1. Introduction
Variola virus (VARV), the causative agent of smallpox, was responsible for over 500 million deaths in the 20th century, with mortality rates for the severe form of smallpox, variola major, greater than 30%. The eradication of smallpox as a human disease was achieved in the 1970s through a concerted vaccination campaign spearheaded by the World Health Organization (WHO) relying on case identification, contact tracing, and focused “ring” vaccination campaigns to stop disease spread [
1]. The success of this campaign existed in part because there were no identifiable animal reservoirs of the disease and the highly effective nature of the vaccine, rapidly providing immunity after a single application. However, the evidence that some countries manufactured smallpox as a bioweapon, the possibility that unknown sources of the virus persist [
2], and the susceptibility of the United States population to smallpox led the United States Government (USG) to reinvest in the development of smallpox countermeasures, resulting in the approval of a new smallpox vaccine (ACAM2000; Acambis Inc., Cambridge, MA, USA) in 1997 [
3]. Although vaccines are extremely effective when administered prior to exposure, the ability of the vaccine to protect against mortality drops rapidly after emergence of the disease and appears to have no ameliorative effect after smallpox symptoms are present. In order to provide treatment options for patients already exposed to smallpox, the USG is investing in the development of small molecule antivirals [
4].
In 2007, the Food and Drug Administration (FDA) issued guidance concerning animal models for testing the efficacy of medical countermeasures (MCMs) against VARV, the etiologic agent for smallpox. The FDA directed that antiviral activity should be first evaluated in vitro, prior to in vivo studies. Since clinical trials with smallpox are impossible, the FDA is evaluating the efficacy of countermeasures against smallpox under the FDA Animal Rule (21 CFR 314.600 for drugs and 21 CFR 601.90 for biological products), where animal models are used in lieu of traditional clinical trials to evaluate efficacy. As there are no animal reservoirs for smallpox, there are no susceptible species that can be infected with variola to test the activity of countermeasures. In order to solicit input for identification of models for evaluating smallpox MCMs, the FDA Center for Drug Evaluation and Research (CDER) held an Antiviral Drugs Advisory Committee Meeting to address smallpox concerns in December 2011, resulting in guidance specifying that rabbits challenged with rabbitpox virus (RPXV) and mice challenged with ectromelia virus (ECTV) were acceptable models for product testing. In order to evaluate the relevancy of these models for evaluation of therapeutic efficacy against smallpox, complete characterization of the model to determine the course of disease and specific biomarkers resulting from infection should be identified. Similarly, in human smallpox, a patient develops high fever and characteristic lesional rash after infection, providing clear, unambiguous signs for medical intervention [
1]. As studies using rabbit or mouse models had measured the efficacy of a drug or countermeasure when administered based on a delay after exposure to infection, it was not immediately evident if these models would provide symptoms appropriate for countermeasure intervention.
Ectromelia is a member of the genus
Orthopoxvirus, which includes VARV, vaccinia virus, monkeypox virus (MPXV), and RPXV. ECTV is responsible for mousepox disease and the original Hampstead strain was isolated approximately 85 years ago in a laboratory mouse colony [
5]. Since this discovery, various ECTV strains have been isolated from naturally-occurring outbreaks with differences in disease severity. ECTV is infectious at low doses and readily transmitted among wild and laboratory mice populations [
6]. The pathogenesis of ECTV infection and the availability of resistant and susceptible mouse strains to ECTV infection has led to many studies investigating the role of protection against infection in the host immune system and the pathogenesis of viral infection [
6,
7]. Genetic and disease presentation similarities between ECTV and VARV have led to the use of ECTV infection in mice as a laboratory model to test smallpox countermeasures [
