
Human Beta Defensin 2 Selectively Inhibits HIV-1 in Highly Permissive CCR6+CD4+ T Cells
by

Mark K. Lafferty
1,2, 
Lingling Sun
1,
Aaron Christensen-Quick
1,2,
Wuyuan Lu
1,3 and
Alfredo Garzino-Demo
1,2,4,* 1
Division of Basic Science, Institute of Human Virology, University of Maryland School of Medicine, Baltimore, MD 21201, USA
2
Department of Microbiology and Immunology, University of Maryland School of Medicine, Baltimore, MD 21201, USA
3
Department of Biochemistry, University of Maryland School of Medicine, Baltimore, MD 21201, USA
4
Department of Molecular Medicine, University of Padova, Padova 35121, Italy
*
Author to whom correspondence should be addressed.
Viruses 2017, 9(5), 111; https://0-doi-org.brum.beds.ac.uk/10.3390/v9050111
Submission received: 14 March 2017
/
Revised: 9 May 2017
/
Accepted: 10 May 2017
/
Published: 16 May 2017
(This article belongs to the Section Viral Immunology, Vaccines, and Antivirals)
Abstract
:Chemokine receptor type 6 (CCR6)+CD4+ T cells are preferentially infected and depleted during HIV disease progression, but are preserved in non-progressors. CCR6 is expressed on a heterogeneous population of memory CD4+ T cells that are critical to mucosal immunity. Preferential infection of these cells is associated, in part, with high surface expression of CCR5, CXCR4, and α4β7. In addition, CCR6+CD4+ T cells harbor elevated levels of integrated viral DNA and high levels of proliferation markers. We have previously shown that the CCR6 ligands MIP-3α and human beta defensins inhibit HIV replication. The inhibition required CCR6 and the induction of APOBEC3G. Here, we further characterize the induction of apolipoprotein B mRNA editing enzyme (APOBEC3G) by human beta defensin 2. Human beta defensin 2 rapidly induces transcriptional induction of APOBEC3G that involves extracellular signal-regulated kinases 1/2 (ERK1/2) activation and the transcription factors NFATc2, NFATc1, and IRF4. We demonstrate that human beta defensin 2 selectively protects primary CCR6+CD4+ T cells infected with HIV-1. The selective protection of CCR6+CD4+ T cell subsets may be critical in maintaining mucosal immune function and preventing disease progression.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
HIV preferentially infects and depletes chemokine receptor type 6 (CCR6)+CD4+ T cells despite suppressive antiretroviral therapy [1,2]. The chemokine receptor CCR6 is critical for mucosal immunity [3,4,5,6,7,8,9]. CCR6 is a marker of memory CD4+ T cells that express the HIV co-receptors, CCR5 and CXC chemokine receptors type 4 (CXCR4), and the integrin α4β7 [2,10]. In addition, CCR6 is a marker of Th17 cells which express CXCR4 and often express CCR5 [11,12,13]. Th17 cells and the CCR6+CCR4+CD4+ T cell subset, which are enriched for interleukin 17 (IL-17), produce lower levels of the CCR5 ligands CCL3, CCL4, and CCL5 [2,13]. The high proportion and increased expression of CCR5 and α4β7 enhances entry and HIV envelope binding to CCR6+CD4+ T cells [2,13,14]. Further, lower expression of CCR5 ligands by CCR6+CD4+ T cells reduces self-protection and increases susceptibility [14,15]. In addition to increased susceptibility, CCR6+CD4+ T cells are more permissive to HIV-1 [16]. Independent of entry, memory CCR6+CD4+ T cells have higher levels of replication when infected with single-cycle VSVG-GFP pseudotyped HIV which was further enhanced only in CCR6+ cells treated with retinoic acid [17]. CCR6+CD4+ T cells, upon HIV infection are prone to apoptosis, which may contribute to their preferential depletion [1,2,13,14,17]. However, some subsets of CCR6+ cells may contribute to the persistent HIV reservoir [18,19,20].
