamage, K.G.; Narayanan, K.; Huang, C.; Makino, S. Severe acute respiratory syndrome coronavirus protein nsp1 is a novel eukaryotic translation inhibitor that represses multiple steps of translation initiation. J. Virol. 2012, 86, 13598–13608. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Kamitani, W.; DeDiego, M.L.; Enjuanes, L.; Matsuura, Y. Severe acute respiratory syndrome coronavirus nsp1 facilitates efficient propagation in cells through a specific translational shutoff of host mRNA. J. Virol. 2012, 86, 11128–11137. [Google Scholar] [CrossRef] [PubMed]
- Lokugamage, K.G.; Narayanan, K.; Nakagawa, K.; Terasaki, K.; Ramirez, S.I.; Tseng, C.T.; Makino, S. Middle east respiratory syndrome coronavirus nsp1 inhibits host gene expression by selectively targeting mRNAs transcribed in the nucleus while sparing mRNAs of cytoplasmic origin. J. Virol. 2015, 89, 10970–10981. [Google Scholar] [CrossRef] [PubMed]
- Rottier, P.J.; Horzinek, M.C.; van der Zeijst, B.A. Viral protein synthesis in mouse hepatitis virus strain A59-infected cells: Effect of tunicamycin. J. Virol. 1981, 40, 350–357. [Google Scholar] [PubMed]
- Hilton, A.; Mizzen, L.; MacIntyre, G.; Cheley, S.; Anderson, R. Translational control in murine hepatitis virus infection. J. Gen. Virol. 1986, 67, 923–932. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Lokugamage, K.G.; Rozovics, J.M.; Narayanan, K.; Semler, B.L.; Makino, S. Alphacoronavirus transmissible gastroenteritis virus nsp1 protein suppresses protein translation in mammalian cells and in cell-free HeLa cell extracts but not in rabbit reticulocyte lysate. J. Virol. 2011, 85, 638–643. [Google Scholar] [CrossRef] [PubMed]
- Jansson, A.M. Structure of alphacoronavirus transmissible gastroenteritis virus nsp1 has implications for coronavirus nsp1 function and evolution. J. Virol. 2013, 87, 2949–2955. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Shi, K.; Yoo, D. Suppression of type I interferon production by porcine epidemic diarrhea virus and degradation of CREB-binding protein by nsp1. Virology 2016, 489, 252–268. [Google Scholar] [CrossRef] [PubMed]
- Tohya, Y.; Narayanan, K.; Kamitani, W.; Huang, C.; Lokugamage, K.; Makino, S. Suppression of host gene expression by nsp1 proteins of group 2 bat coronaviruses. J. Virol. 2009, 83, 5282–5288. [Google Scholar] [CrossRef] [PubMed]
- Brostrom, C.O.; Brostrom, M.A. Regulation of translational initiation during cellular responses to stress. Prog. Nucl. Acid Res. Mol. Biol. 1998, 58, 79–125. [Google Scholar]
- Clemens, M.J. Initiation factor eIF2αphosphorylation in stress responses and apoptosis. Prog. Mol. Subcell. Biol. 2001, 27, 57–89. [Google Scholar] [PubMed]
- Beattie, E.; Denzler, K.L.; Tartaglia, J.; Perkus, M.E.; Paoletti, E.; Jacobs, B.L. Reversal of the interferon-sensitive phenotype of a vaccinia virus lacking E3L by expression of the reovirus S4 gene. J. Virol. 1995, 69, 499–505. [Google Scholar] [PubMed]
- Proud, C.G. eIF2 and the control of cell physiology. Semin. Cell Dev. Biol. 2005, 16, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Vattem, K.M.; Staschke, K.A.; Wek, R.C. Mechanism of activation of the double-stranded-RNA-dependent protein kinase, PKR: Role of dimerization and cellular localization in the stimulation of PKR phosphorylation of eukaryotic initiation factor-2 (eIF2). Eur. J. Biochem. 2001, 268, 3674–3684. [Google Scholar] [CrossRef] [PubMed]
- Sood, R.; Porter, A.C.; Ma, K.; Quilliam, L.A.; Wek, R.C. Pancreatic eukaryotic initiation factor-2α kinase (PEK) homologues in humans, Drosophila melanogaster and Caenorhabditis elegans that mediate translational control in response to endoplasmic reticulum stress. Biochem. J. 2000, 346, 281–293. [Google Scholar] [CrossRef] [PubMed]
