
Foamy Virus Protein—Nucleic Acid Interactions during Particle Morphogenesis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
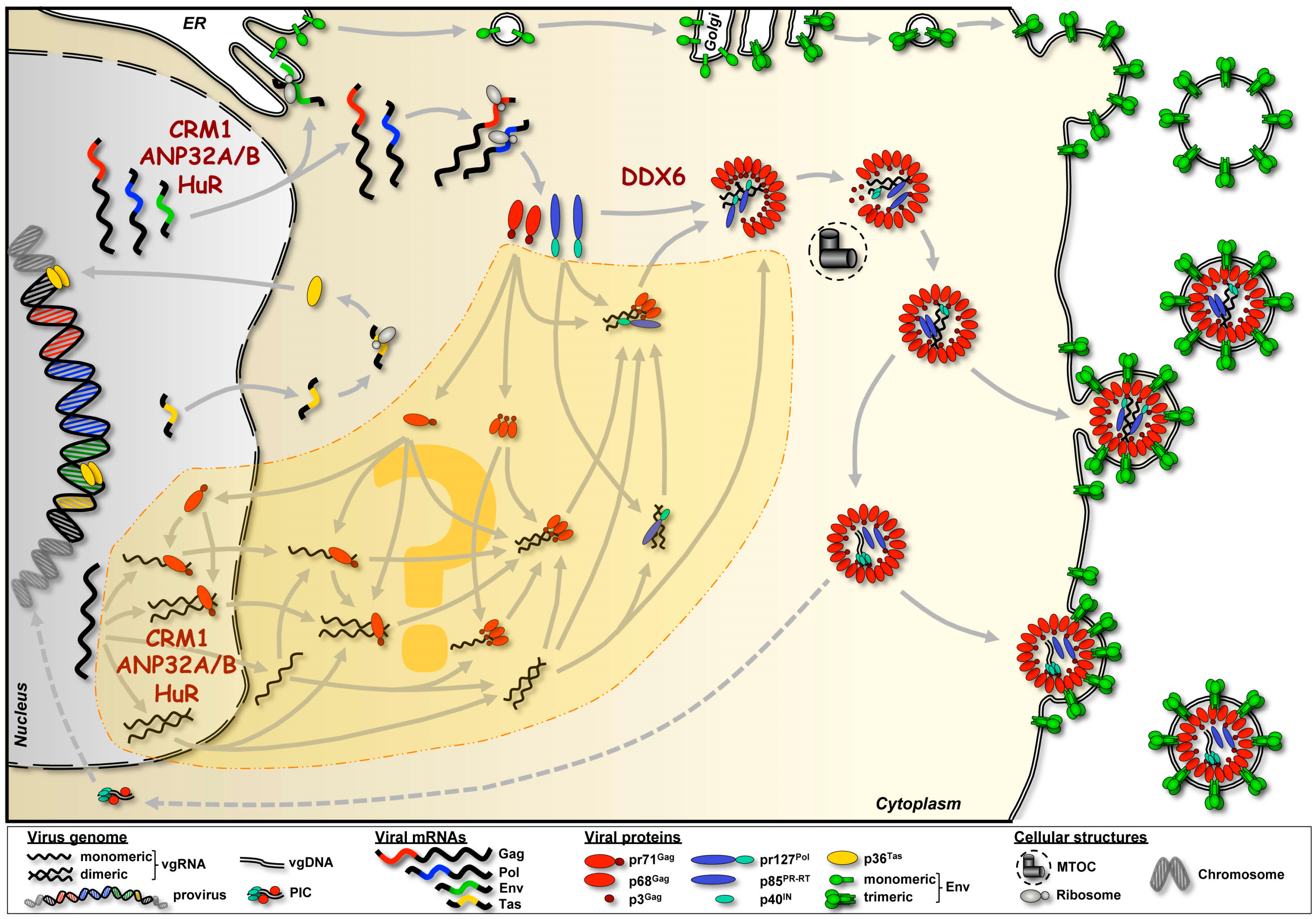
:1. Introduction
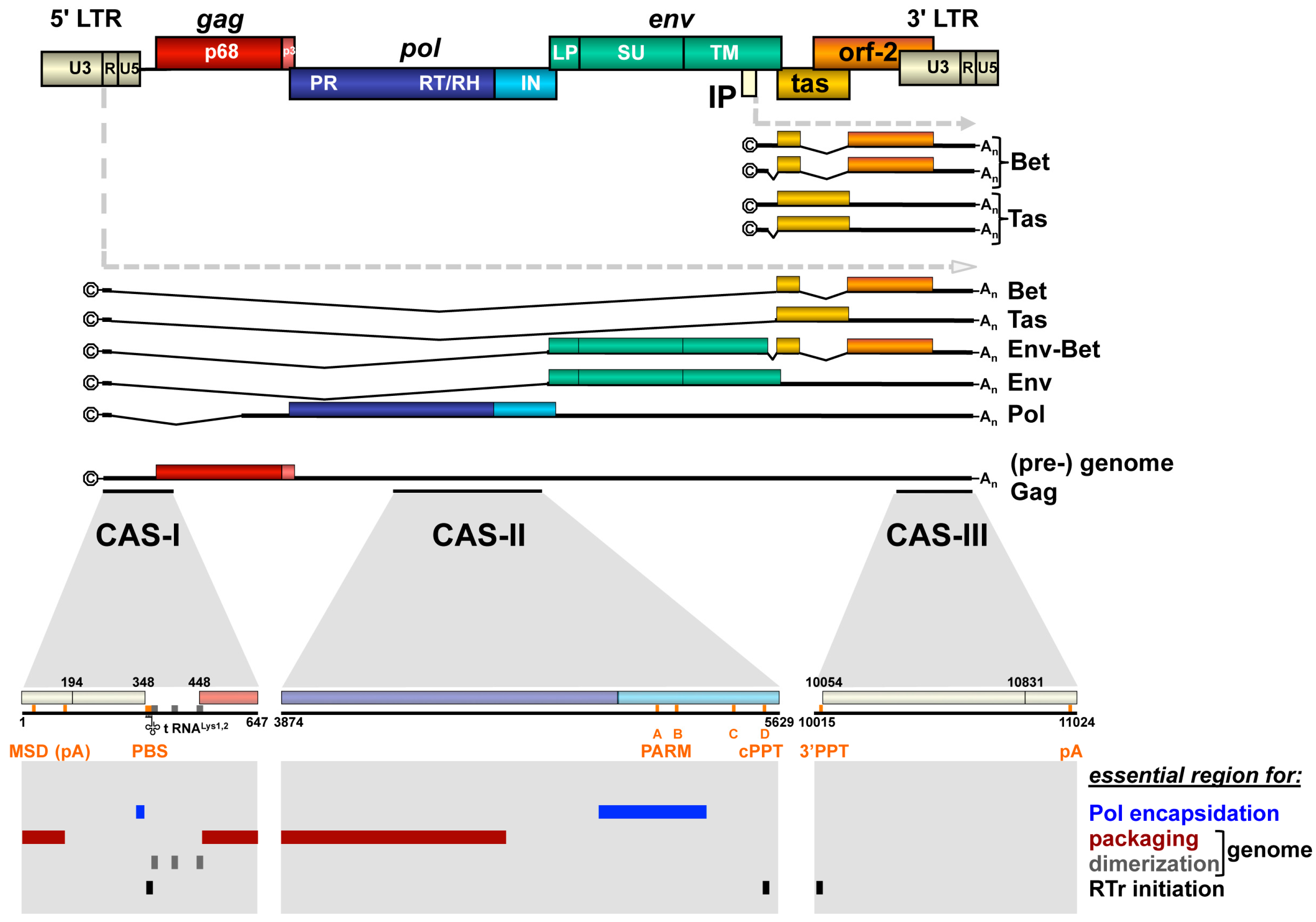
2. FV Transcription and Cis-Acting Viral Genomic RNA Sequences
2.1. Transcription
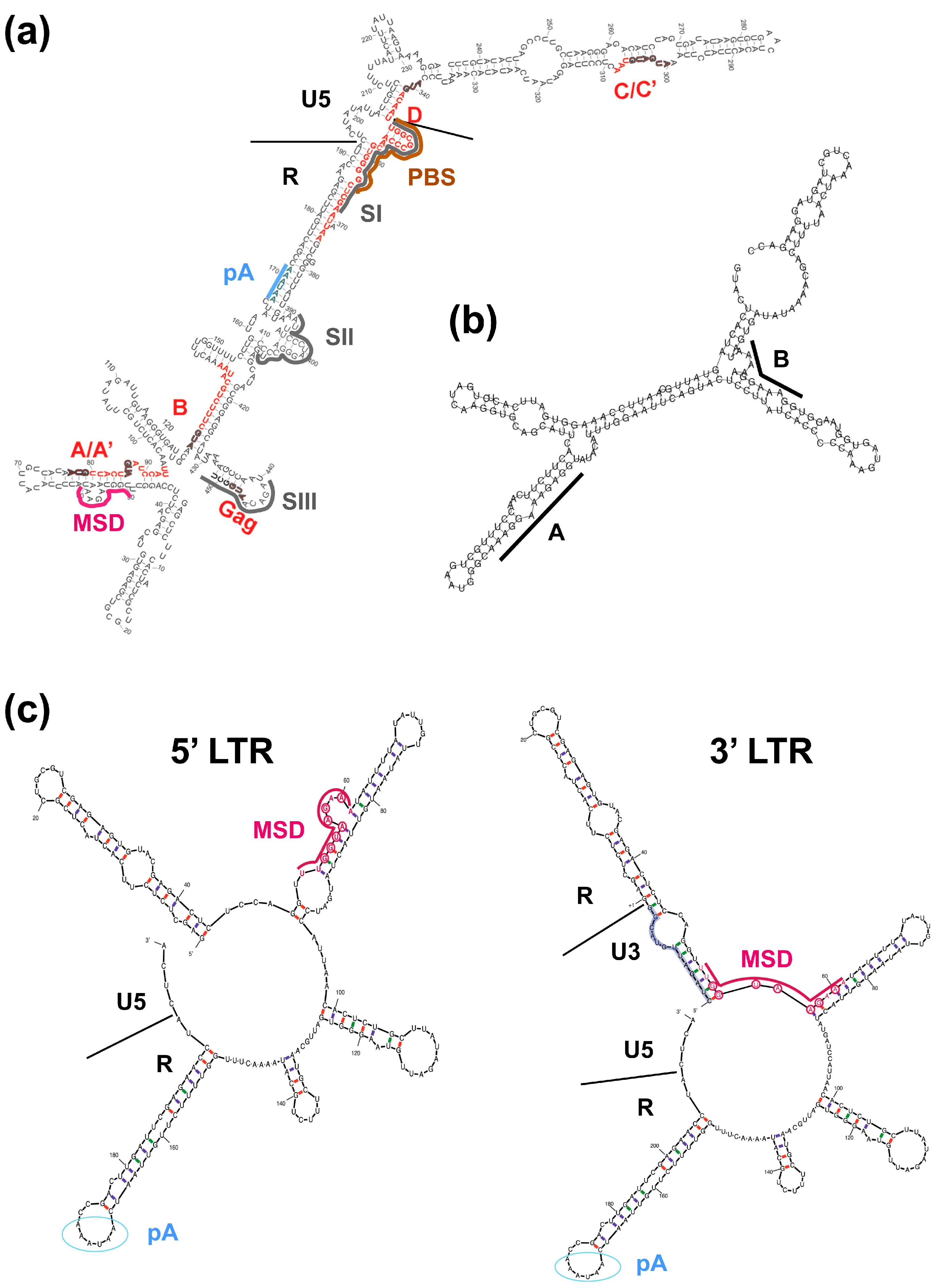
2.2. Cis-Acting Sequences
3. FV Gag Protein Biogenesis and Viral Genome Encapsidation
3.1. Transcription and Translation
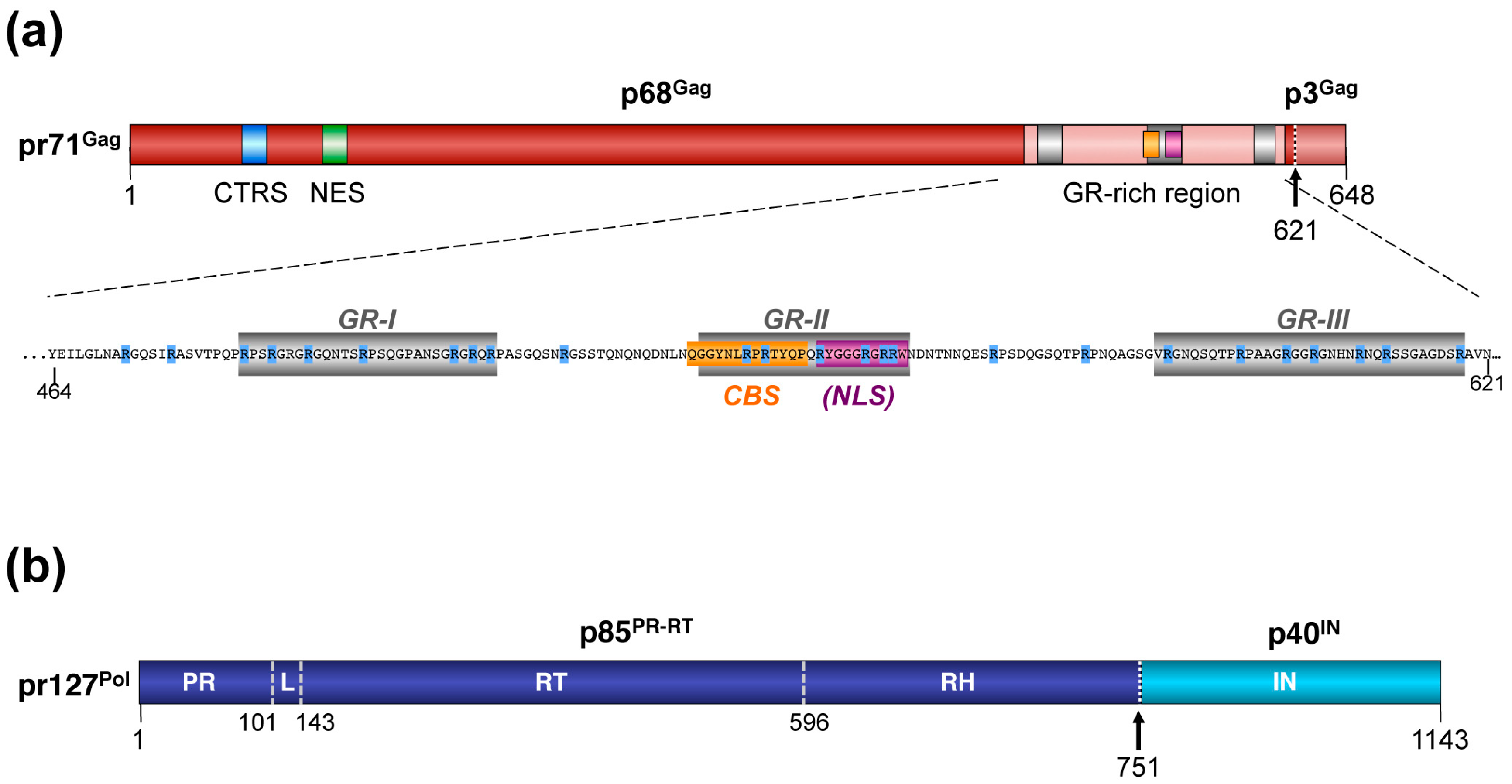
3.2. Post-translational Processing
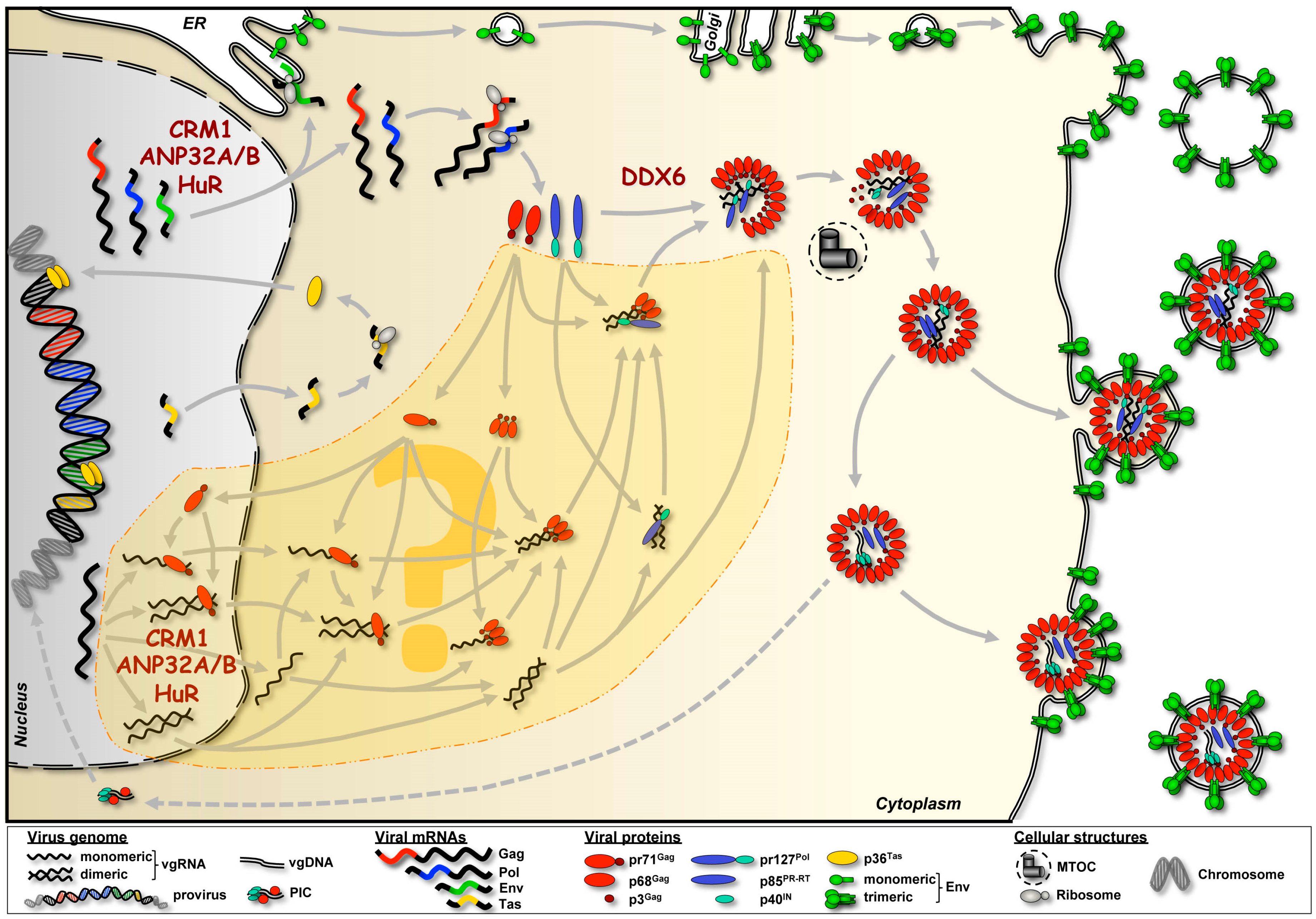
3.3. Trafficking and Intracellular Distribution
3.4. Nucleic Acid Interactions
4. FV Pol Biogenesis and Viral Genome-Dependent Encapsidation
4.1. Transcription and Translation
4.2. Posttranslational Processing and Nucleic Acid Interactions
5. Cellular Cofactors Interacting with FV RNAs
6. Conclusions and Perspectives
Acknowledgments
Conflicts of Interest
References
- Linial, M.L.; Fan, H.; Hahn, B.; Lwer, R.; Neil, J.; Quackenbush, S.; Rethwilm, A.; Sonigo, P.; Stoye, J.; Tristem, M. Retroviridae. In Virus Taxonomy, 2nd ed.; Fauquet, C., Mayo, M., Maniloff, J., Desselsberger, U., Ball, L., Eds.; Elsevier Inc.: Oxford, UK, 2005; pp. 421–440. [Google Scholar]
- Rethwilm, A.; Lindemann, D. Foamy Viruses. In Fields Virology, 6th ed.; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams & Wilkins, a Wolters Kluwer business: Philadelphia, PA, USA, 2013; Volume 2, pp. 1613–1632. [Google Scholar]
- Glebe, D.; Bremer, C.M. The molecular virology of hepatitis B virus. Semin. Liver Dis. 2013, 33, 103–112. [Google Scholar]
- Fischer, N.; Heinkelein, M.; Lindemann, D.; Enssle, J.; Baum, C.; Werder, E.; Zentgraf, H.; Müller, J.G.; Rethwilm, A. Foamy virus particle formation. J. Virol. 1998, 72, 1610–1615. [Google Scholar]
- Yu, S.F.; Eastman, S.W.; Linial, M.L. Foamy virus capsid assembly occurs at a pericentriolar region through a cytoplasmic targeting/retention signal in Gag. Traffic 2006, 7, 966–977. [Google Scholar]
- Bodem, J.; Löchelt, M.; Winkler, I.; Flower, R.P.; Delius, H.; Flügel, R.M. Characterization of the spliced pol transcript of feline foamy virus: the splice acceptor site of the pol transcript is located in gag of foamy viruses. J. Virol. 1996, 70, 9024–9027. [Google Scholar]
- Enssle, J.; Jordan, I.; Mauer, B.; Rethwilm, A. Foamy virus reverse transcriptase is expressed independently from the Gag protein. Proc. Natl. Acad. Sci. USA 1996, 93, 4137–4141. [Google Scholar]
- Jordan, I.; Enssle, J.; Guttler, E.; Mauer, B.; Rethwilm, A. Expression of human foamy virus reverse transcriptase involves a spliced pol mRNA. Virology 1996, 224, 314–319. [Google Scholar]
