
Rapid Construction of Complex Plant RNA Virus Infectious cDNA Clones for Agroinfection Using a Yeast-E. coli-Agrobacterium Shuttle Vector


Abstract
:1. Introduction
2. Materials and Methods
2.1. Virus and Viral RNA
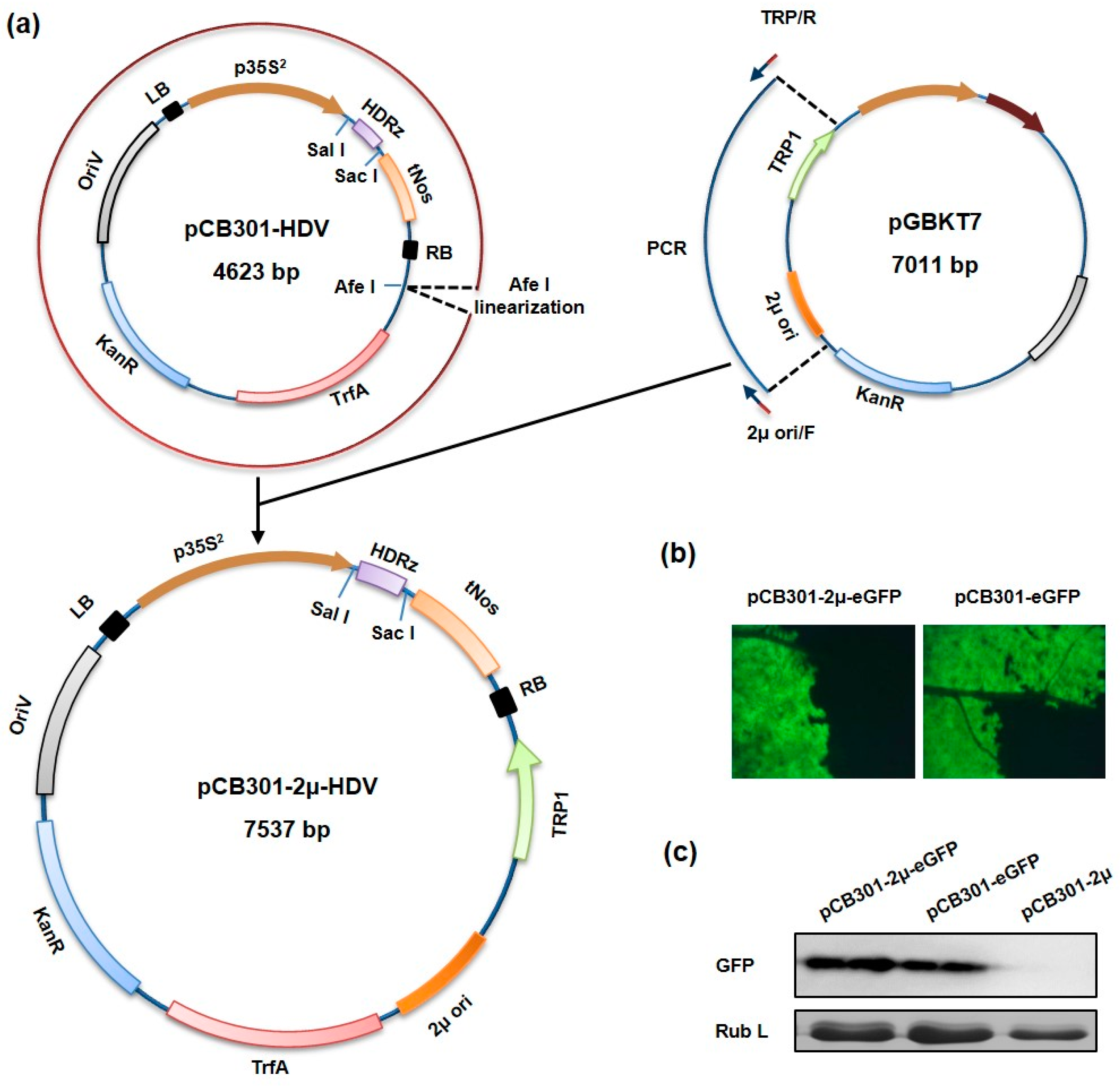
2.2. Construction of Yeast-E. Coli-Agrobacterium Shuttle Vector
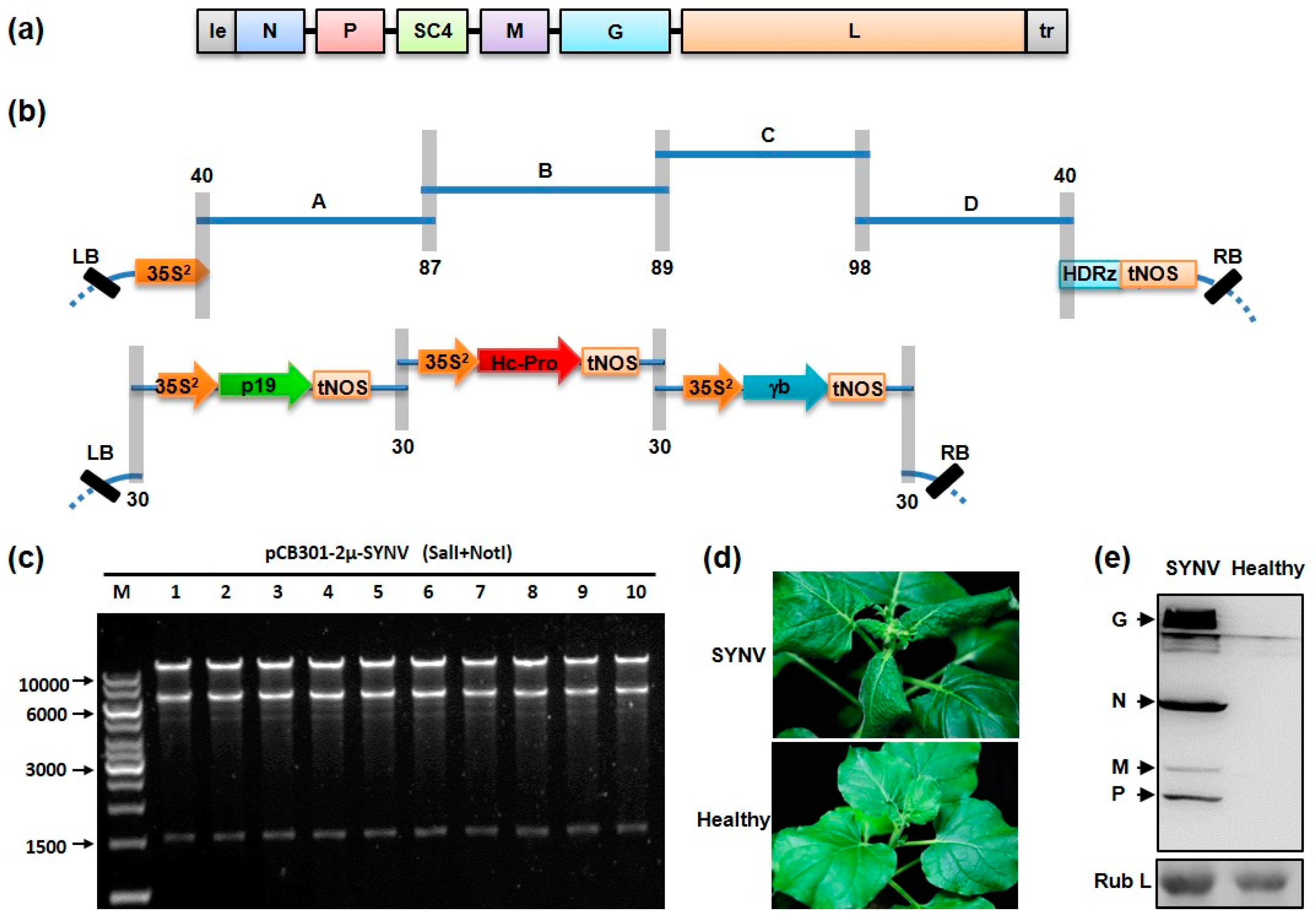
2.3. Assembly of SYNV and PVY cDNA Clones by Yeast Homologous Recombination
2.4. Yeast Plasmid Purification
2.5. Agrobacterium Infiltration
2.6. Immunoblotting
3. Results
3.1. Construction of a Yeast-E. coli-Agrobacterium Shuttle Vector
3.2. Assembly of SYNV cDNA Clones by Yeast Homologous Recombination
3.3. One-Step Construction of Intron-Less and Intron-Containing PVY Infectious cDNA Clones
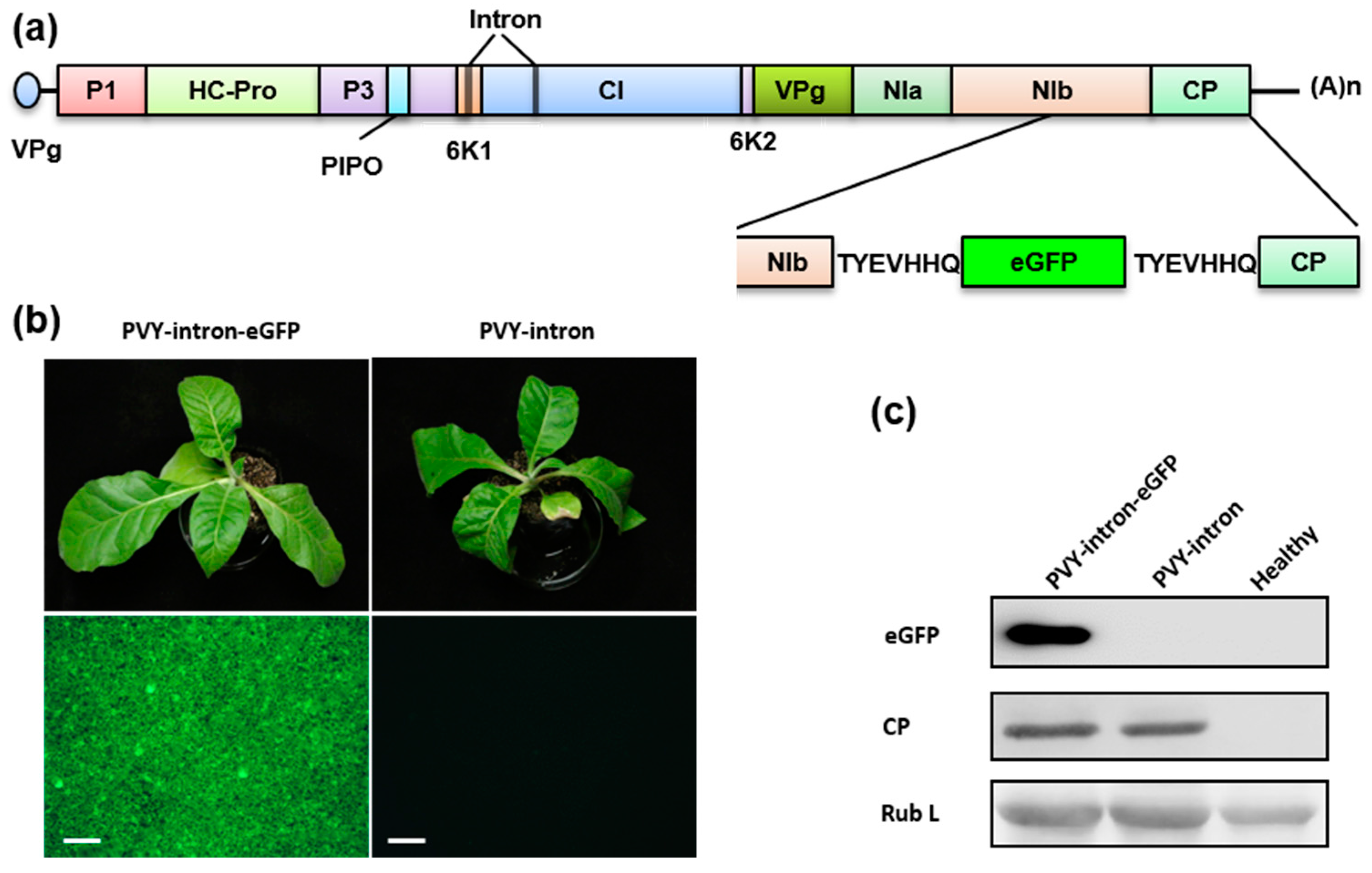
3.4. Construction of an eGFP-Tagged PVY Infectious Clones
4. Discussion
Acknowledgments
Author Contributions
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer | Sequence 1 |
|---|---|
| 2μ ori/F | agatctcagtaaagcGAAAAGTGCCACCTGAACGA |
| TRP/R | gggggttcagccagcAAGGATCTGGCGTAATAGCG |
| eGFP/SalI/F | GCGTCGACATGGTGAGCAAGGGCGAGGA |
| eGFP/SacI/R | GCGAGCTCTTACTTGTACAGCTCGTCCAT |
| Backbone/LB/R | AAAACCACCCCAGTACATTAAAAAC |
| Backbone/RB/F | GAATTCAGATTGTCGTTTCCCG |
| 35S-p19/F | CGGACGTTTTTAATGTACTGGGGTGGTTTTACGCGTCATGGTGGAGCACG |
| NOS-p19/R | GTCTAGAGAATCTCCTGCAATCATTCCGGAAACACTGATAGTTTAATTCCCGAT |
| 35S-HCPro/F | GGAGGGACTGTATTCATGTTAAATGATAAGACGCGTCATGGTGGAGCACG |
| NOS-HCPro/R | ACTGAAGGCGGGAAACGACAATCTGAATTCAACACTGATAGTTTAATTCCCGAT |
| 35S-γb/F | TCCGGAATGATTGCAGGAGATTCTCTAGACACGCGTCATGGTGGAGCACG |
| NOS-γb/R | CTTATCATTTAACATGAATACAGTCCCTCCAACACTGATAGTTTAATTCCCGAT |
| oligo(dT)17 | GACTCGAGTCGACATCGATTTTTTTTTTTTTTTTT |
| PVY backbone/R | GTATTGAGTTGTTTTAATTTCCTCTCCAAATGAAATGAACTTCCT |
| PVY backbone/F | GATGTCGACTCGAGTCGAATTTCCCCGATCGTTCAAACATTTG |
| PVY a/F | CATTTCATTTGGAGAGGAAATTAAAACAACTCAATACAACATAAG |
| PVY a/R | TATTGCCTGACACACTGCCA |