8,
9,
10]. As previously reported, the first signs of overt disease, such as lethargy and ruffled fur, normally occur 3–6 days post-challenge in the animals infected with high ECTV titers and 6–8 days post-challenge in animals infected with low ECTV titers [
11]. Primary viremia occurs in the mouse after localized replication in regional lymph nodes, resulting in infection of the liver and spleen [
12,
13]. Mousepox disease severity is largely dependent on both mouse and virus strain, along with the route of infection [
7,
9].
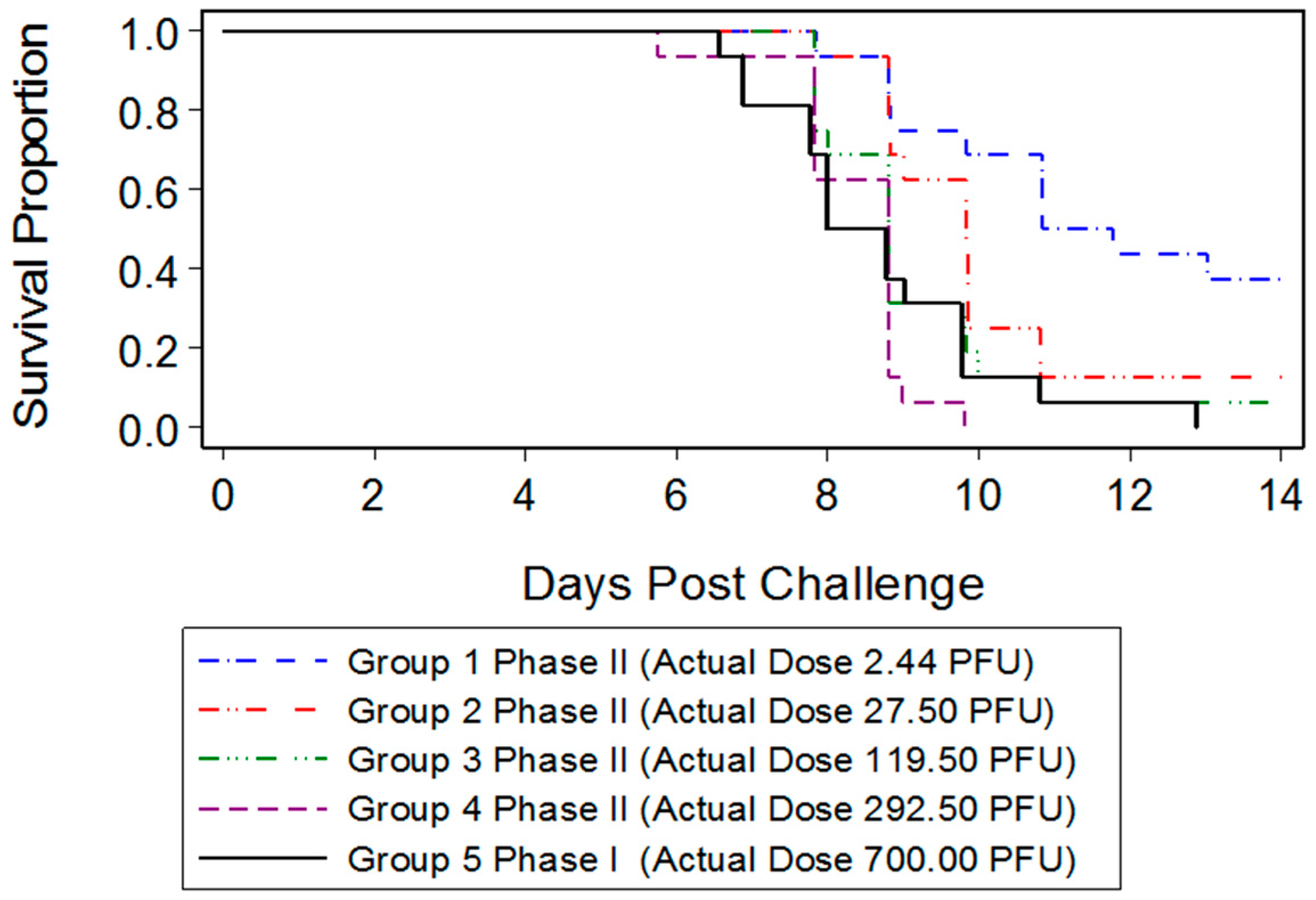
The ECTV model preferred by the FDA for product testing would resemble smallpox in humans as having high mortality, a low infectious dose, and disease progression through the respiratory system. The ECTV model that best recapitulates these characteristics is the intranasal (IN) challenge model [
14] using BALB/c mice at low infectious doses [
15]. Therefore, the objective of this study was to provide data supporting use of an IN ECTV challenge in the BALB/c mouse model to test the efficacy of anti-viral MCMs. In this study, a well-characterized stock of ECTV was produced and further characterized in vivo with a natural history of infection following IN challenge, including clinical and pathological response to infection. This model is sufficiently well-characterized to provide data supporting the efficacy of MCMs and dose justification.
4. Discussion
The regulatory evaluation of MCMs against smallpox requires the use of animal models challenged with the cognate poxvirus to serve as surrogates for natural VARV infection in humans. The countermeasure intended for therapeutic intervention requires an animal model with unambiguous and reliable symptoms of the disease to serve as triggers for intervention. Although scientific literature contains many publications demonstrating the ability of drugs and antibodies to block poxvirus infections, studies are typically described in which drug intervention is based on a time delay after exposure. These models of post-exposure prophylaxis reflect a scenario where a drug is given after a putative exposure of virus, prior to disease symptoms.
Prior to the WHO Advisory Committee Meeting on VARV in 2012, the only animal model used in the therapeutic evaluation of smallpox countermeasures was the MPXV intravenous challenge of cynomologus macaques [
20]. Although intervention in numerous MPXV studies was based on time delay, investigators were consistently able to document that all animals exhibited symptoms of disease (temperature elevation, viremia) prior to drug intervention [
21]. Since regulatory evaluation of smallpox countermeasures would require demonstration of effectiveness in multiple animal models, two animal models proposed by the FDA for development of a therapeutic model for countermeasure evaluation include the New Zealand White rabbit infected with RPXV and the BALB/c mouse infected with ECTV.
Inconsistencies with mouse strain variation, titer determination, and inoculation route in previous studies have led to difficulties in developing an effective treatment model [
22]. In this study, we describe an investigation into the ability of the IN BALB/c mouse challenge model to serve as an animal model to evaluate the therapeutic efficacy of smallpox countermeasures. Data were collected and biomarkers evaluated for suitability of use for clinical intervention to test the therapeutic efficacy of MCMs for human smallpox disease. The characterization of ECTV infection was investigated in this study through delineation of viral potency, lethal infective dose, mortality, alterations in temperature and weight, viral burden, pathological changes, and clinical observations. There were no unambiguous, reproducible biomarkers associated with infection until late in disease progression in which the earliest and most reliable indicator of ECTV infection was the accumulation of replicating virus in the liver and spleen, consistent with earlier observations in ECTV-infected mice [
8]. The concentration of viruses in the liver and spleen may indicate tissue necrosis as the primary causes of death, differing from humans infected with smallpox as hepatic and splenic necrosis are not considered primary causes of mortality [
8,
9].
