
Influenza A Virus Hemagglutinin is Required for the Assembly of Viral Components Including Bundled vRNPs at the Lipid Raft

Abstract
:1. Introduction
2. Materials and Methods
2.1. Vectors and Antibodies
2.2. Cells and Viruses
2.3. Virus Concentration
2.4. Western Blotting
2.5. Primer Extension Assay
2.6. qPCR Assay
2.7. Indirect Immunofluorescence
2.8. Electron Microscopy
2.9. Lipid Raft Fractionation
3. Results
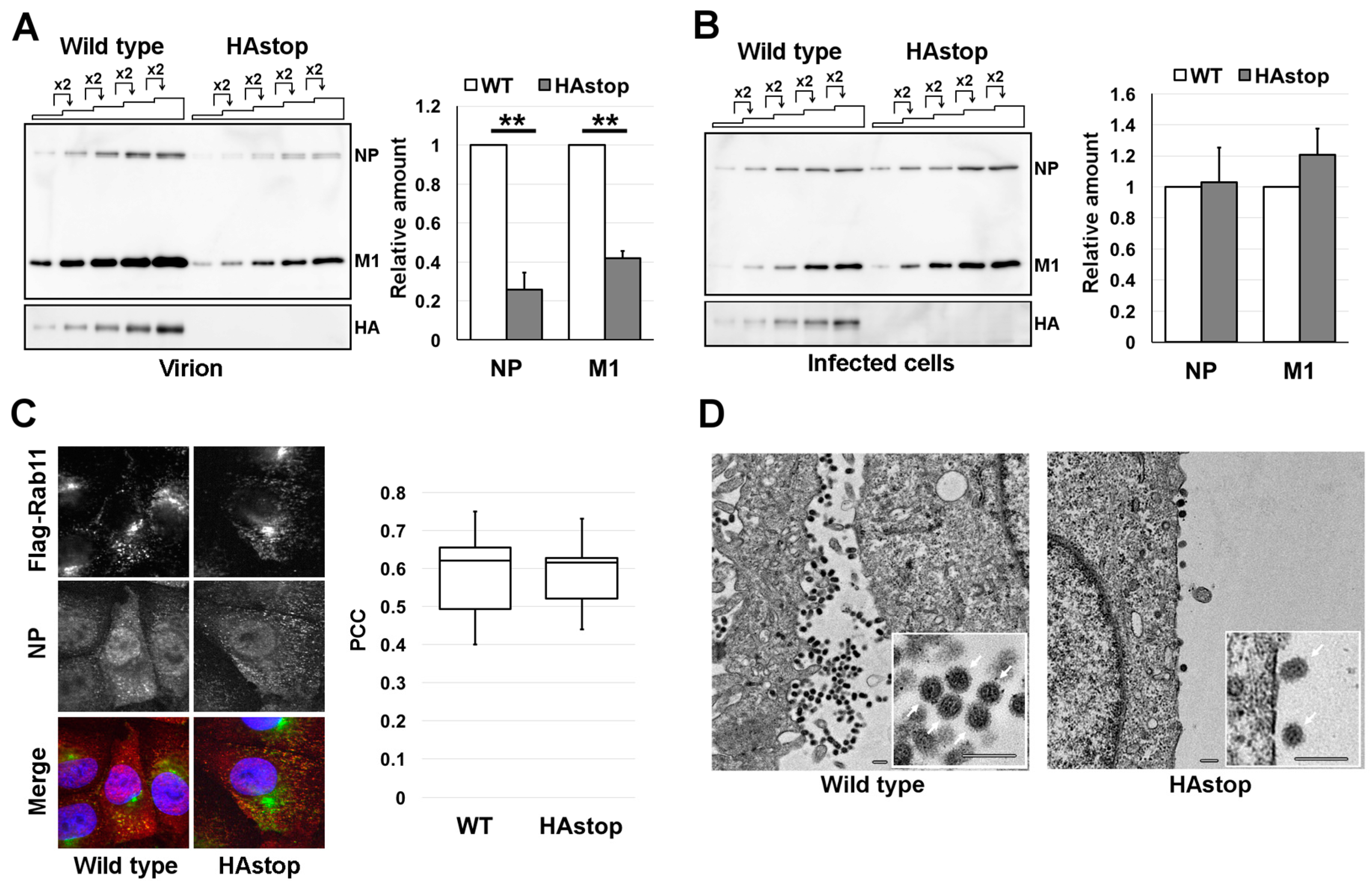
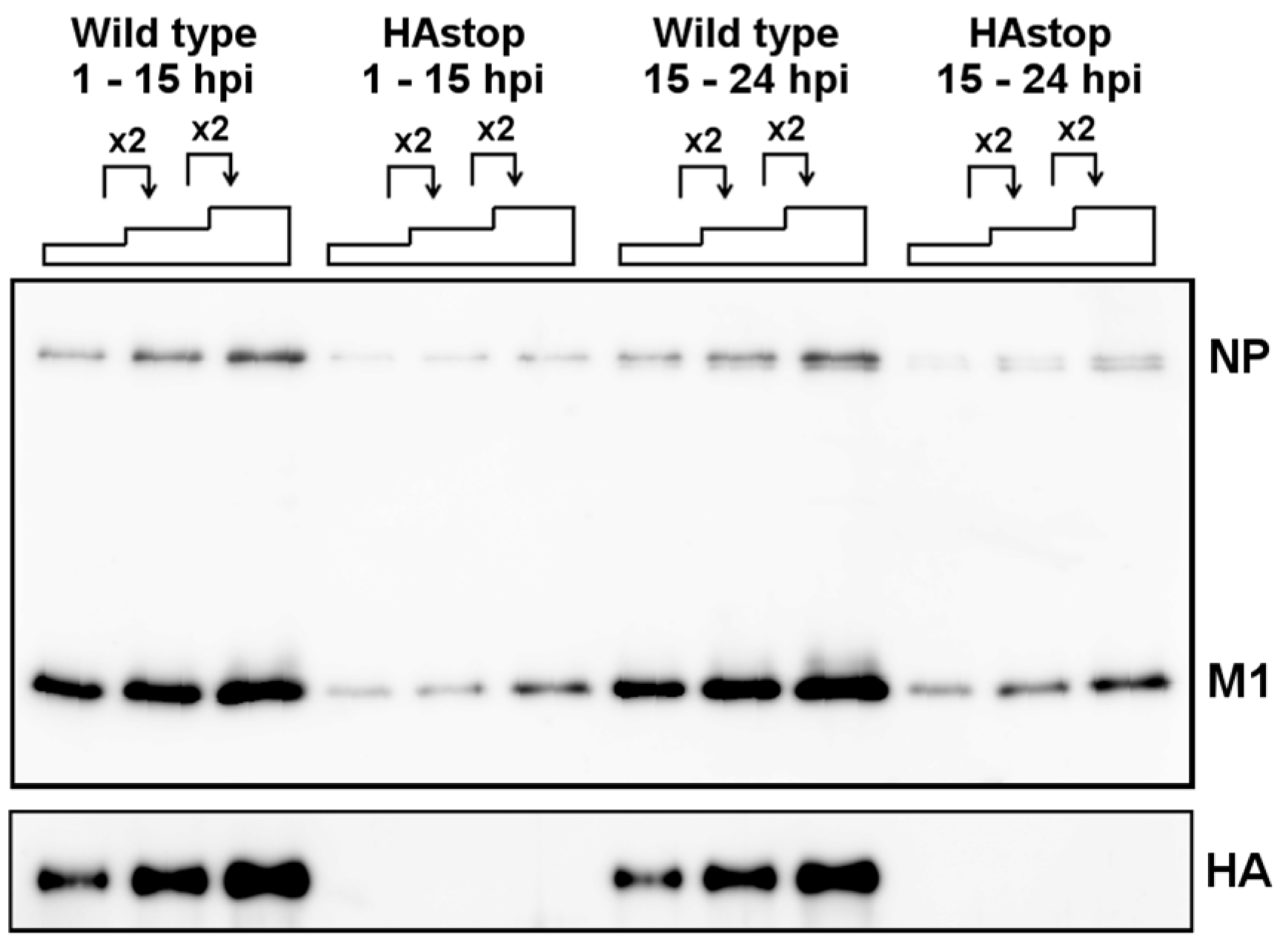
3.1. HA Is Necessary for Efficient Virion Production
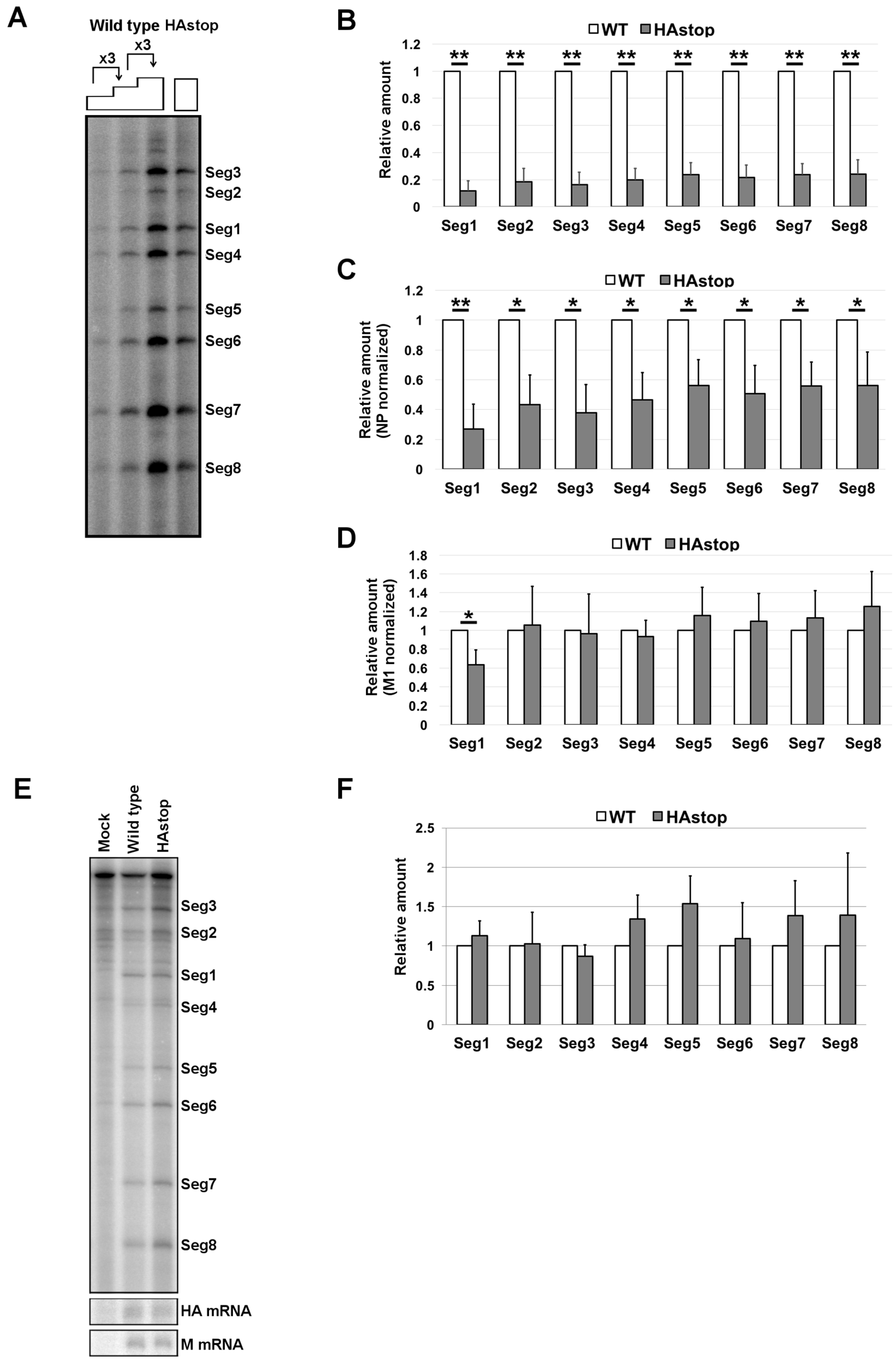
3.2. HA Is Required for Efficient vRNA Packaging into Progeny Virions
3.3. The Bundling of vRNP Segments Is Not Impaired in Cells Infected with the HAstop Virus
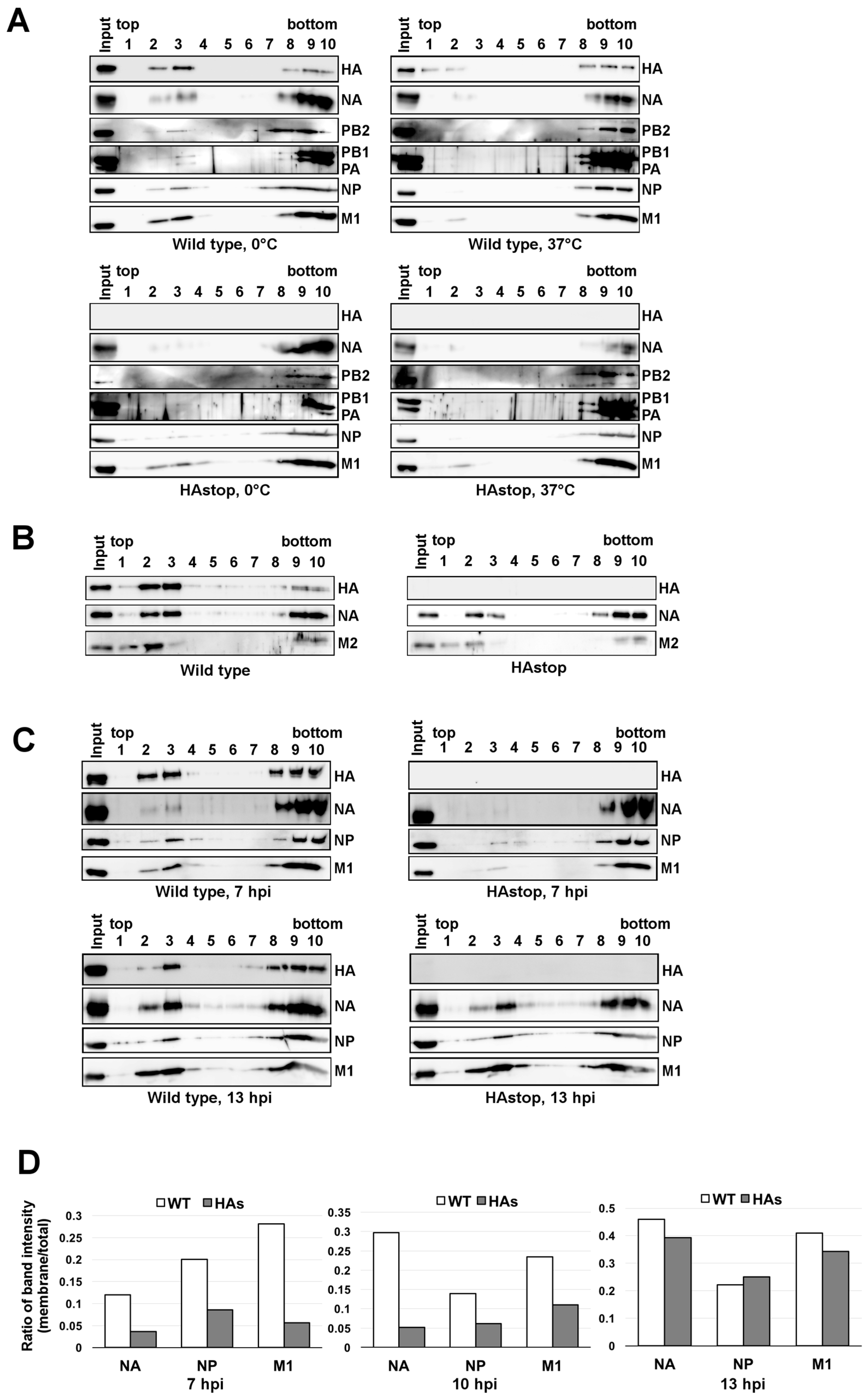
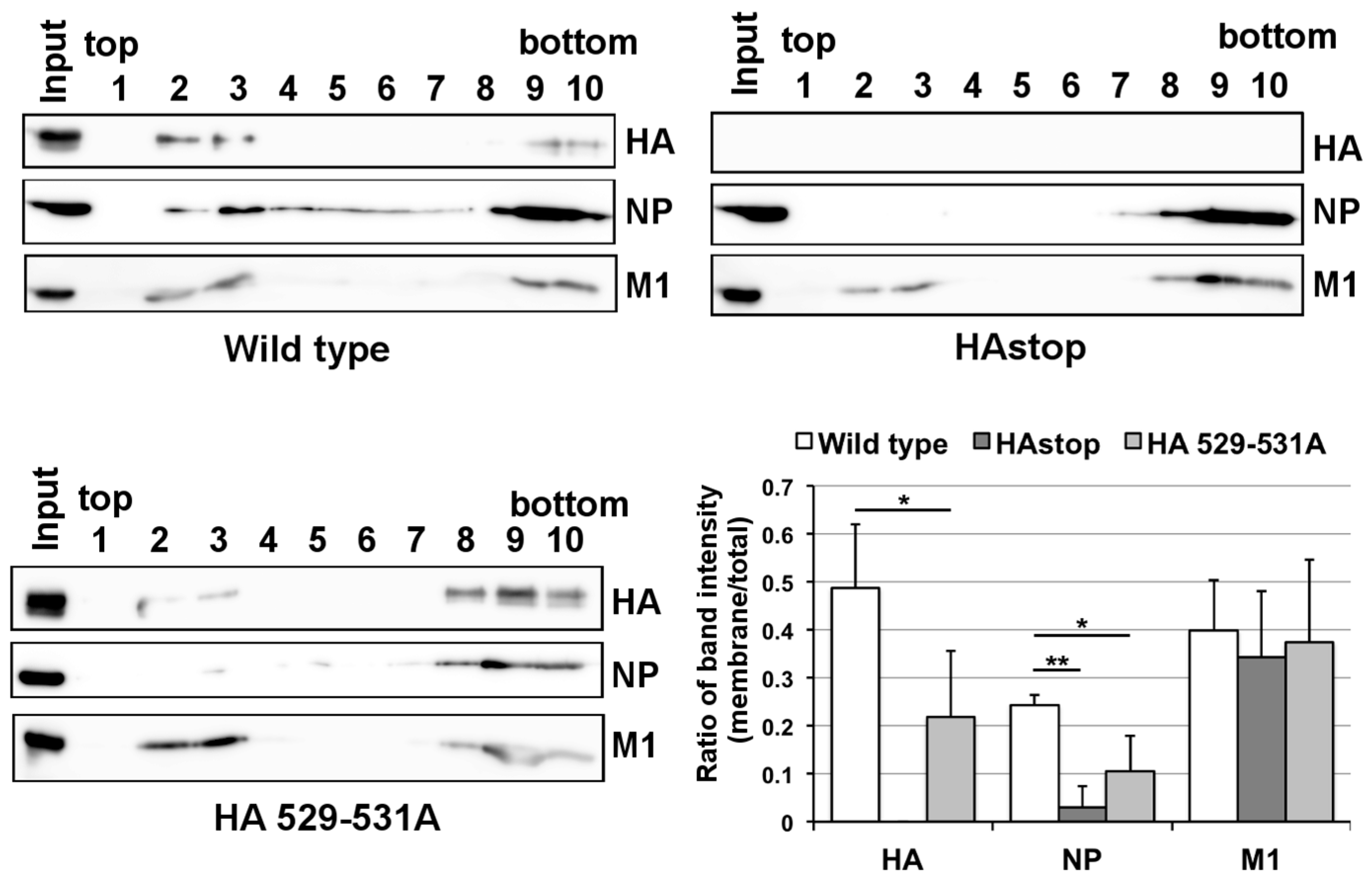
3.4. The Lipid Raft Association of NP Is Reduced in Cells Infected with the HAstop Virus
4. Discussion
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Watanabe, K.; Takizawa, N.; Katoh, M.; Hoshida, K.; Kobayashi, N.; Nagata, K. Inhibition of nuclear export of ribonucleoprotein complexes of influenza virus by leptomycin B. Virus Res. 2001, 77, 31–42. [Google Scholar] [CrossRef]
- O’Neill, R.E.; Talon, J.; Palese, P. The influenza virus NEP (NS2 protein) mediates the nuclear export of viral ribonucleoproteins. EMBO J. 1998, 17, 288–296. [Google Scholar] [CrossRef] [PubMed]