| PVY b/F | ATCTTTAGGCGTTTGCCAAC |
| PVY b/R | ACAGGGAAATCCTTTGGCATC |
| PVY c/F | TTTTACCGATCAAAGGCAGAGAC |
| PVY c/R | TTGAACGATCGGGGAAATTCGACTCGAGTCGACATCGATT |
| PVY/b2/F | TTTGTTGATATGCAGGTGTTAAAAATTTGGAACAAGTGGT |
| PVY/b1/R | GAAGCAGAAACTTACCTGGTGTGGAACGCTGGTGT |
| PVY/b2/R | TAAGTGCATACTTACCTCTCTTCCCACTGGAGTAGCT |
| PVY/b3/F | TTTGAAATTATGCAGGTTGAATTTACAACACAGCAACCA |
| Intron1/modified/R | CTGCATATCAACAAATTTTGGTCATATATTAGAAAAGTTATAAATTAAAAT |
| Intron1/F | AGCGTTCCACACCAGGTAAGTTTCTGCTTCTACCTTTGAT |
| Intron1/R | CCAAATTTTTAACACCTGCATATCAACAAATTTTGGTCAT |
| Intron2/F | CCAGTGGGAAGAGAGGTAAGTATGCACTTAAAGAGTATGTGT |
| Intron2/R | TGTTGTAAATTCAACCTGCATAATTTCAAAGATTGAACCTA |
| SYNV backbone/F | TGTTCTGAGCTTTTGTCTCTGGGTCGGCATGGCATCTCCA |
| SYNV backbone/R | TTTTCTGAGTTTCTGTCTCTCCTCTCCAAATGAAATGAACTTC |
| SYNV a/F | GTTCATTTCATTTGGAGAGGAGAGACAGAAACTCAGAAAATACAAT |
| SYNV a/R | CACATCCTCAAGAAACATGCT |
| SYNV b/F | CCATTTGCCATGATCAGACG |
| SYNV b/R | GATGGGTCGTTTGATCGATG |
| SYNV c/F | GCTTCTGAACGACATCTGAATC |
| SYNV c/R | CCGATAGATCTCGCAAATATTGAT |
| SYNV d/F | CAAAGCAGAGGCCGTAATGAG |
| SYNV d/R | TGGAGATGCCATGCCGACCCAGAGACAAAAGCTCAGAACAAT |
| PVY/NIb/BglII/20/F | tcacaacatttctc agatcTTGGTTTG |
| PVY/NIb/eGFP15/R | gcccttgctcaccatTGCTTGATGGTGTACTTCATAAGTATCGC |
| PVY/CP/eGFP15/F | gacgagctgtacaagACTTATGAAGTACACCATCAAGCAAAT |
| PCB/BglII/15/R | cgctttactg agatcTCCTGTG |
| eGFP/F | ATGGTGAGCAAGGGCGAGGA |
| eGFP/R | TTACTTGTACAGCTCGTCCATGCCGA |
References
- Ahlquist, P.; French, R.; Janda, M.; Loesch-Fries, L.S. Multicomponent RNA plant virus infection derived from cloned viral cDNA. Proc. Natl. Acad. Sci. USA 1984, 81, 7066–7070. [Google Scholar] [CrossRef] [PubMed]
- Boyer, J.-C.; Haenni, A.-L. Infectious Transcripts and cDNA Clones of RNA Viruses. Virology 1994, 198, 415–426. [Google Scholar] [CrossRef] [PubMed]
- Leiser, R.M.; Ziegler-Graff, V.; Reutenauer, A.; Herrbach, E.; Lemaire, O.; Guilley, H.; Richards, K.; Jonard, G. Agroinfection as an alternative to insects for infecting plants with beet western yellows luteovirus. Proc. Natl. Acad. Sci. USA 1992, 89, 9136–9140. [Google Scholar] [CrossRef] [PubMed]