The ECTV murine model mimics many features similar to that of human smallpox, including acute systemic disease caused by a low viral infectious dose, lack of pulmonary involvement early in the disease progression, viremia, and fever [
6]. A consistent, high mortality rate in BALB/c mice challenged IN with a high dose (10
3 PFU) of ECTV allows for a disease progression that begins 3–4 days PI and results in complete mortality within 10 days after challenge [
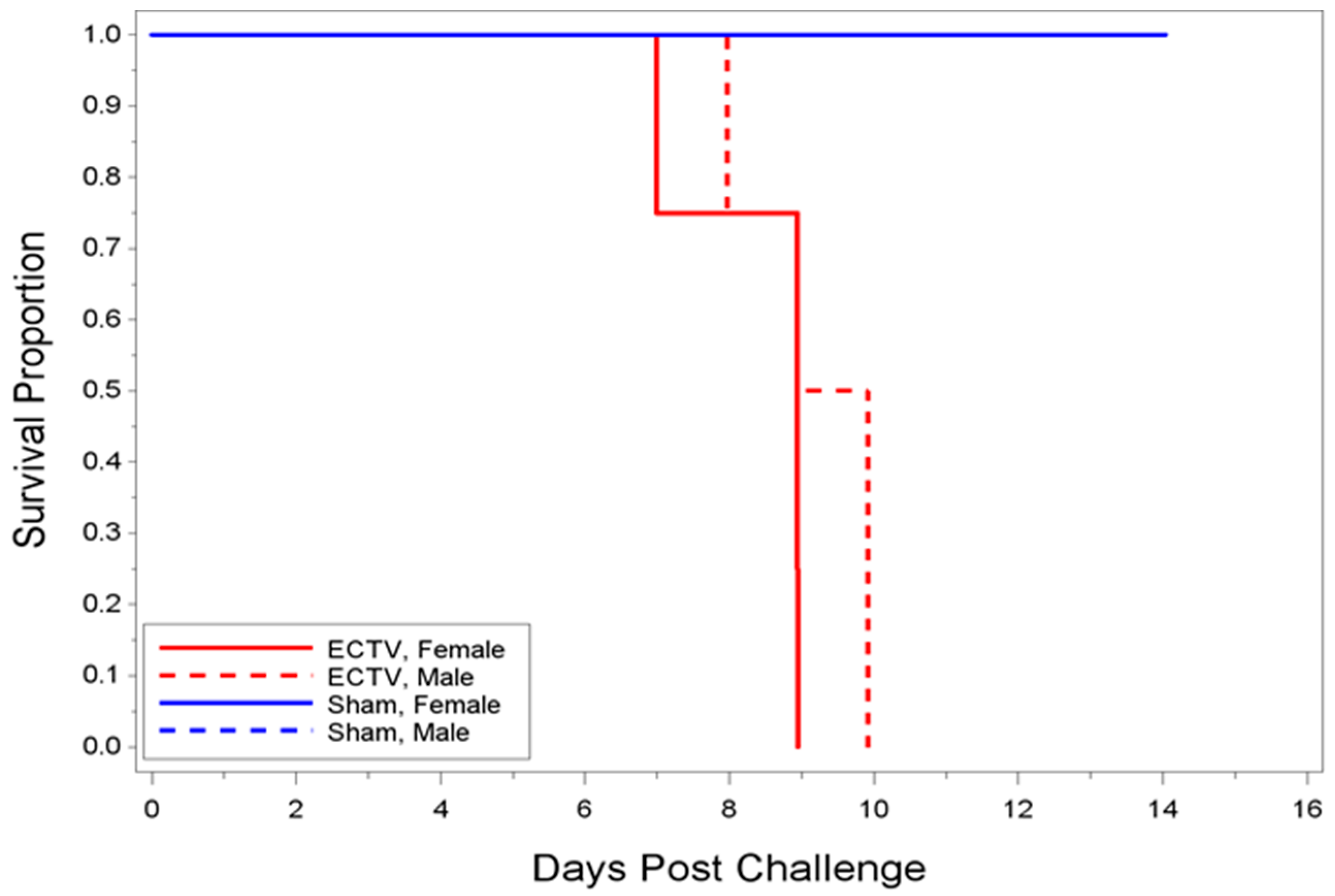
23]. In the present study, we were interested in developing a model more reflective of the moderate or low dose exposure needed for natural infection, while retaining high mortality in order to measure the statistical significance of MCM intervention. Mice infected with 700 PFU ECTV-Mos succumbed to disease as early as six days after challenge, with all mice dead by 13 days PI. The mean time-to-death was inversely proportional to the dose as mice infected with 700 PFU (eight days) died sooner than mice infected with a lower PFU (2.44 PFU, 10.84 days). A statistically significant difference in the median time-to-death between males and females following infection could not be determined, indicating the sex of the mouse did not appear to influence mortality.
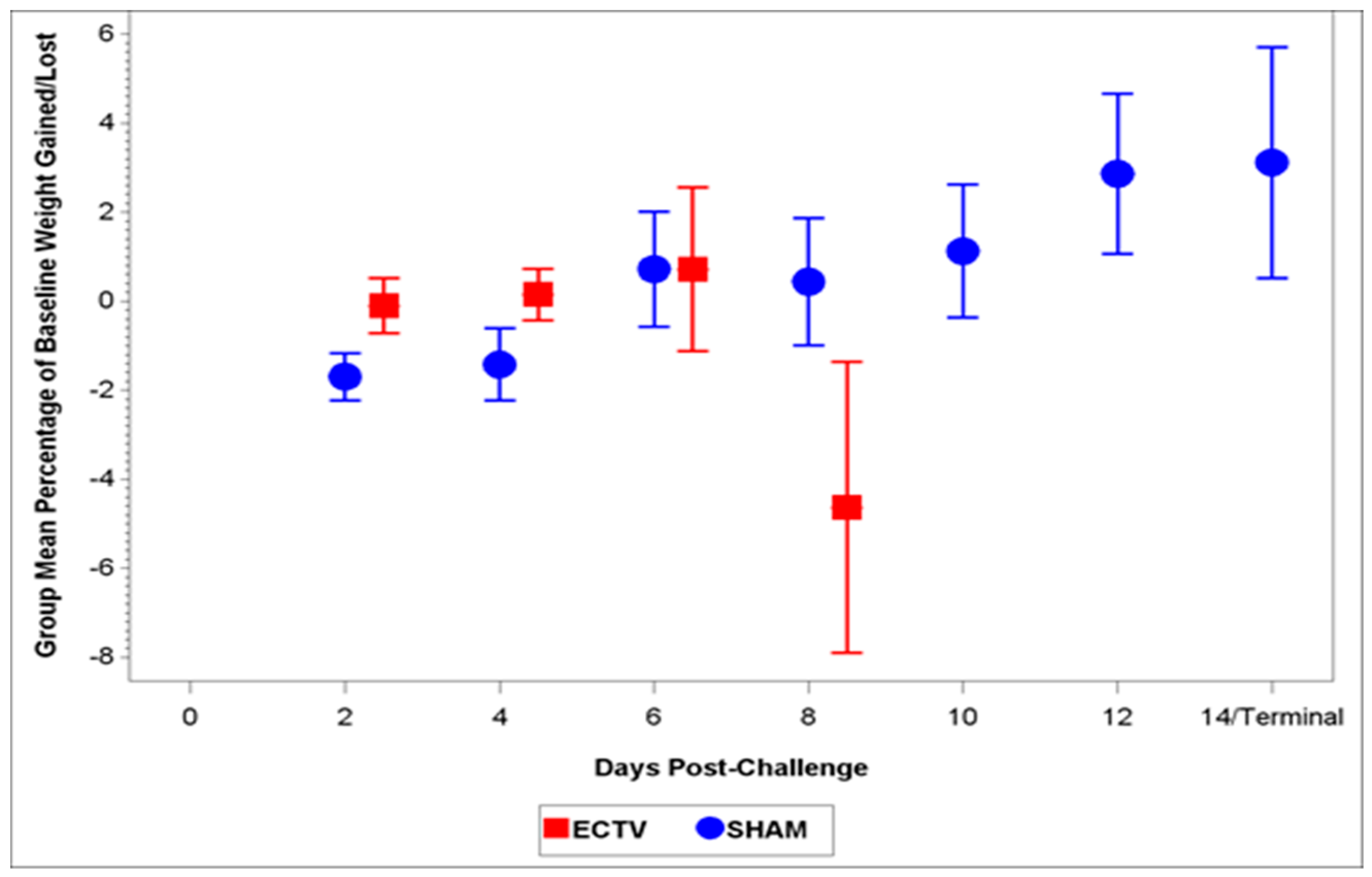
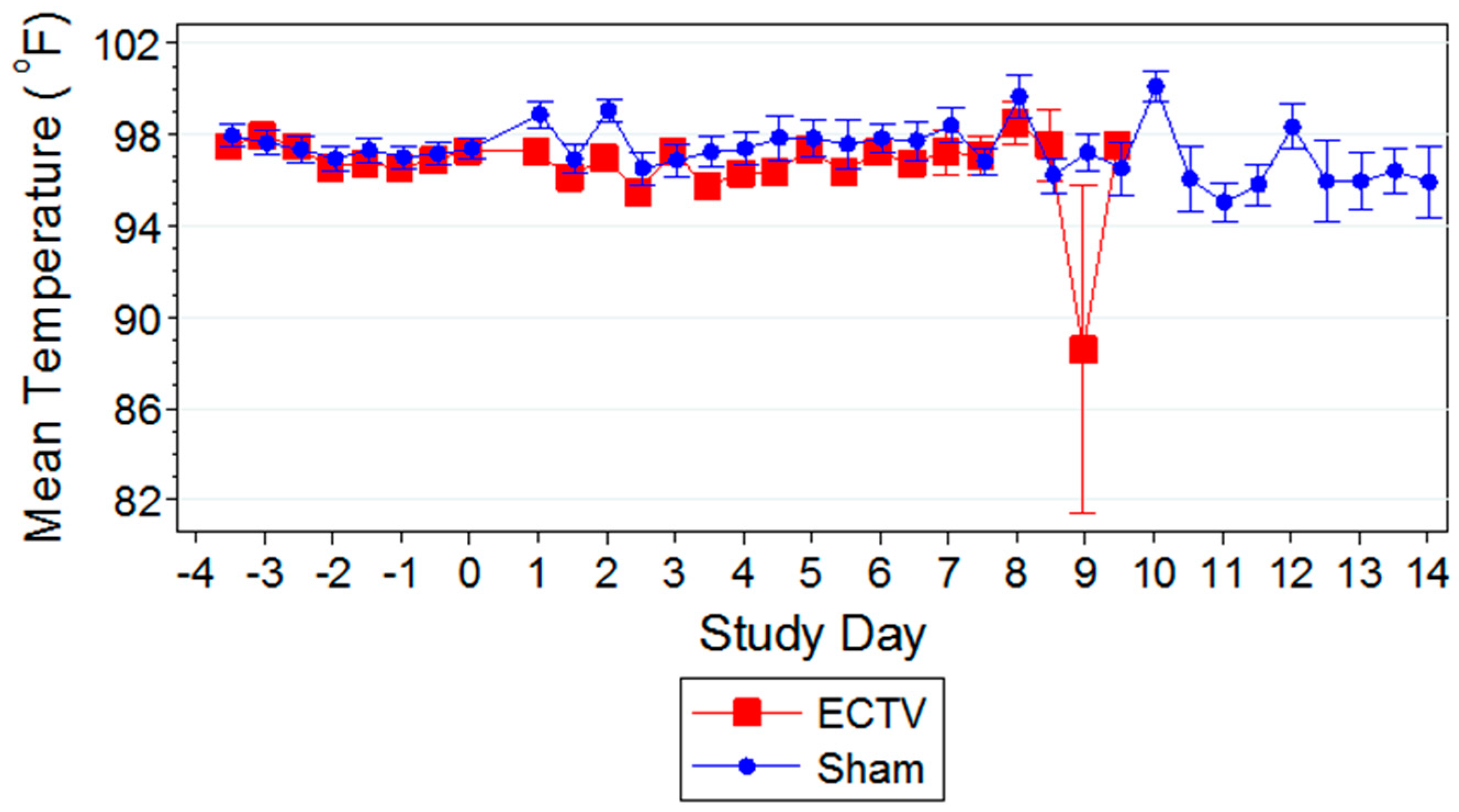
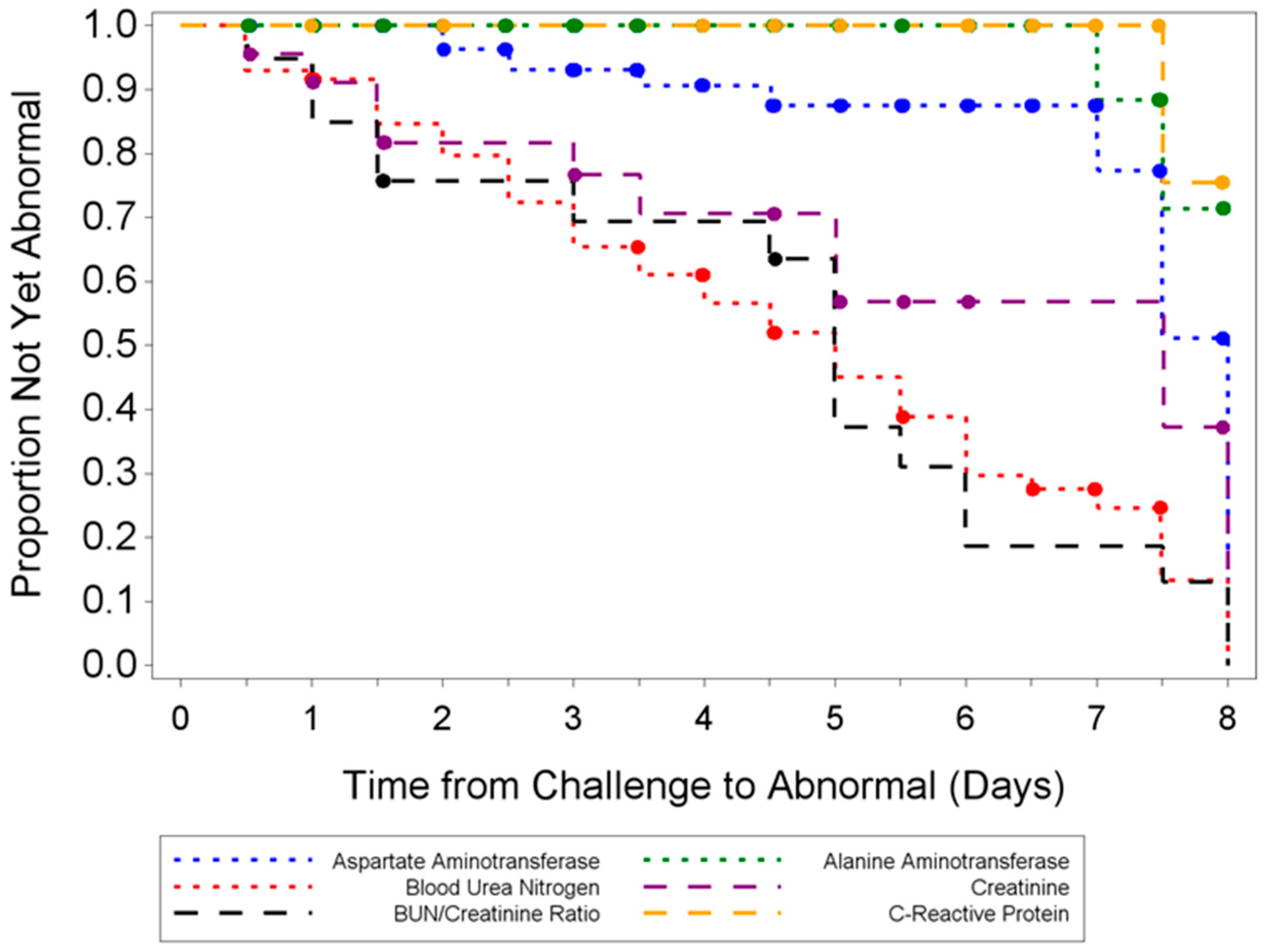
Classic symptoms routinely observed after high dose challenge in mouse models were also observed. Significant weight loss, commonly noted in high dose challenge models in animals infected with ECTV-Mos, was only initially observed eight days following challenge. Statistically significant body temperature changes were not observed until nine days post-challenge. The appearance of weight loss and temperature changes very late in the disease course suggests these biomarkers may not be useful “trigger-to-treat” parameters used for drug intervention in therapeutic efficacy studies. Historically, temperature change has not been a reliable biomarker as an early indication of ECTV disease infection [