- Ma, K.; Roy, A.M.; Whittaker, G.R. Nuclear export of influenza virus ribonucleoproteins: Identification of an export intermediate at the nuclear periphery. Virology 2001, 282, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Eisfeld, A.J.; Kawakami, E.; Watanabe, T.; Neumann, G.; Kawaoka, Y. RAB11A is essential for transport of the influenza virus genome to the plasma membrane. J. Virol. 2011, 85, 6117–6126. [Google Scholar] [CrossRef] [PubMed]
- Momose, F.; Sekimoto, T.; Ohkura, T.; Jo, S.; Kawaguchi, A.; Nagata, K.; Morikawa, Y. Apical Transport of Influenza A Virus Ribonucleoprotein Requires Rab11-positive Recycling Endosome. PLoS ONE 2011, 6, e21123. [Google Scholar] [CrossRef] [PubMed]
- Amorim, M.J.; Bruce, E.A.; Read, E.K.C.; Foeglein, A.; Mahen, R.; Stuart, A.D.; Digard, P. A Rab11- and Microtubule-Dependent Mechanism for Cytoplasmic Transport of Influenza A Virus Viral RNA. J. Virol. 2011, 85, 4143–4156. [Google Scholar] [CrossRef] [PubMed]
- Takizawa, N.; Kumakura, M.; Takeuchi, K.; Kobayashi, N.; Nagata, K. Sorting of influenza A virus RNA genome segments after nuclear export. Virology 2010, 401, 248–256. [Google Scholar] [CrossRef] [PubMed]
- Lakdawala, S.S.; Wu, Y.; Wawrzusin, P.; Kabat, J.; Broadbent, A.J.; Lamirande, E.W.; Fodor, E.; Altan-Bonnet, N.; Shroff, H.; Subbarao, K. Influenza A Virus Assembly Intermediates Fuse in the Cytoplasm. PLoS Pathog. 2014, 10, e1003971. [Google Scholar] [CrossRef] [PubMed]