- Jackson, A.O.; Li, Z. Developments in Plant Negative-Strand RNA Virus Reverse Genetics. Annu. Rev. Phytopathol. 2016, 54, 469–498. [Google Scholar] [CrossRef] [PubMed]
- Mori, M.; Mise, K.; Kobayashi, K.; Okuno, T.; Furusawa, I. Infectivity of plasmids containing brome mosaic virus cDNA linked to the cauliflower mosaic virus 35S RNA promoter. J. Gen. Virol. 1991, 72, 243–246. [Google Scholar] [CrossRef] [PubMed]
- Grimsley, N.; Hohn, T.; Davies, J.W.; Hohn, B. Agrobacterium-mediated delivery of infectious maize streak virus into maize plants. Nature 1987, 325, 177–179. [Google Scholar] [CrossRef]
- Grimsley, N.; Hohn, B.; Hohn, T.; Walden, R. “Agroinfection”, an alternative route for viral infection of plants by using the Ti plasmid. Proc. Natl. Acad. Sci. USA 1986, 83, 3282–3286. [Google Scholar] [CrossRef] [PubMed]
- Peyret, H.; Lomonossoff, G.P. When plant virology met Agrobacterium: The rise of the deconstructed clones. Plant Biotechnol. J. 2015, 13, 1121–1135. [Google Scholar] [CrossRef] [PubMed]
- Grimsley, N. Agroinfection. Methods Mol. Biol. 1995, 44, 325–342. [Google Scholar] [PubMed]
- Tuo, D.; Shen, W.; Yan, P.; Li, X.; Zhou, P. Rapid Construction of Stable Infectious Full-Length cDNA Clone of Papaya Leaf Distortion Mosaic Virus Using In-Fusion Cloning. Viruses 2015, 7, 6241–6250. [Google Scholar] [CrossRef] [PubMed]
- Jakab, G.; Droz, E.; Malno, P.; Brigneti, G.; Baulcombe, D. Infectious in vivo and in vitro transcripts from a full-length cDNA clone of PVY-N605, a Swiss necrotic isolate of potato virus Y. J. Gen. Virol. 1997, 78, 3141–3145. [Google Scholar] [CrossRef] [PubMed]
- Chikh Ali, M.; Said Omar, A.; Natsuaki, T. An infectious full-length cDNA clone of potato virus YNTN-NW, a recently reported strain of PVY that causes potato tuber necrotic ringspot disease. Arch. Virol. 2011, 156, 2039–2043. [Google Scholar] [CrossRef] [PubMed]