- Löchelt, M.; Flügel, R.M. The human foamy virus pol gene is expressed as a Pro-Pol polyprotein and not as a Gag-Pol fusion protein. J. Virol. 1996, 70, 1033–1040. [Google Scholar]
- Yu, S.F.; Baldwin, D.N.; Gwynn, S.R.; Yendapalli, S.; Linial, M.L. Human foamy virus replication: a pathway distinct from that of retroviruses and hepadnaviruses. Science 1996, 271, 1579–1582. [Google Scholar]
- Moebes, A.; Enssle, J.; Bieniasz, P.D.; Heinkelein, M.; Lindemann, D.; Bock, M.; McClure, M.O.; Rethwilm, A. Human foamy virus reverse transcription that occurs late in the viral replication cycle. J. Virol. 1997, 71, 7305–7311. [Google Scholar]
- Roy, J.; Rudolph, W.; Juretzek, T.; Gärtner, K.; Bock, M.; Herchenröder, O.; Lindemann, D.; Heinkelein, M.; Rethwilm, A. Feline foamy virus genome and replication strategy. J. Virol. 2003, 77, 11324–11331. [Google Scholar]
- Heinkelein, M.; Pietschmann, T.; Jarmy, G.; Dressler, M.; Imrich, H.; Thurow, J.; Lindemann, D.; Bock, M.; Moebes, A.; Roy, J.; et al. Efficient intracellular retrotransposition of an exogenous primate retrovirus genome. EMBO J. 2000, 19, 3436–3445. [Google Scholar]
- Baldwin, D.N.; Linial, M.L. The roles of Pol and Env in the assembly pathway of human foamy virus. J. Virol. 1998, 72, 3658–3665. [Google Scholar]
- Pietschmann, T.; Heinkelein, M.; Heldmann, M.; Zentgraf, H.; Rethwilm, A.; Lindemann, D. Foamy virus capsids require the cognate envelope protein for particle export. J. Virol. 1999, 73, 2613–2621. [Google Scholar]
- Lindemann, D.; Pietschmann, T.; Picard-Maureau, M.; Berg, A.; Heinkelein, M.; Thurow, J.; Knaus, P.; Zentgraf, H.; Rethwilm, A. A particle-associated glycoprotein signal peptide essential for virus maturation and infectivity. J. Virol. 2001, 75, 5762–5771. [Google Scholar]
- Wilk, T.; Geiselhart, V.; Frech, M.; Fuller, S.D.; Flügel, R.M.; Löchelt, M. Specific interaction of a novel foamy virus Env leader protein with the N-terminal Gag domain. J. Virol. 2001, 75, 7995–8007. [Google Scholar]
- Lindemann, D.; Rethwilm, A. Foamy virus biology and its application for vector development. Viruses 2011, 3, 561–585. [Google Scholar]
- Löchelt, M. Foamy virus transactivation and gene expression. Curr. Top. Microbiol. Immunol. 2003, 277, 27–61. [Google Scholar]
- Löchelt, M.; Romen, F.; Bastone, P.; Muckenfuss, H.; Kirchner, N.; Kim, Y.B.; Truyen, U.; Rösler, U.; Battenberg, M.; Saib, A.; et al. The antiretroviral activity of APOBEC3 is inhibited by the foamy virus accessory Bet protein. Proc. Natl. Acad. Sci. USA 2005, 102, 7982–7987. [Google Scholar]
- Cain, D.; Erlwein, O.; Grigg, A.; Russell, R.A.; McClure, M.O. Palindromic sequence plays a critical role in human foamy virus dimerization. J. Virol. 2001, 75, 3731–3739. [Google Scholar] [PubMed]
- Erlwein, O.; Bieniasz, P.D.; McClure, M.O. Sequences in pol are required for transfer of human foamy virus-based vectors. J. Virol. 1998, 72, 5510–5516. [Google Scholar] [PubMed]