The gut associated lymphoid tissue (GALT) is enriched in CCR6+CD4+ T cells including Th17 cells which are preferentially depleted in HIV-1 infection and pathogenic simian immunodeficiency virus (SIV) infection of rhesus macaques, starting during acute infection [21,22,23,24]. The severe depletion of GALT Th17 cells is not restored despite uninterrupted and suppressive highly active antiretroviral therapy which results in defects in mucosal immunity and barrier function that can lead to impaired bacterial control and bacterial dissemination [25,26,27,28,29]. In contrast, gut Th17 cells in HIV-1-infected long-term non-progressors are preserved and gut Th17 cells in non-pathogenic SIV infection are depleted but subsequently restored, and these animals do not develop AIDS or chronic immune activation [21,28,30]. These findings suggest that preservation of mucosal Th17 cells is critical to controlling microbial infections and maintaining intestinal barrier function, which may prevent microbial translocation and immune activation [21,28,31].
IL-17 producing cells are an important component of mucosal immune defenses against extracellular and intracellular bacteria and fungi, many of which are opportunistic infections observed in AIDS patients [32,33,34,35]. IL-17 plays an essential role in epithelial and mucosal defenses through the induction of pro-inflammatory cytokines, chemokines, factors involved in wound repair and enterocyte homeostasis, and is a potent inducer of human beta defensin 2 (hBD2) [36,37,38,39,40,41,42,43]. In addition, Th17 cells produce IL-22, which stimulates the production of hBD2 by epithelial cells [44,45]. Defensins are expressed at mucosal sites and exhibit broad antimicrobial activity against Gram-positive and Gram-negative bacteria, mycobacteria, fungi, enveloped viruses, and non-enveloped viruses [46,47,48,49,50,51,52,53,54,55,56,57]. In addition to direct antimicrobial properties, beta defensins recruit innate and adaptive effector cells to sites of inflammation, induce cytokines and mast cell degranulation, and aid in wound healing [58,59,60,61,62,63,64,65,66,67,68]. We and others have previously shown reduced infectivity of HIV-1 virions treated with hBDs [69,70]. We have also reported that hBD2 inhibits HIV-1 post-entry requiring the upregulation of the host restriction factor apolipoprotein B mRNA editing enzyme (APOBEC3G) [71]. APOBEC3G inhibits HIV replication involving deamination and deamination independent mechanisms and is associated with HIV control in vivo [72,73,74,75,76,77,78]. The hBD2-mediated increase in APOBEC3G depended on the expression of CCR6, the receptor used by hBD2 to induce chemotaxis of memory T cells [58,71]. We sought to identify the signaling pathway(s) that resulted in the upregulation of APOBEC3G in CCR6+ cells given their importance to mucosal immunity. Here, we report that hBD2 induced APOBEC3G transcripts via extracellular signal-regulated kinases 1/2 (ERK1/2) activation in concomitance with increased binding of transcription factors NFATc2, NFATc1, and IRF-4 on the APOBEC3G promoter. Further, hBD2 selectively inhibited HIV-1 replication in CCR6+CD4+ T cells, suggesting CCR6 as a target for preventive and therapeutic approaches against HIV.
2. Materials and Methods
2.1 Isolation and Culture of Primary Cells and Cell Lines
Human peripheral blood mononuclear cells (PBMCs) were isolated from leukapheresis-processed blood (NY Blood Center, New York, NY, USA) using Lymphoprep™. CD4+ T cells (purity of >95% as determined by flow cytometry) were isolated from unstimulated PBMCs using the Human CD4+ T Cell Enrichment Kit (Stem Cell Technologies Inc., Vancouver, BC, Canada). CCR6+CD4+ T cells were isolated from unstimulated CD4+ T cells using a CCR6-APC antibody (BD Biosciences, San Jose, CA, USA) and BD FACSAria II (BD Biosciences) performed at the Institute of Human Virology Flow cytometry core. PBMCs, CD4+ T cells, CCR6+CD4+ T cells, and CCR6−CD4+ T cells were maintained in complete RPMI-1640 media. Cells were activated with 10 ng/mL IL-2 and 2.5 μg/mL phytohemagglutinin (PHA) or with 5μg/mL anti-CD3 coated plates and 1 μg/mLanti-CD28 (eBioscience, San Diego, CA, USA) for 48 h and then maintained in complete RPMI-1640 media and 10 ng/mL IL-2 at a density of 1 × 106 cells/mL. JKT-FT7 and JKT-FT7 CCR6 GFP cells, derivatives of the Jurkat CD4+ T lymphoblastoid cell line, were maintained in complete RPMI-1640 media. The JKT-FT7 and JKT-FT7 CCR6 GFP cell lines were a gift from Dr. Sam Hwang.