- Maiss, E.; Timpe, U.; Brisske-Rode, A.; Lesemann, D.-E.; Casper, R. Infectious in vivo transcripts of a plum pox potyvirus full-length cDNA clone containing the cauliflower mosaic virus 35S RNA promoter. J. Gen. Virol. 1992, 73, 709–713. [Google Scholar] [CrossRef] [PubMed]
- López-Moya, J.J.; García, J.A. Construction of a stable and highly infectious intron-containing cDNA clone of plum pox potyvirus and its use to infect plants by particle bombardment. Virus Res. 2000, 68, 99–107. [Google Scholar] [CrossRef]
- Yang, S.J.; Revers, F.; Souche, S.; Lot, H.; Le Gall, O.; Candresse, T.; Dunez, J. Construction of full-length cDNA clones of lettuce mosaic virus (LMV) and the effects of intron-insertion on their viability in Escherichia coli and on their infectivity to plants. Arch. Virol. 1998, 143, 2443–2451. [Google Scholar] [CrossRef] [PubMed]
- Johansen, I.E. Intron insertion facilitates amplification of cloned virus cDNA in Escherichia coli while biological activity is reestablished after transcription in vivo. Proc. Natl. Acad. Sci. USA 1996, 93, 12400–12405. [Google Scholar] [CrossRef] [PubMed]
- Bukovinszki, Á.; Götz, R.; Johansen, E.; Maiss, E.; Balázs, E. The role of the coat protein region in symptom formation on Physalis floridana varies between PVY strains. Virus Res. 2007, 127, 122–125. [Google Scholar] [CrossRef] [PubMed]
- Fakhfakh, H.; Vilaine, F.; Makni, M.; Robaglia, C. Cell-free cloning and biolistic inoculation of an infectious cDNA of potato virus Y. J. Gen. Virol. 1996, 77, 519–523. [Google Scholar] [CrossRef] [PubMed]
- Desbiez, C.; Chandeysson, C.; Lecoq, H.; Moury, B. A simple, rapid and efficient way to obtain infectious clones of potyviruses. J. Virol. Methods 2012, 183, 94–97. [Google Scholar] [CrossRef] [PubMed]
- Gao, R.; Tian, Y.-P.; Wang, J.; Yin, X.; Li, X.-D.; Valkonen, J.P.T. Construction of an infectious cDNA clone and gene expression vector of Tobacco vein banding mosaic virus (genus Potyvirus). Virus Res. 2012, 169, 276–281. [Google Scholar] [CrossRef] [PubMed]