- Chou, Y.Y.; Heaton, N.S.; Gao, Q.; Palese, P.; Singer, R.; Lionnet, T. Colocalization of Different Influenza Viral RNA Segments in the Cytoplasm before Viral Budding as Shown by Single-molecule Sensitivity FISH Analysis. PLoS Pathog. 2013, 9, e1003358. [Google Scholar] [CrossRef]
- Chou, Y.Y.; Vafabakhsh, R.; Doganay, S.; Gao, Q.; Ha, T.; Palese, P. One influenza virus particle packages eight unique viral RNAs as shown by FISH analysis. Proc. Natl. Acad. Sci. USA 2012, 109, 9101–9106. [Google Scholar] [CrossRef] [PubMed]
- Noda, T.; Sagara, H.; Yen, A.; Takada, A.; Kida, H.; Cheng, R.H.; Kawaoka, Y. Architecture of ribonucleoprotein complexes in influenza A virus particles. Nature 2006, 439, 490–492. [Google Scholar] [CrossRef] [PubMed]
- Gerber, M.; Isel, C.; Moules, V.; Marquet, R. Selective packaging of the influenza A genome and consequences for genetic reassortment. Trends Microbiol. 2014, 22, 446–455. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.J.; Takeda, M.; Lamb, R.A. Influenza virus hemagglutinin (H3 subtype) requires palmitoylation of its cytoplasmic tail for assembly: M1 proteins of two subtypes differ in their ability to support assembly. J. Virol. 2005, 79, 13673–13684. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.J.; Leser, G.P.; Morita, E.; Lamb, R.A. Influenza virus hemagglutinin and neuraminidase, but not the matrix protein, are required for assembly and budding of plasmid-derived virus-like particles. J. Virol. 2007, 81, 7111–7123. [Google Scholar] [CrossRef] [PubMed]