2.2. Flow Cytometry
Cells were surface stained with fluorochrome conjugated antibodies and isotype matched anti-IgG controls for 20 min. Stained cells were acquired on a FACSAria II (BD Biosciences) with a minimum of 10,000 gated events. Lymphocytes were gated based upon live/dead staining and forward and side scatter profiles. Gated populations were analyzed using FlowJo software (Tree Star, Inc., St. Ashland, OR, USA). FITC- or PE-anti-CD4, APC-or PE-anti-CCR6 were purchased from BD Biosciences.
2.3. Virus Production
HIV-1BaL (R5 tropic) was prepared from monocyte-derived macrophages in RPMI-1640 media/human AB serum. Pseudotyped virions were generated by calcium phosphate cotransfection of 293T cells with an HIV-1 reporter virus, pNL4-3-deltaE-enhanced green fluorescent protein (EGFP) (the following reagent was obtained through the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH: pNL4-3-deltaE-EGFP (Cat# 11,100) from Drs. Haili Zhang, Yan Zhou, and Robert Siliciano) and an amphotropic murine leukemia virus (AMLV) envelope expressing plasmid.
2.4. Infectivity Assays
Activated PBMC, CCR6+CD4+ T cells and CCR6−CD4+ T cells (105 cells/well) were infected for 2 h with 100 TCID50 of HIV-1BaL, or AMLV pNL4-3 ΔE-EGFP. After 2 h, cells were washed with phosphate-buffered saline (PBS, Arlington, TX, USA), and complete media was added with the appropriate treatment. Infection was monitored by p24 enzyme-linked immunoabsorbent assay (ELISA) with the use of a commercially available kit (PerkinElmer Life and Analytical Sciences, Boston, MA, USA) or by flow cytometry for GFP.
2.5. Immunoblotting
Cells were lysed with Radioimmunoprecipitation (RIPA) buffer (Sigma-Aldrich, Saint Louis, MO, USA) containing 1× ethylenediaminetetraacetic acid (EDTA)-free protease inhibitor cocktail (Calbiochem, Billerica, CA, USA), 0.1 mM phenylmethane sulfonyl fluoride (PMSF), and clarified by centrifugation. Total protein concentration was determined by detergent-compatible (DC) Protein Assay (Bio-Rad, Hercules, CA, USA), and equal amounts of total protein were subjected to sodium dodecyl sulfate–polyacrylamide gel electrophoresis analysis. The primary antibodies included APOBEC3G rabbit antisera (the following reagent was obtained through the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH: anti-ApoC17 from Dr. Klaus Strebel) and mouse anti–β-actin (Abcam, Cambridge, UK).
2.6. Quantitative RT-PCR
Activated CD4+ T cells, JKT-FT7, and JKT-FT7 CCR6 GFP cells (0.5 × 106 cells per timepoint) were untreated or pre-treated with actinomycin D (10 μg/mL) and subsequently incubated in the presence or absence of hBD2 (20 μg/mL). RNA was extracted with the use of the RNeasy Kit with On-Column DNase digestion (Qiagen, Hilden, Germany) at the indicated timepoints. First-strand cDNA was synthesized from 500 ng total RNA with the use of the iScript cDNA Synthesis Kit (Bio-Rad). cDNA was analyzed by real-time qPCR with iQSYBR green supermix (Bio-Rad) with the following primers: APOBEC3G forward 5′-CGCAGCCTGTGTCAGAAAAG-3′ and reverse 5′-CCAACAGTGCTGAAATTCGTCATA-3′, and 18S forward 5′-ATCAACTTTCGATGGTAGTCG-3′ and reverse: 5′-TCCTTGGATGTGGTAGCCG-3′. ΔΔCt method was used to calculate fold change between untreated and treated cells normalized to 18S ribosomal RNA [79].