- Moury, B.; Morel, C.; Johansen, E.; Guilbaud, L.; Souche, S.; Ayme, V.; Caranta, C.; Palloix, A.; Jacquemond, M. Mutations in Potato virus Y Genome-Linked Protein Determine Virulence Toward Recessive Resistances in Capsicum annuum and Lycopersicon hirsutum. Mol. Plant-Microbe Interact. 2004, 17, 322–329. [Google Scholar] [CrossRef] [PubMed]
- Olsen, B.S.; Johansen, I.E. Nucleotide sequence and infectious cDNA clone of the L1 isolate of Pea seed-borne mosaic potyvirus. Arch. Virol. 2001, 146, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Ma, X.; Qian, S.; Zhou, X.; Sun, K.; Chen, X.; Zhou, X.; Jackson, A.O.; Li, Z. Rescue of a Plant Negative-Strand RNA Virus from Cloned cDNA: Insights into Enveloped Plant Virus Movement and Morphogenesis. PLoS Pathog. 2015, 11, e1005223. [Google Scholar] [CrossRef] [PubMed]
- Ganesan, U.; Bragg, J.N.; Deng, M.; Marr, S.; Lee, M.Y.; Qian, S.; Shi, M.; Kappel, J.; Peters, C.; Lee, Y.; et al. Construction of a Sonchus Yellow Net Virus Minireplicon: A Step toward Reverse Genetic Analysis of Plant Negative-Strand RNA Viruses. J. Virol. 2013, 87, 10598–10611. [Google Scholar] [CrossRef] [PubMed]
- Qian, S.; Chen, X.; Sun, K.; Zhang, Y.; Li, Z. Capped antigenomic RNA transcript facilitates rescue of a plant rhabdovirus. Virol. J. 2017, 14, 113. [Google Scholar] [CrossRef] [PubMed]
- Larionov, V.; Kouprina, N.; Graves, J.; Chen, X.N.; Korenberg, J.R.; Resnick, M.A. Specific cloning of human DNA as yeast artificial chromosomes by transformation-associated recombination. Proc. Natl. Acad. Sci. USA 1996, 93, 491–496. [Google Scholar] [CrossRef] [PubMed]
- Kouprina, N.; Graves, J.; Cancilla, M.R.; Resnick, M.A.; Larionov, V. Specific isolation of human rDNA genes by TAR cloning. Gene 1997, 197, 269–276. [Google Scholar] [CrossRef]
- Oldenburg, K.R.; Vo, K.T.; Michaelis, S.; Paddon, C. Recombination-mediated PCR-directed plasmid construction in vivo in yeast. Nucleic Acids Res. 1997, 25, 451–452. [Google Scholar] [CrossRef] [PubMed]