17].
Clinical signs of disease including lethargy, ruffled fur, hunched posture, abnormal breathing, labored breathing, lacrimation, ocular abnormalities, and lesions were monitored to determine if an unambiguous indication for disease intervention could be presented. The mild clinical signs, such as lethargy and ruffled fur, were not good indications of disease as they were observed, albeit rarely, in sham infected animals, implying that the identification of these symptoms was not unambiguous for infected animals. The most common lesions appeared on the tail of mice in a small percentage of animals. However, group-housed mice could provoke tail biting, creating difficulty in discerning a pox lesion from a lesion caused by biting. Therefore, it is inconclusive as to whether all lesions observed in this study were directly related to ECTV infection or inflammation. Several mice did exhibit ear lesions that appeared at the base of the ears; however, these lesions were sporadic and relatively uncommon. As fur was not removed from mice, it was difficult to evaluate whether animals exhibited lesions or an ECTV-induced rash on the skin. Thus, utilization of rash or lesions as a potential trigger-to-treat in future mouse studies may be difficult without removal of hair on the BALB/c model. Some hairless mouse strains, such as the SKH1 strain, are sensitive to ECTV when the virus is administered via the IN route [
9]. Mouse models utilizing lesion presentation as a treatment trigger may not be advantageous since the presentation of lesions occurs late in the disease process, which, in turn, does not allow for a proper therapeutic window to observe product efficacy. The difference in clinical signs for