2.7. Fast Protein Liquid Chromatography
Cells were lysed in ice cold lysis buffer (50 mM (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), pH 7.4, 125 mM NaCl, 0.2% NP-40, 0.1 mM PMSF and 1× EDTA-Free protease inhibitor cocktail (Calbiochem)) and clarified by centrifugation. Total protein concentration was determined by DC Protein Assay (Bio-Rad), and equal amounts of total protein were subjected to gel filtration using a Superose 6 10/300 GL gel filtration column and AKTA Explorer (GE Healthcare, Little Chalfont, UK). Molecular weight was determined using a Gel Filtration Markers Kit for gel filtration chromatography (Sigma-Aldrich). Twenty-four 1 mL fractions were collected and equal volumes were subjected to sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) analysis and probed with APOBEC3G rabbit antisera.
2.8. Chromatin Immunoprecipitation (ChIP) Assays
Chromatin was prepared according to the ChIP-IT Express Enzymatic protocol (Active Motif, Carlsbad, CA, USA). Immunoprecipitation was performed overnight using 2 μg of anti-NFATc1, anti-NFATc2, anti-IRF4 (Santa Cruz Biotechnology, Dallas, TX, USA), and anti-RNA Pol II and anti-IgG (Active Motif). Following incubation with antibody, DNA was eluted according to the Active Motif protocol and DNA was purified using the QIAquick PCR purification kit (Qiagen). Real-time qPCR was performed using iQSYBR green supermix (Bio-Rad) with the following primers: APOBEC3G ChIP-F 5′-GGG GAG GGG CTT GTG C-3′ and APOBEC3G ChIP-R 5′-AAG GCA ATT GCA AAG GGA A-3′. PCR was performed in triplicate with reaction conditions of 95 °C for 10 min followed by 40 cycles of 95 °C for 30 s, 60 °C for 1 min. Fold enrichment was calculated for each ChIP antibody used as quantity of ChIP DNA divided by amount of IgG DNA.
2.9. Total Chemical Synthesis of hBD2 and MIP-3α
hBD2 was chemically synthesized by solid-phase peptide synthesis with the use of a custom-modified procedure tailored from the previously published in situ neutralization protocol developed for Boc chemistry [59]. The β connectivity of three disulfide bonds (Cys1–Cys5, Cys2–Cys4, Cys3–Cys6) in highly pure synthetic hBD2 was independently verified by mass mapping of peptide fragments generated by enzymatic digestion and Edman degradation [59]. Correct folding of synthetic macrophage Inflammatory Protein-3 (MIP-3α) was demonstrated by the solution of its high-resolution x-ray crystal structure. Protein concentrations were determined by absorbance measurements at 280 nm with the use of molar extinction coefficients.
2.10. Statistical Analysis
All statistical analysis were performed using GraphPad Prism software version 5 (GraphPad Software, La Jolla, CA, USA). Statistical significance between paired samples (p values < 0.05 were considered significant) was calculated using a 2-tailed Student t test.
3. Results
3.1. CCR6+ Cells Are Protected by hBD2
CCR6+CD4+ T cells are preferentially infected by HIV-1 and depleted [1,2]. The frequency of CD4+ T cells that express CCR6 varies by subtype. CCR6 is prominently expressed on peripheral blood CD45RO+, CCR5+, and IL-17 producing CD4+ T cells [10,11,12]. PBMC and peripheral blood CD4+ T cells were isolated and infected, as described in Section 2 (Materials and Methods). Consistent with reported findings, CCR6+ PBMC and CCR6+CD4+ T cells have higher levels of viral replication compared to CCR6− cells when infected with single-cycle AMLV pseudotyped virions, which suggests the enhanced replication in CCR6+ cells is independent of co-receptor expression (Figure 1a). We previously reported that the CCR6 ligand hBD2 inhibits HIV-1 directly and by a post-entry mechanism during reverse transcription [70,71]. Using CCR6+ and CCR6− Jurkat-derived cell lines, we showed that the post-entry inhibition required the expression of CCR6 [71]. To evaluate the requirement of CCR6 for inhibition in primary cells, peripheral blood CD4+ T cells were isolated, separated into CCR6 positive and negative fractions, and activated with anti-CD3 and anti-CD28. Cells were treated with 20 μg/mL of hBD2 for 4 h and subsequently washed three times with PBS to remove the hBD2 and infected with HIV-1BaL. We observed inhibition in the CCR6+CD4+ T cells but not in the CCR6−CD4+ T cells (Figure 1b).