- Mizutani, K. High-throughput plasmid construction using homologous recombination in yeast: Its mechanisms and application to protein production for X-ray crystallography. Biosci. Biotechnol. Biochem. 2015, 79, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Kouprina, N.; Larionov, V. Transformation-associated recombination (TAR) cloning for genomics studies and synthetic biology. Chromosoma 2016, 125, 621–632. [Google Scholar] [CrossRef] [PubMed]
- Noskov, V.N.; Chuang, R.-Y.; Gibson, D.G.; Leem, S.-H.; Larionov, V.; Kouprina, N. Isolation of circular yeast artificial chromosomes for synthetic biology and functional genomics studies. Nat. Protoc. 2010, 6, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Gibson, D.G.; Glass, J.I.; Lartigue, C.; Noskov, V.N.; Chuang, R.Y.; Algire, M.A.; Benders, G.A.; Montague, M.G.; Ma, L.; Moodie, M.M.; et al. Creation of a bacterial cell controlled by a chemically synthesized genome. Science 2010, 329, 52–56. [Google Scholar] [CrossRef] [PubMed]
- Youssef, F.; Marais, A.; Faure, C.; Gentit, P.; Candresse, T. Strategies to facilitate the development of uncloned or cloned infectious full-length viral cDNAs: Apple chlorotic leaf spot virus as a case study. Virol. J. 2011, 8, 488. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Delmond, I.; Pierrugues, O.; de Wispelaere, M.; Guilbaud, L.; Gaubert, S.; Divéki, Z.; Godon, C.; Tepfer, M.; Jacquemond, M. A novel strategy for creating recombinant infectious RNA virus genomes. J. Virol. Methods 2004, 121, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Klebe, R.J.; Harriss, J.V.; Sharp, Z.D.; Douglas, M.G. A general method for polyethylene-glycol-induced genetic transformation of bacteria and yeast. Gene 1983, 25, 333–341. [Google Scholar] [CrossRef]