- Erlwein, O.; Cain, D.; Fischer, N.; Rethwilm, A.; McClure, M.O. Identification of sites that act together to direct dimerization of human foamy virus RNA in vitro. Virology 1997, 229, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Maurer, B.; Bannert, H.; Darai, G.; Flügel, R.M. Analysis of the primary structure of the long terminal repeat and the gag and pol genes of the human spumaretrovirus. J. Virol. 1988, 62, 1590–1597. [Google Scholar] [PubMed]
- Muranyi, W.; Flügel, R.M. Analysis of splicing patterns of human spumaretrovirus by polymerase chain reaction reveals complex RNA structures. J. Virol. 1991, 65, 727–735. [Google Scholar] [PubMed]
- Heinkelein, M.; Schmidt, M.; Fischer, N.; Moebes, A.; Lindemann, D.; Enssle, J.; Rethwilm, A. Characterization of a cis-acting sequence in the pol region required to transfer human foamy virus vectors. J. Virol. 1998, 72, 6307–6314. [Google Scholar] [PubMed]
- Heinkelein, M.; Thurow, J.; Dressler, M.; Imrich, H.; Neumann-Haefelin, D.; McClure, M.O.; Rethwilm, A. Complex effects of deletions in the 5' untranslated region of primate foamy virus on viral gene expression and RNA packaging. J. Virol. 2000, 74, 3141–3148. [Google Scholar] [CrossRef] [PubMed]
- Heinkelein, M.; Dressler, M.; Jarmy, G.; Rammling, M.; Imrich, H.; Thurow, J.; Lindemann, D.; Rethwilm, A. Improved primate foamy virus vectors and packaging constructs. J. Virol. 2002, 76, 3774–3783. [Google Scholar] [CrossRef] [PubMed]
- Russell, R.A.; Zeng, Y.; Erlwein, O.; Cullen, B.R.; McClure, M.O. The R region found in the human foamy virus long terminal repeat is critical for both Gag and Pol protein expression. J. Virol. 2001, 75, 6817–6824. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Backes, P.; Löchelt, M. Importance of the major splice donor and redefinition of cis-acting sequences of gutless feline foamy virus vectors. Virology 2009, 394, 208–217. [Google Scholar] [CrossRef] [PubMed]
- Schrom, E.M.; Moschall, R.; Hartl, M.J.; Weitner, H.; Fecher, D.; Langemeier, J.; Bohne, J.; Wöhrl, B.M.; Bodem, J. U1snRNP-mediated suppression of polyadenylation in conjunction with the RNA structure controls poly (A) site selection in foamy viruses. Retrovirology 2013, 10. [Google Scholar] [CrossRef] [PubMed]
- Schepetilnikov, M.; Schott, G.; Katsarou, K.; Thiebeauld, O.; Keller, M.; Ryabova, L.A. Molecular dissection of the prototype foamy virus (PFV) RNA 5'-UTR identifies essential elements of a ribosomal shunt. Nucleic Acids Res. 2009, 37, 5838–5847. [Google Scholar] [CrossRef] [PubMed]
- Hartl, M.J.; Bodem, J.; Jochheim, F.; Rethwilm, A.; Rösch, P.; Wöhrl, B.M. Regulation of foamy virus protease activity by viral RNA: a novel and unique mechanism among retroviruses. J. Virol. 2011, 85, 4462–4469. [Google Scholar] [CrossRef] [PubMed]