- Leser, G.P.; Lamb, R.A. Influenza virus assembly and budding in raft-derived microdomains: A quantitative analysis of the surface distribution of HA, NA and M2 proteins. Virology 2005, 342, 215–227. [Google Scholar] [CrossRef] [PubMed]
- Takeda, M.; Leser, G.P.; Russell, C.J.; Lamb, R.A. Influenza virus hemagglutinin concentrates in lipid raft microdomains for efficient viral fusion. Proc. Natl. Acad. Sci. USA 2003, 100, 14610–14617. [Google Scholar] [CrossRef] [PubMed]
- Ohkura, T.; Momose, F.; Ichikawa, R.; Takeuchi, K.; Morikawa, Y. Influenza A virus hemagglutinin and neuraminidase mutually accelerate their apical targeting through clustering of lipid rafts. J. Virol. 2014, 88, 10039–10055. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Harmon, A.; Jin, J.; Francis, D.H.; Christopher-Hennings, J.; Nelson, E.; Montelaro, R.C.; Li, F. The lack of an inherent membrane targeting signal is responsible for the failure of the matrix (M1) protein of influenza A virus to bud into virus-like particles. J. Virol. 2010, 84, 4673–4681. [Google Scholar] [CrossRef] [PubMed]
- Lai, J.C.C.; Chan, W.W.L.; Kien, F.; Nicholls, J.M.; Peiris, J.S.M.; Garcia, J.-M. Formation of virus-like particles from human cell lines exclusively expressing influenza neuraminidase. J. Gen. Virol. 2010, 91, 2322–2330. [Google Scholar] [CrossRef] [PubMed]