- Vancanneyt, G.; Schmidt, R.; O’Connor-Sanchez, A.; Willmitzer, L.; Rocha-Sosa, M. Construction of an intron-containing marker gene: Splicing of the intron in transgenic plants and its use in monitoring early events in Agrobacterium-mediated plant transformation. MGG Mol. Gen. Genet. 1990, 220. [Google Scholar] [CrossRef]
- Singh, M.V.; Anthony Weil, P. A method for plasmid purification directly from yeast. Anal. Biochem. 2002, 307, 13–17. [Google Scholar] [CrossRef]
- Jackson, A.O.; Christie, S.R. Purification and some physicochemical properties of sonchus yellow net virus. Virology 1977, 77, 344–355. [Google Scholar] [CrossRef]
- Walpita, P.; Flick, R. Reverse genetics of negative-stranded RNA viruses: A global perspective. FEMS Microbiol. Lett. 2005, 244, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Boyer, J.C.; Haenni, A.L. Infectious transcripts and cDNA clones of RNA viruses. Virology 1994, 198, 415–426. [Google Scholar] [CrossRef] [PubMed]
- Bordat, A.; Houvenaghel, M.C.; German-Retana, S. Gibson assembly: An easy way to clone potyviral full-length infectious cDNA clones expressing an ectopic Vpg. Virol. J. 2015, 12, 89. [Google Scholar] [CrossRef] [PubMed]
- Tuo, D.; Fu, L.; Shen, W.; Li, X.; Zhou, P.; Yan, P. Generation of stable infectious clones of plant viruses by using Rhizobium radiobacter for both cloning and inoculation. Virology 2017, 510, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Blawid, R.; Nagata, T. Construction of an infectious clone of a plant RNA virus in a binary vector using one-step Gibson assembly. J. Virol. Methods 2015, 222, 11–15. [Google Scholar] [CrossRef] [PubMed]
- Polo, S.; Ketner, G.; Levis, R.; Falgout, B. Infectious RNA transcripts from full-length dengue virus type 2 cDNA clones made in yeast. J. Virol. 1997, 71, 5366–5374. [Google Scholar] [PubMed]
- Shang, Y.; Wang, M.; Xiao, G.; Wang, X.; Hou, D.; Pan, K.; Liu, S.; Li, J.; Wang, J.; Arif, B.M.; et al. Construction and rescue of a functional synthetic baculovirus. ACS Synth. Biol. 2017, 6, 1393–1402. [Google Scholar] [CrossRef] [PubMed]