- Mathews, D.H.; Sabina, J.; Zuker, M.; Turner, D.H. Expanded sequence dependence of thermodynamic parameters improves prediction of RNA secondary structure. J. Mol. Biol. 1999, 288, 911–940. [Google Scholar] [CrossRef] [PubMed]
- Gruber, A.R.; Lorenz, R.; Bernhart, S.H.; Neubock, R.; Hofacker, I.L. The Vienna RNA websuite. Nucleic Acids Res. 2008, 36, W70–W74. [Google Scholar] [CrossRef] [PubMed]
- Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003, 31, 3406–3415. [Google Scholar] [CrossRef] [PubMed]
- Zuker, M.; Jacobson, A.B. Using reliability information to annotate RNA secondary structures. RNA 1998, 4, 669–679. [Google Scholar] [CrossRef] [PubMed]
- Waugh, A.; Gendron, P.; Altman, R.; Brown, J.W.; Case, D.; Gautheret, D.; Harvey, S.C.; Leontis, N.; Westbrook, J.; Westhof, E.; et al. RNAML: A standard syntax for exchanging RNA information. RNA 2002, 8, 707–717. [Google Scholar] [CrossRef] [PubMed]
- Trobridge, G.; Josephson, N.; Vassilopoulos, G.; Mac, J.; Russell, D. Improved foamy virus vectors with minimal viral sequences. Mol. Ther. 2002, 6, 321. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Chari, S.; Yanchis, T.; Mergia, A. cis-Acting sequences required for simian foamy virus type 1 vectors. J. Virol. 1998, 72, 3451–3454. [Google Scholar] [PubMed]
- Kupiec, J.J.; Tobaly-Tapiero, J.; Canivet, M.; Santillana-Hayat, M.; Flügel, R.M.; Peries, J.; Emanoil-Ravier, R. Evidence for a gapped linear duplex DNA intermediate in the replicative cycle of human and simian spumaviruses. Nucleic Acids Res. 1988, 16, 9557–9565. [Google Scholar] [CrossRef] [PubMed]
- Peters, K.; Barg, N.; Gärtner, K.; Rethwilm, A. Complex effects of foamy virus central purine-rich regions on viral replication. Virology 2008, 373, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Peters, K.; Wiktorowicz, T.; Heinkelein, M.; Rethwilm, A. RNA and protein requirements for incorporation of the Pol protein into foamy virus particles. J. Virol. 2005, 79, 7005–7013. [Google Scholar] [CrossRef] [PubMed]
- Wiktorowicz, T.; Peters, K.; Armbruster, N.; Steinert, A.F.; Rethwilm, A. Generation of an improved foamy virus vector by dissection of cis-acting sequences. J. Gen. Virol. 2009, 90, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Goff, S.P. Retroviridae. In Fields Virology, 6th ed.; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams & Wilkins, a Wolters Kluwer business: Philadelphia, PA, USA, 2013; Volume 2, pp. 1424–1473. [Google Scholar]
- Konvalinka, J.; Kräusslich, H.G.; Müller, B. Retroviral proteases and their roles in virion maturation. Virology 2015, 479–480, 403–417. [Google Scholar] [CrossRef] [PubMed]