moderate or severe symptoms between the infected and sham infected animals was significant by day 7 post exposure, just prior to mortality, demonstrating that clinical signs could be used to identify infected animals and potentially serve as triggers for intervention. Since observations consistent with mousepox disease were only apparent very late after infection, using clinical signs to inform drug intervention suggests that the time of intervention in the mouse model will be very late in disease course and not analogous to time of intervention and therapeutic window for smallpox disease in humans.
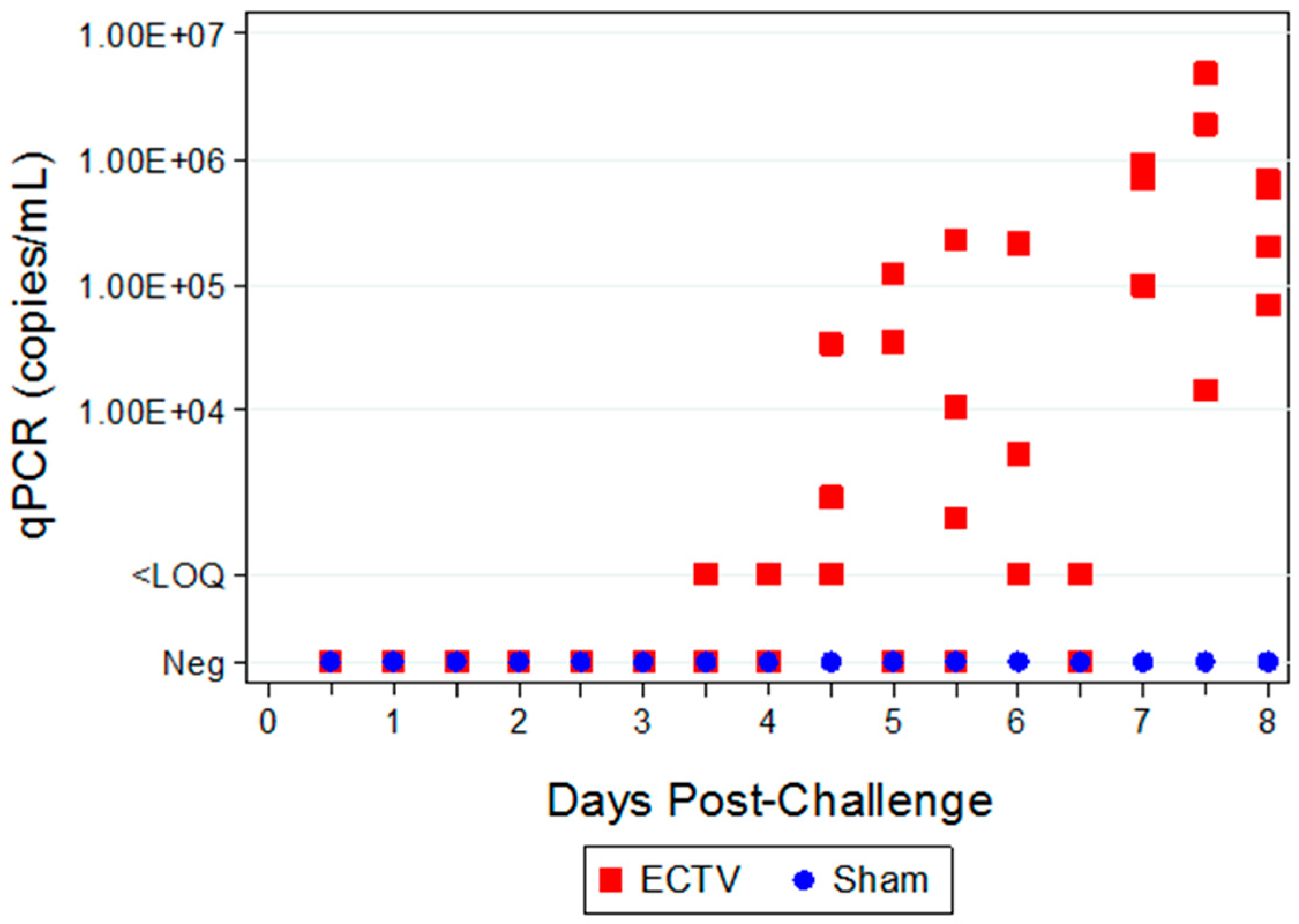
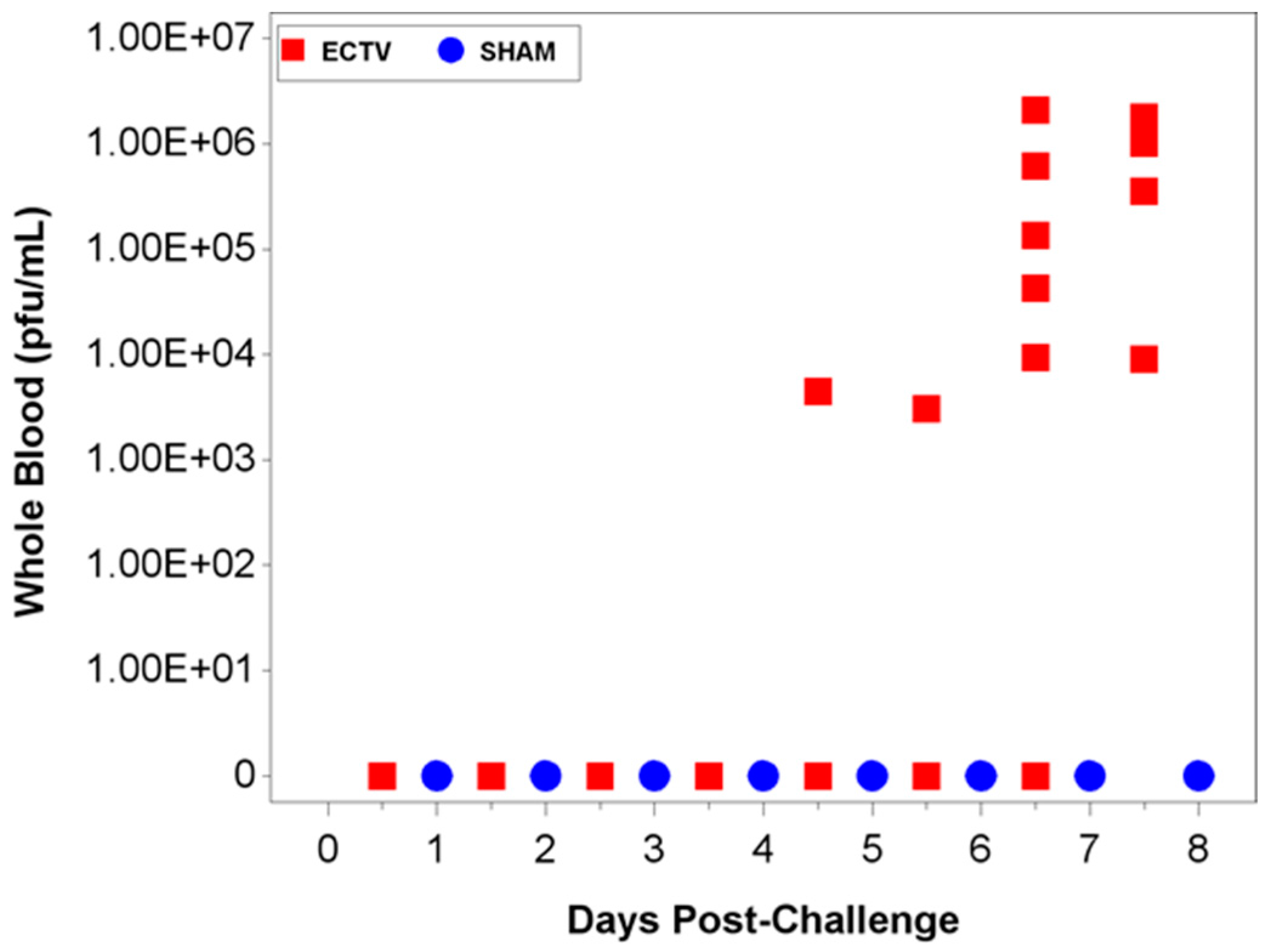
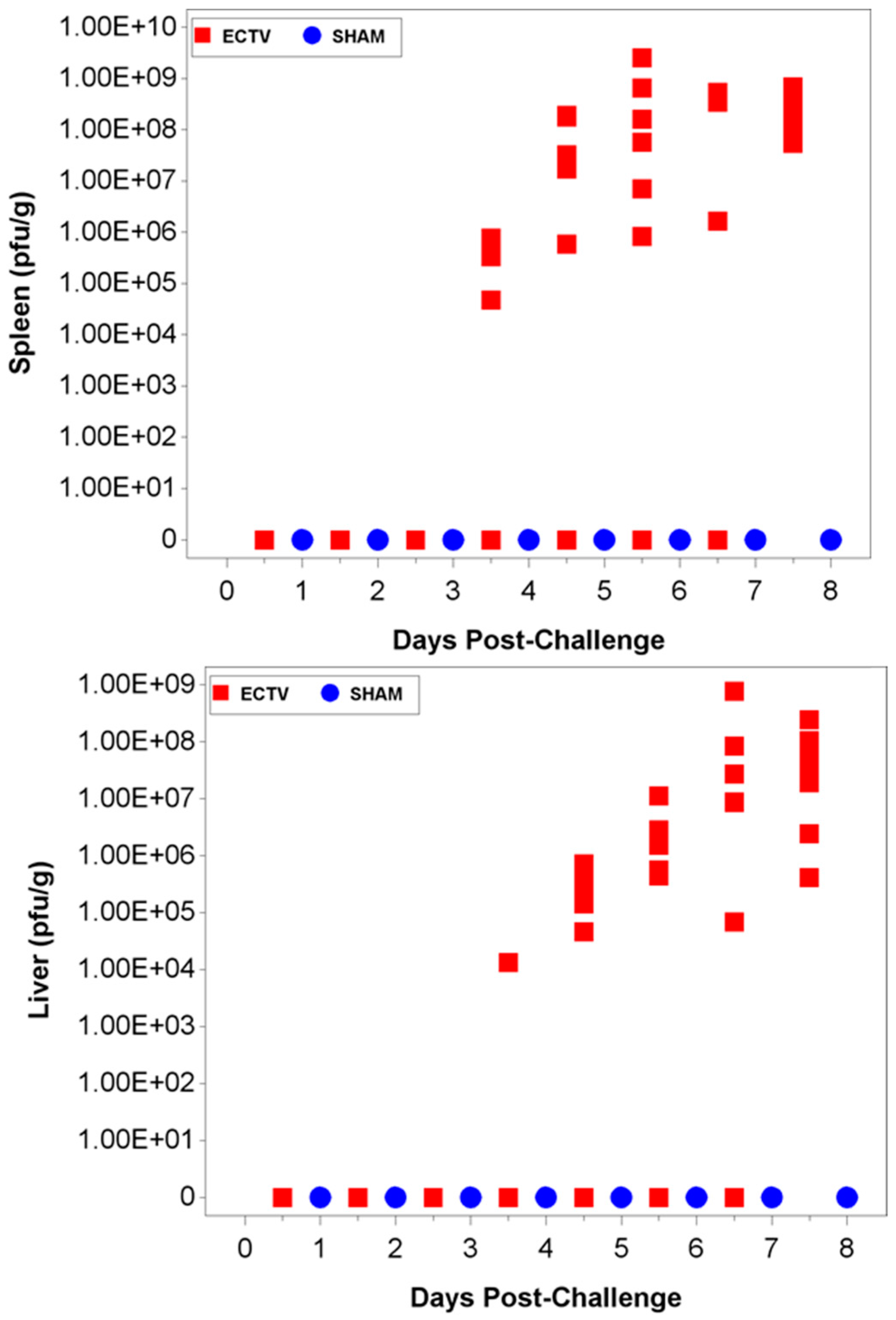
Two attempts were made to measure disseminated virus after infection. Blood was drawn at specified times and viral copy number (using PCR) and infectious virus (using plaque assay) were measured. In addition, as a non-invasive method that could be adaptable for real-time and multiple observations, oropharyngeal swabs from the oral cavity were collected and tested for viral copy number by qPCR. However, the viral copy numbers were below the assay LOQ in all swab samples from infected animals, with only a small percentage of animals having detectable viral genome copy numbers. Therefore, oropharyngeal swabs are not a reliable biomarker for countermeasure intervention. For blood samples, viral genomic material and infectious virus were observed at quantifiable levels in most, but not all infected animals starting at 4.5 days PI and the values increased with disease progression.
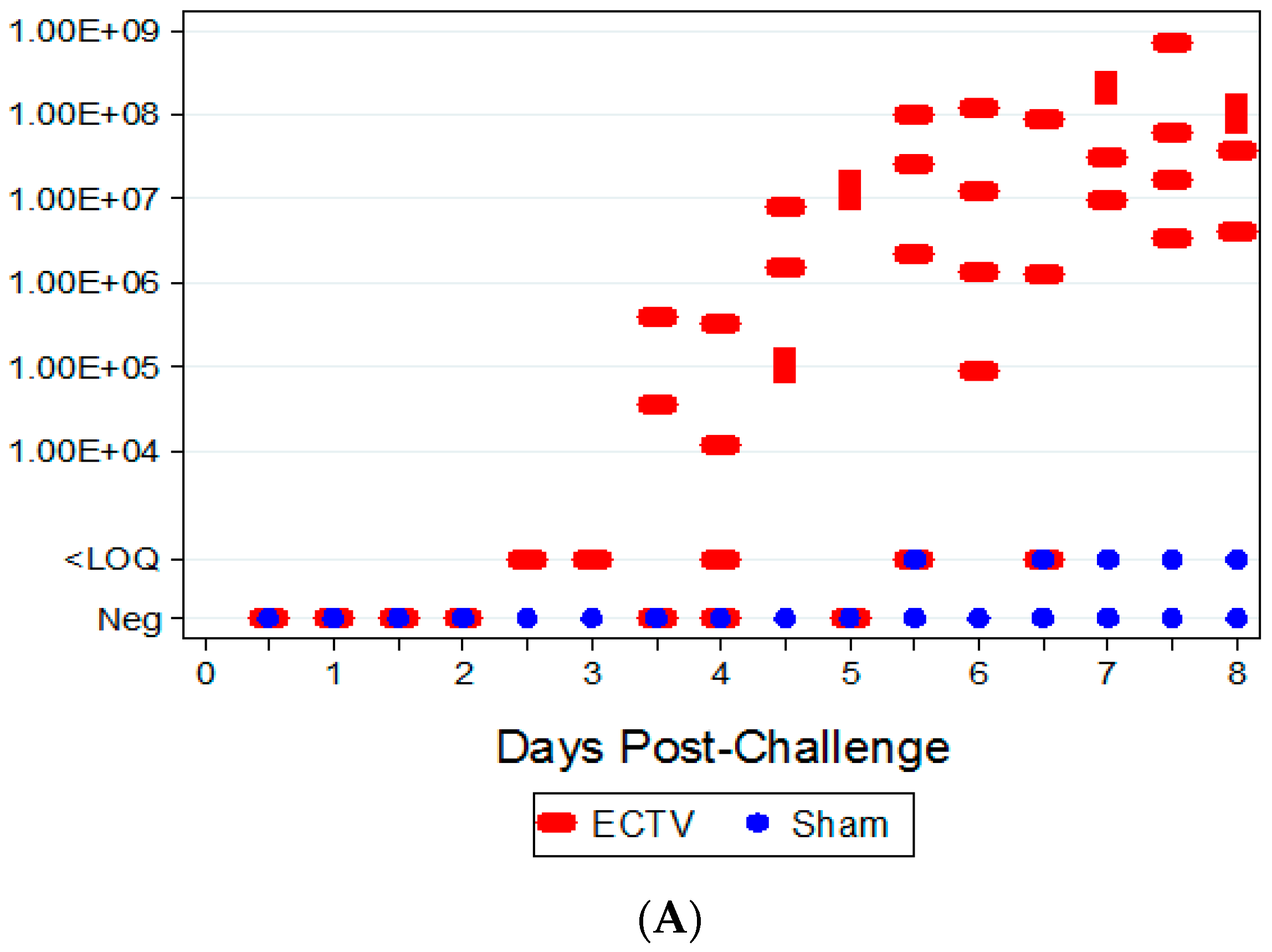