- Kormelink, R.; Garcia, M.L.; Goodin, M.; Sasaya, T.; Haenni, A.L. Negative-strand RNA viruses: The plant-infecting counterparts. Virus Res. 2011, 162, 184–202. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Albina, E.; Gil, P.; Minet, C.; de Almeida, R.S. Two-plasmid system to increase the rescue efficiency of paramyxoviruses by reverse genetics: The example of rescuing Newcastle disease virus. Virology 2017, 509, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Almeida, R.S.; Gil, P.; Albina, E. Comparison of the efficiency of different Newcastle disease virus reverse genetics systems. J. Virol. Methods 2017, 249, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Kong, W.; Ashraf, S.; Curtiss, R., III. A one-plasmid system to generate influenza virus in cultured chicken cells for potential use in influenza vaccine. J. Virol. 2009, 83, 9296–9303. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Curtiss, R., III. Efficient generation of influenza virus with a mouse RNA polymerase I-driven all-in-one plasmid. Virol. J. 2015, 12, 95. [Google Scholar] [CrossRef] [PubMed]




© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, K.; Zhao, D.; Liu, Y.; Huang, C.; Zhang, W.; Li, Z. Rapid Construction of Complex Plant RNA Virus Infectious cDNA Clones for Agroinfection Using a Yeast-E. coli-Agrobacterium Shuttle Vector. Viruses 2017, 9, 332. https://0-doi-org.brum.beds.ac.uk/10.3390/v9110332
Sun K, Zhao D, Liu Y, Huang C, Zhang W, Li Z. Rapid Construction of Complex Plant RNA Virus Infectious cDNA Clones for Agroinfection Using a Yeast-E. coli-Agrobacterium Shuttle Vector. Viruses. 2017; 9(11):332. https://0-doi-org.brum.beds.ac.uk/10.3390/v9110332
Chicago/Turabian StyleSun, Kai, Danyang Zhao, Yong Liu, Changjun Huang, Wei Zhang, and Zhenghe Li. 2017. "Rapid Construction of Complex Plant RNA Virus Infectious cDNA Clones for Agroinfection Using a Yeast-E. coli-Agrobacterium Shuttle Vector" Viruses 9, no. 11: 332. https://0-doi-org.brum.beds.ac.uk/10.3390/v9110332