- Chlanda, P.; Schraidt, O.; Kummer, S.; Riches, J.; Oberwinkler, H.; Prinz, S.; Kräusslich, H.-G.; Briggs, J.A.G. Structural analysis of the roles of influenza A virus membrane-associated proteins in assembly and morphology. J. Virol. 2015, 89, 8957–8966. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.; Avalos, R.T.; Ponimaskin, E.; Nayak, D.P. Influenza virus assembly: Effect of influenza virus glycoproteins on the membrane association of M1 protein. J. Virol. 2000, 74, 8709–8719. [Google Scholar] [CrossRef] [PubMed]
- Rossman, J.S.; Jing, X.; Leser, G.P.; Balannik, V.; Pinto, L.H.; Lamb, R.A. Influenza virus m2 ion channel protein is necessary for filamentous virion formation. J. Virol. 2010, 84, 5078–5088. [Google Scholar] [CrossRef] [PubMed]
- McCown, M.F.; Pekosz, A. Distinct domains of the influenza a virus M2 protein cytoplasmic tail mediate binding to the M1 protein and facilitate infectious virus production. J. Virol. 2006, 80, 8178–8189. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.J.; Leser, G.P.; Jackson, D.; Lamb, R.A. The influenza virus M2 protein cytoplasmic tail interacts with the M1 protein and influences virus assembly at the site of virus budding. J. Virol. 2008, 82, 10059–10070. [Google Scholar] [CrossRef] [PubMed]
- Rossman, J.S.; Lamb, R.A. Influenza virus assembly and budding. Virology 2011, 411, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Leser, G.P.; Pekosz, A.; Lamb, R.A. The cytoplasmic tails of the influenza virus spike glycoproteins are required for normal genome packaging. Virology 2000, 269, 325–334. [Google Scholar] [CrossRef] [PubMed]