- Rojas, M.; Vasconcelos, G.; Dever, T.E. An eIF2α-binding motif in protein phosphatase 1 subunit GADD34 and its viral orthologs is required to promote dephosphorylation of eIF2α. Proc. Natl. Acad. Sci. USA 2015, 112, E3466–E3475. [Google Scholar] [CrossRef] [PubMed]
- Raaben, M.; Groot Koerkamp, M.J.; Rottier, P.J.; de Haan, C.A. Mouse hepatitis coronavirus replication induces host translational shutoff and mRNA decay, with concomitant formation of stress granules and processing bodies. Cell. Microbiol. 2007, 9, 2218–2229. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; An, S.; Zhou, A.; Silverman, R.H.; Makino, S. RNase L-independent specific 28S rRNA cleavage in murine coronavirus-infected cells. J. Virol. 2000, 74, 8793–8802. [Google Scholar] [CrossRef] [PubMed]
- Cruz, J.L.; Sola, I.; Becares, M.; Alberca, B.; Plana, J.; Enjuanes, L.; Zuniga, S. Coronavirus gene 7 counteracts host defenses and modulates virus virulence. PLoS Pathog. 2011, 7, e1002090. [Google Scholar] [CrossRef] [PubMed]
- Kamitani, W.; Huang, C.; Narayanan, K.; Lokugamage, K.G.; Makino, S. A two-pronged strategy to suppress host protein synthesis by SARS coronavirus Nsp1 protein. Nat. Struct. Mol. Biol. 2009, 16, 1134–1140. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Lokugamage, K.G.; Rozovics, J.M.; Narayanan, K.; Semler, B.L.; Makino, S. SARS coronavirus nsp1 protein induces template-dependent endonucleolytic cleavage of mRNAs: Viral mRNAs are resistant to nsp1-induced RNA cleavage. PLoS Pathog. 2011, 7, e1002433. [Google Scholar] [CrossRef] [PubMed]
- Wathelet, M.G.; Orr, M.; Frieman, M.B.; Baric, R.S. Severe acute respiratory syndrome coronavirus evades antiviral signaling: Role of nsp1 and rational design of an attenuated strain. J. Virol. 2007, 81, 11620–11633. [Google Scholar] [CrossRef] [PubMed]
- Zust, R.; Cervantes-Barragan, L.; Kuri, T.; Blakqori, G.; Weber, F.; Ludewig, B.; Thiel, V. Coronavirus non-structural protein 1 is a major pathogenicity factor: Implications for the rational design of coronavirus vaccines. PLoS Pathog. 2007, 3, e109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, R.; Li, Y.; Cowley, T.J.; Steinbrenner, A.D.; Phillips, J.M.; Yount, B.L.; Baric, R.S.; Weiss, S.R. The nsp1, nsp13, and M proteins contribute to the hepatotropism of murine coronavirus JHM.WU. J. Virol. 2015, 89, 3598–3609. [Google Scholar] [CrossRef] [PubMed]
- Lei, L.; Ying, S.; Baojun, L.; Yi, Y.; Xiang, H.; Wenli, S.; Zounan, S.; Deyin, G.; Qingyu, Z.; Jingmei, L.; et al. Attenuation of mouse hepatitis virus by deletion of the LLRKxGxKG region of Nsp1. PLoS ONE 2013, 8, e61166. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Shi, H.; Rigolet, P.; Wu, N.; Zhu, L.; Xi, X.G.; Vabret, A.; Wang, X.; Wang, T. Nsp1 proteins of group I and SARS coronaviruses share structural and functional similarities. Infect. Genet. Evol. 2010, 10, 919–924. [Google Scholar] [CrossRef] [PubMed]


© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fung, T.S.; Liao, Y.; Liu, D.X. Regulation of Stress Responses and Translational Control by Coronavirus. Viruses 2016, 8, 184. https://0-doi-org.brum.beds.ac.uk/10.3390/v8070184
Fung TS, Liao Y, Liu DX. Regulation of Stress Responses and Translational Control by Coronavirus. Viruses. 2016; 8(7):184. https://0-doi-org.brum.beds.ac.uk/10.3390/v8070184
Chicago/Turabian StyleFung, To Sing, Ying Liao, and Ding Xiang Liu. 2016. "Regulation of Stress Responses and Translational Control by Coronavirus" Viruses 8, no. 7: 184. https://0-doi-org.brum.beds.ac.uk/10.3390/v8070184