The ability to detect genomic copy numbers and infectious virus in spleen and liver tissue was achieved through qPCR and plaque assay. Quantifiable viral genomic material measured by qPCR and viral titers by plaque assay were first measured in the spleen and liver at 3.5 days PI, increasing in both tissues as the disease progressed. Overall, the level of viral genomic material in the liver was slightly less than observed in the spleen; however, all infected animals had measureable levels of genomic copies. Not surprisingly, the plaque assay for detecting infectious virus was less sensitive than qPCR as fewer ECTV-Mos infected animals exhibited detectable viral titers by plaque assay. In contrast to the ability to detect copy numbers or infectious virus in the blood, which was not universal for infected animals until at least seven days post exposure, splenic tissue samples did reproducibly and unambiguously identify infected animals by 5 to 5.5 days post exposure. Consideration must be given to this method for confirmation of viral detection, as sacrificing the animal to determine the level of infection is an evident drawback of tissue collection.
Hematological and clinical chemistry parameters were measured and screened for statistically significant differences compared to sham-infected animals and baseline. Several parameters demonstrated abnormal results in a majority of infected animals; however, no distinguishable trend for a reliable and unambiguous biomarker was observed. Pathological findings from tissues of challenged animals exhibited microscopic evidence of virus in the organs examined. Gross observations included pocks on the tail and discoloration of the spleen and liver. Tissue lesions were first noted at five days PI. Following ECTV-Mos infection, the time at which animals succumbed to disease correlated with the onset of the most severe microscopic findings.