- McCown, M.; Pekosz, A. The Influenza A Virus M2 Cytoplasmic Tail Is Required for Infectious Virus Production and Efficient Genome Packaging. J. Virol. 2005, 79, 3595–3605. [Google Scholar] [CrossRef] [PubMed]
- Neumann, G.; Watanabe, T.; Ito, H.; Watanabe, S.; Goto, H.; Gao, P.; Hughes, M.; Perez, D.R.; Donis, R.; Hoffmann, E.; et al. Generation of influenza A viruses entirely from cloned cDNAs. Proc. Natl. Acad. Sci. USA 1999, 96, 9345–9350. [Google Scholar] [CrossRef] [PubMed]
- Goto, H.; Kawaoka, Y. A novel mechanism for the acquisition of virulence by a human influenza A virus. Proc. Natl. Acad. Sci. USA 1998, 95, 10224–10228. [Google Scholar] [CrossRef] [PubMed]
- Goto, H.; Wells, K.; Takada, A.; Kawaoka, Y. Plasminogen-Binding Activity of Neuraminidase Determines the Pathogenicity of Influenza A Virus. J. Virol. 2001, 75, 9297–9301. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, A.; Momose, F.; Nagata, K. Replication-coupled and host factor-mediated encapsidation of the influenza virus genome by viral nucleoprotein. J. Virol. 2011, 85, 6197–6204. [Google Scholar] [CrossRef] [PubMed]
- Naito, T.; Momose, F.; Kawaguchi, A.; Nagata, K. Involvement of Hsp90 in assembly and nuclear import of influenza virus RNA polymerase subunits. J. Virol. 2007, 81, 1339–1349. [Google Scholar] [CrossRef] [PubMed]