- Rein, A.; Datta, S.A.; Jones, C.P.; Musier-Forsyth, K. Diverse interactions of retroviral Gag proteins with RNAs. Trends Biochem. Sci. 2011, 36, 373–380. [Google Scholar] [CrossRef] [PubMed]
- Linial, M.L.; Eastman, S.W. Particle assembly and genome packaging. Curr. Top. Microbiol. Immunol. 2003, 277, 89–110. [Google Scholar] [PubMed]
- Müllers, E. The foamy virus Gag proteins: what makes them different? Viruses 2013, 5, 1023–1041. [Google Scholar] [CrossRef] [PubMed]
- Morozov, V.A.; Copeland, T.D.; Nagashima, K.; Gonda, M.A.; Oroszlan, S. Protein composition and morphology of human foamy virus intracellular cores and extracellular particles. Virology 1997, 228, 307–317. [Google Scholar] [CrossRef] [PubMed]
- Giron, M.L.; Colas, S.; Wybier, J.; Rozain, F.; Emanoil Ravier, R. Expression and maturation of human foamy virus Gag precursor polypeptides. J. Virol. 1997, 71, 1635–1639. [Google Scholar] [PubMed]
- Bodem, J.; Schied, T.; Gabriel, R.; Rammling, M.; Rethwilm, A. Foamy virus nuclear RNA export is distinct from that of other retroviruses. J. Virol. 2011, 85, 2333–2341. [Google Scholar] [CrossRef] [PubMed]
- Cartellieri, M.; Rudolph, W.; Herchenröder, O.; Lindemann, D.; Rethwilm, A. Determination of the relative amounts of Gag and Pol proteins in foamy virus particles. Retrovirology 2005, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hütter, S.; Müllers, E.; Stanke, N.; Reh, J.; Lindemann, D. Prototype foamy virus protease activity is essential for intraparticle reverse transcription initiation but not absolutely required for uncoating upon host cell entry. J. Virol. 2013, 87, 3163–3176. [Google Scholar] [CrossRef] [PubMed]
- Spannaus, R.; Bodem, J. Determination of the protease cleavage site repertoireTthe RNase H but not the RT domain is essential for foamy viral protease activity. Virology 2014, 454–455, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Enssle, J.; Fischer, N.; Moebes, A.; Mauer, B.; Smola, U.; Rethwilm, A. Carboxy-terminal cleavage of the human foamy virus Gag precursor molecule is an essential step in the viral life cycle. J. Virol. 1997, 71, 7312–7317. [Google Scholar] [PubMed]
- Zemba, M.; Wilk, T.; Rutten, T.; Wagner, A.; Flügel, R.M.; Löchelt, M. The carboxy-terminal p3Gag domain of the human foamy virus Gag precursor is required for efficient virus infectivity. Virology 1998, 247, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Konvalinka, J.; Löchelt, M.; Zentgraf, H.; Flügel, R.M.; Kräusslich, H.G. Active foamy virus proteinase is essential for virus infectivity but not for formation of a Pol polyprotein. J. Virol. 1995, 69, 7264–7268. [Google Scholar] [PubMed]
- Spannaus, R.; Schneider, A.; Hartl, M.J.; Wöhrl, B.M.; Bodem, J. Foamy virus Gag p71-p68 cleavage is required for template switch of the reverse transcriptase. J. Virol. 2013, 87, 7774–7776. [Google Scholar] [CrossRef] [PubMed]