The aim of these studies was to characterize the ECTV intranasal challenge model in BALB/c mice as a potential animal model for therapeutic evaluation of smallpox countermeasures. The ECTV model resembles human smallpox in that a low respiratory challenge dose produces high mortality. We intended to identify biomarkers to define the appropriate time for product intervention in order to measure therapeutic efficacy. The onset of clinical signs confirming ECTV infection are apparent relatively late in the course of the disease and do not provide a therapeutic window suitable for product evaluation. The easily measured and non-invasive analyses of weight loss, temperature rise, and genomic copies from oropharyngeal swabs are not reliable and unambiguous until just prior to death. The biomarker with the earliest indication of ECTV infection is genomic copy number as measured by qPCR. These copy numbers are detectable as early as at day 2.5 (organs) and 3.5 (blood). The percentage of infected animals with high copy numbers increases until all animals are positive by days 6 (spleen) or 7 (blood).
In human smallpox there is an extended incubation period (7–17 days) followed by prodromal fever and eruption of lesions, with patient outcome decided approximately two weeks after prodrome [
21]. In the mouse, the analogous disease caused by ECTV is compressed with disease progression and outcome completed in about one and a half weeks [
11]. The extended time from exposure to outcome in humans suggests that a biomarker earlier than one or two days prior to death in the mouse model would be a better analogue to the timing of intervention in a smallpox case. The medical intervention in smallpox would ideally occur near the midpoint of the disease course, as soon as lesions are detected, or after an outbreak is confirmed, and with the onset of fever in exposed patients. The biomarker in the mouse model that appears to correspond to this timeline is the PCR detection of viral genomes in blood and organs. However, the use of these biomarkers as triggers for intervention in a blinded study may require ingenuity in protocol design by culling a portion of each group as sentinel animals for disease detection or, as in the case of MPXV experiments, using a time-based intervention with confirmation of infection post-hoc.