- Takizawa, N.; Watanabe, K.; Nouno, K.; Kobayashi, N.; Nagata, K. Association of functional influenza viral proteins and RNAs with nuclear chromatin and sub-chromatin structure. Microbes Infect. 2006, 8, 823–833. [Google Scholar] [CrossRef] [PubMed]
- Momose, F.; Kikuchi, Y.; Komase, K.; Morikawa, Y. Visualization of microtubule-mediated transport of influenza viral progeny ribonucleoprotein. Microbes Infect. 2007, 9, 1422–1433. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Carrasco, M.; Amorim, M.J.; Digard, P. Lipid Raft-Dependent Targeting of the Influenza A Virus Nucleoprotein to the Apical Plasma Membrane. Traffic 2004, 5, 979–992. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Pekosz, A.; Lamb, R.A. Influenza virus assembly and lipid raft microdomains: A role for the cytoplasmic tails of the spike glycoproteins. J. Virol. 2000, 74, 4634–4644. [Google Scholar] [CrossRef] [PubMed]
- Harris, A.; Cardone, G. Influenza virus pleiomorphy characterized by cryoelectron tomography. Proc. Natl. Acad. Sci. USA 2006, 103, 19123–19127. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Leser, G.P.; Lamb, R.A. The influenza virus hemagglutinin cytoplasmic tail is not essential for virus assembly or infectivity. EMBO J. 1994, 13, 5504–5515. [Google Scholar] [PubMed]
- Hutchinson, E.C.; Curran, M.D.; Read, E.K.; Gog, J.R.; Digard, P. Mutational analysis of cis-acting RNA signals in segment 7 of influenza A virus. J. Virol. 2008, 82, 11869–11879. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Hong, Y.; Parslow, T. cis-Acting packaging signals in the influenza virus PB1, PB2, and PA genomic RNA segments. J. Virol. 2005, 79, 10348–10355. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Correlating Protein Expression (%) | |||||