- Eastman, S.W.; Linial, M.L. Identification of a conserved residue of foamy virus Gag required for intracellular capsid assembly. J. Virol. 2001, 75, 6857–6864. [Google Scholar] [CrossRef]
- Renault, N.; Tobaly-Tapiero, J.; Paris, J.; Giron, M.L.; Coiffic, A.; Roingeard, P.; Saib, A. A nuclear export signal within the structural Gag protein is required for prototype foamy virus replication. Retrovirology 2011, 8, 6. [Google Scholar] [Green Version]
- Fleming, W.A.; Clarke, J.K. Fluorescence assay of foamy virus. J. Gen. Virol. 1970, 6, 277–284. [Google Scholar] [CrossRef] [PubMed]
- Hooks, J.J.; Detrick-Hooks, B. Spumavirinae: foamy virus group infections: comparative aspects and diagnosis. In Comparative Diagnosis of Viral Diseases; Kurstak, K., Kurstak, C., Eds.; Academic Press: New York, 1981; Vol. 4, pp. 599–618. [Google Scholar]
- Schliephake, A.W.; Rethwilm, A. Nuclear localization of foamy virus Gag precursor protein. J. Virol. 1994, 68, 4946–4954. [Google Scholar] [PubMed]
- Yu, S.F.; Edelmann, K.; Strong, R.K.; Moebes, A.; Rethwilm, A.; Linial, M.L. The carboxyl terminus of the human foamy virus Gag protein contains separable nucleic acid binding and nuclear transport domains. J. Virol. 1996, 70, 8255–8262. [Google Scholar] [PubMed]
- Müllers, E.; Stirnnagel, K.; Kaulfuss, S.; Lindemann, D. Prototype foamy virus gag nuclear localization: a novel pathway among retroviruses. J. Virol. 2011, 85, 9276–9285. [Google Scholar] [CrossRef] [PubMed]
- Tobaly-Tapiero, J.; Bittoun, P.; Lehmann-Che, J.; Delelis, O.; Giron, M.L.; de The, H.; Saib, A. Chromatin tethering of incoming foamy virus by the structural Gag protein. Traffic 2008, 9, 1717–1727. [Google Scholar] [CrossRef] [PubMed]
- Roy, J.; Linial, M.L. Role of the foamy virus Pol cleavage site in viral replication. J. Virol. 2007, 81, 4956–4962. [Google Scholar] [CrossRef] [PubMed]
- Stirnnagel, K.; Schupp, D.; Dupont, A.; Kudryavtsev, V.; Reh, J.; Müllers, E.; Lamb, D.C.; Lindemann, D. Differential pH-dependent cellular uptake pathways among foamy viruses elucidated using dual-colored fluorescent particles. Retrovirology 2012, 9, 71. [Google Scholar] [CrossRef] [PubMed]
- Winkler, I.; Bodem, J.; Haas, L.; Zemba, M.; Delius, H.; Flower, R.; Flügel, R.M.; Löchelt, M. Characterization of the genome of feline foamy virus and its proteins shows distinct features different from those of primate spumaviruses. J. Virol. 1997, 71, 6727–6741. [Google Scholar] [PubMed]
- Tobaly-Tapiero, J.; Bittoun, P.; Neves, M.; Guillemin, M.C.; Lecellier, C.H.; Puvion-Dutilleul, F.; Gicquel, B.; Zientara, S.; Giron, M.L.; de The, H.; et al. Isolation and characterization of an equine foamy virus. J. Virol. 2000, 74, 4064–4073. [Google Scholar] [CrossRef] [PubMed]