The ECTV mouse model has afforded a powerful tool to study poxvirus-host interactions since there are genetic mouse strains that are sensitive and others that are resistant to infection. The cell-mediated arm of the immune response appears to be critical in determining the sensitivity of the mouse to ECTV infection [
7,
12,
24,
25]. The ability of the mouse to generate an early and robust cell-mediated immune response to clear the initial infection is the primary factor in surviving ECTV infection in resistant mice. The cell-mediated response limits the virus to the regional lymph nodes, prevents extensive liver and spleen damage, and prevents systemic spread. However, in sensitive strains of mice, such as BALB/c, the lack of an effective cell-mediated response leads to extensive replication in the liver and spleen and, just prior to death, systemic spread [
11]. The data from our studies are consistent with these observations. We detected early viral replication in the liver and spleen as well as the upper respiratory tract, while dyspnea and ocular discharges were noted late in infection.
The use of this BALB/c ECTV challenge model provides another method for the evaluation of smallpox countermeasures. The low cost and small size of mice allow for conducting large, statistically powered studies. In addition, use of a BSL-2 virus allows for greater accessibility compared to the rigid select agent practices necessary when working with BSL-3 agents, such as MPXV. Thus, ECTV infection in the BALB/c mouse provides a potential model for assessment of therapeutics and development of effective medical countermeasures of disease.


 and
and 










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}