|---|---|---|---|---|---|
| Virus | HA | NP | NA | M1 | |
| NP | WT | 74.6 ± 8.35 | |||
| HA-low a | n.t. | ||||
| NA | WT | 89.5 ± 8.87 | 85.0 ± 4.50 | ||
| HA-low | n.t. | 84.8 ± 5.65 | |||
| M1 | WT | 83.8 ± 7.13 | 73.1 ± 9.83 | 86.1 ± 6.84 | |
| HA-low | n.t. | 76.7 ± 7.21 | 87.4 ± 4.20 | ||
| NS1 | WT | n.t. | 78.6 ± 9.61 | 88.1 ± 6.46 | 91.8 ± 5.43 |
| HA-low | n.t. | 80.3 ± 7.00 | 88.9 ± 3.46 | 93.2 ± 3.11 | |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Takizawa, N.; Momose, F.; Morikawa, Y.; Nomoto, A. Influenza A Virus Hemagglutinin is Required for the Assembly of Viral Components Including Bundled vRNPs at the Lipid Raft. Viruses 2016, 8, 249. https://0-doi-org.brum.beds.ac.uk/10.3390/v8090249
Takizawa N, Momose F, Morikawa Y, Nomoto A. Influenza A Virus Hemagglutinin is Required for the Assembly of Viral Components Including Bundled vRNPs at the Lipid Raft. Viruses. 2016; 8(9):249. https://0-doi-org.brum.beds.ac.uk/10.3390/v8090249
Chicago/Turabian StyleTakizawa, Naoki, Fumitaka Momose, Yuko Morikawa, and Akio Nomoto. 2016. "Influenza A Virus Hemagglutinin is Required for the Assembly of Viral Components Including Bundled vRNPs at the Lipid Raft" Viruses 8, no. 9: 249. https://0-doi-org.brum.beds.ac.uk/10.3390/v8090249