- Müllers, E.; Uhlig, T.; Stirnnagel, K.; Fiebig, U.; Zentgraf, H.; Lindemann, D. Novel functions of prototype foamy virus Gag glycine-arginine-rich boxes in reverse transcription and particle morphogenesis. J. Virol. 2011, 85, 1452–1463. [Google Scholar] [CrossRef] [PubMed]
- Stenbak, C.R.; Linial, M.L. Role of the C terminus of foamy virus Gag in RNA packaging and Pol expression. J. Virol. 2004, 78, 9423–9430. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.G.; Linial, M.L. The C terminus of foamy retrovirus Gag contains determinants for encapsidation of Pol protein into virions. J. Virol. 2008, 82, 10803–10810. [Google Scholar] [CrossRef] [PubMed]
- Hamann, M.V.; Müllers, E.; Reh, J.; Stanke, N.; Effantin, G.; Weissenhorn, W.; Lindemann, D. The cooperative function of arginine residues in the Prototype Foamy Virus Gag C-terminus mediates viral and cellular RNA encapsidation. Retrovirology 2014, 11. [Google Scholar] [CrossRef] [PubMed]
- Muriaux, D.; Mirro, J.; Harvin, D.; Rein, A. RNA is a structural element in retrovirus particles. Proc. Natl. Acad. Sci. USA 2001, 98, 5246–5251. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.G.; Kuppers, D.; Horn, M.; Roy, J.; May, C.; Linial, M.L. A premature termination codon mutation at the C terminus of foamy virus Gag downregulates the levels of spliced pol mRNA. J. Virol. 2008, 82, 1656–1664. [Google Scholar] [CrossRef] [PubMed]
- Jackson, D.L.; Lee, E.G.; Linial, M.L. Expression of prototype foamy virus pol as a Gag-Pol fusion protein does not change the timing of reverse transcription. J. Virol. 2013, 87, 1252–1254. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.G.; Sinicrope, A.; Jackson, D.L.; Yu, S.F.; Linial, M.L. Foamy virus Pol protein expressed as a Gag-Pol fusion retains enzymatic activities, allowing for infectious virus production. J. Virol. 2012, 86, 5992–6001. [Google Scholar] [CrossRef] [PubMed]
- Swiersy, A.; Wiek, C.; Reh, J.; Zentgraf, H.; Lindemann, D. Orthoretroviral-like prototype foamy virus Gag-Pol expression is compatible with viral replication. Retrovirology 2011, 8. [Google Scholar] [CrossRef] [PubMed]
- Pfrepper, K.I.; Rackwitz, H.R.; Schnolzer, M.; Heid, H.; Löchelt, M.; Flügel, R.M. Molecular characterization of proteolytic processing of the Pol proteins of human foamy virus reveals novel features of the viral protease. J. Virol. 1998, 72, 7648–7652. [Google Scholar] [PubMed]
- Flügel, R.M.; Pfrepper, K.I. Proteolytic processing of foamy virus Gag and Pol proteins. Curr. Top. Microbiol. Immunol. 2003, 277, 63–88. [Google Scholar] [PubMed]
- Schneider, A.; Peter, D.; Schmitt, J.; Leo, B.; Richter, F.; Rösch, P.; Wöhrl, B.M.; Hartl, M.J. Structural requirements for enzymatic activities of foamy virus protease-reverse transcriptase. Proteins 2014, 82, 375–385. [Google Scholar] [CrossRef] [PubMed]
- Hare, S.; Gupta, S.S.; Valkov, E.; Engelman, A.; Cherepanov, P. Retroviral intasome assembly and inhibition of DNA strand transfer. Nature 2010, 464, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Hartl, M.J.; Schweimer, K.; Reger, M.H.; Schwarzinger, S.; Bodem, J.; Rösch, P.; Wöhrl, B.M. Formation of transient dimers by a retroviral protease. Biochem. J. 2010, 427, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.G.; Roy, J.; Jackson, D.; Clark, P.; Boyer, P.L.; Hughes, S.H.; Linial, M.L. Foamy retrovirus integrase contains a Pol dimerization domain required for protease activation. J. Virol. 2011, 85, 1655–1661. [Google Scholar] [CrossRef] [PubMed]
- Boyer, P.L.; Stenbak, C.R.; Clark, P.K.; Linial, M.L.; Hughes, S.H. Characterization of the polymerase and RNase H activities of human foamy virus reverse transcriptase. J. Virol. 2004, 78, 6112–6121. [Google Scholar] [CrossRef]
- Hartl, M.J.; Kretzschmar, B.; Frohn, A.; Nowrouzi, A.; Rethwilm, A.; Wöhrl, B.M. AZT resistance of simian foamy virus reverse transcriptase is based on the excision of AZTMP in the presence of ATP. Nucleic Acids Res. 2008, 36, 1009–1016. [Google Scholar]
- Yu, S.F.; Lujan, P.; Jackson, D.L.; Emerman, M.; Linial, M.L. The DEAD-box RNA helicase DDX6 is required for efficient encapsidation of a retroviral genome. PLoS Pathog. 2011, 7, e1002303. [Google Scholar]
- Johnson, S.F.; Telesnitsky, A. Retroviral RNA dimerization and packaging: the what, how, when, where, and why. PLoS Pathog. 2010, 6, e1001007. [Google Scholar]
- Jouvenet, N.; Laine, S.; Pessel-Vivares, L.; Mougel, M. Cell biology of retroviral RNA packaging. RNA Biol. 2011, 8, 572–580. [Google Scholar]
- Kuzembayeva, M.; Dilley, K.; Sardo, L.; Hu, W.S. Life of psi: how full-length HIV-1 RNAs become packaged genomes in the viral particles. Virology 2014, 454–455, 362–370. [Google Scholar] [PubMed]
- Lu, K.; Heng, X.; Summers, M.F. Structural determinants and mechanism of HIV-1 genome packaging. J. Mol. Biol. 2011, 410, 609–633. [Google Scholar] [CrossRef] [PubMed]




© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hamann, M.V.; Lindemann, D. Foamy Virus Protein—Nucleic Acid Interactions during Particle Morphogenesis. Viruses 2016, 8, 243. https://0-doi-org.brum.beds.ac.uk/10.3390/v8090243
Hamann MV, Lindemann D. Foamy Virus Protein—Nucleic Acid Interactions during Particle Morphogenesis. Viruses. 2016; 8(9):243. https://0-doi-org.brum.beds.ac.uk/10.3390/v8090243
Chicago/Turabian StyleHamann, Martin V., and Dirk Lindemann. 2016. "Foamy Virus Protein—Nucleic Acid Interactions during Particle Morphogenesis" Viruses 8, no. 9: 243. https://0-doi-org.brum.beds.ac.uk/10.3390/v8090243