3.2. hBD2 Enhances LMM and HMM APOBEC3G Expression
Our previously published data showed that the post-entry inhibition occurred at an early stage of infection and required induction of the host restriction factor APOBEC3G [71]. We next investigated which form of APOBEC3G was present after treatment with hBD2. APOBEC3G exists in a range of molecular weight forms from a low-molecular-mass (LMM) form that restricts HIV-1 to a high molecular mass (HMM) form [80,81,82,83]. The form of APOBEC3G induced by hBD2 was determined using size exclusion chromatography. As expected, LMM APOBEC3G predominates in unstimulated CD4+ T cells while HMM APOBEC3G predominates in PHA stimulated CD4+ T cells [83,84]. Both the LMM and HMM forms of APOBEC3G exist in CD4+ T cells treated with hBD2 (20 μg/mL) for 8 h that were previously stimulated with PHA (Figure 2a). In JKT-FT7 CCR6 GFP cells, the predominant form was the HMM, but after treatment with hBD2 (20 μg/mL), APOBEC3G was detected only in the LMM form (Figure 2b).
3.3. Induction of APOBEC3G by hBD2 Requires ERK1/2 Phosphorylation
Mitogen induction of APOBEC3G expression requires ERK1/2 activation [85,86]. To assess whether hBD2 induction of APOBEC3G involves signaling through the mitogen-activated protein kinases (MAPK) pathway, we treated activated PBMC and CD4+ T cells, JKT-FT7 CCR6 GFP cells, and JKT-FT7 cells, which are CCR6−, with hBD2 (20 μg/mL) and measured phosphorylated and total ERK1/2 by Western blot. Treatment with hBD2 increased the amount of phosphorylated ERK1/2 in PBMC, CD4+ T cells, and the JKT-FT7 CCR6 GFP cells (Figure 3a). Treatment with MIP-3α, the cognate ligand for CCR6, activated ERK1/2 in PBMC, CD4+ T cells, and JKT-FT7 CCR6 GFP cells (Figure 3b) which further supports that signaling through CCR6 activates the ERK1/2 MAPK pathway. We next determined whether signaling through CCR6 activates the ERK1/2 pathway in primary CD4+ T cells. Both hBD2 and MIP3α increased the phosphorylation in primary CCR6+CD4+ T cells but not in the CCR6−CD4+ T cells (Figure 3c). We then tested whether the activation of ERK1/2 by hBD2 is required for APOBEC3G induction. CD4+ T cells were pre-treated with U0126, a specific inhibitor of MEK1/2 activation; the only known downstream substrate of phosphorylated MEK1/2 is ERK1/2 [87]. By comparing CD4+ T cells and JKT-FT7 CCR6 GFP cells (Figure 3a) with U0126 pre-treated cells, we find that pre-treatment with U0126 inhibited hBD2-mediated phosphorylation of ERK1/2 in both CD4+ T cells and JKT-FT7 CCR6 GFP cells (Figure 3d) and induction of APOBEC3G in activated CD4+ T cells (Figure 3e).
3.4. hBD2 Induces APOBEC3G Transcription
We have demonstrated that hBD2 increases APOBEC3G protein expression and requires phosphorylation of ERK1/2. We previously showed that the post-entry inhibition of HIV-1 by hBD2 required the induction of APOBEC3G and CCR6 expression [71]. To determine whether the increase in APOBEC3G by hBD2 is attributable to enhanced transcription, PHA activated primary CD4+ T cells were treated with hBD2 (20 µg/mL). hBD2 induced both a rapid transient increase in APOBEC3G mRNA as well as a 2-fold increase at 8 h (Figure 4a). To distinguish between transcription and RNA stability, cells were pre-treated with actinomycin D (ActD). Pre-treatment with ActD abrogated the increase in APOBEC3G mRNA at all timepoints (Figure 4a). These results suggest that hBD2 induces transcription of APOBEC3G mRNA. The post-entry inhibition and induction of APOBEC3G protein by hBD2 requires CCR6, so we next tested whether the same is true for the RNA expression. JKT-FT7 cells and JKT-FT7 CCR6 GFP cells were treated with hBD2 (20 μg/mL) and APOBEC3G mRNA levels were measured by qPCR. APOBEC3G mRNA expression was compared with untreated cells at matched timepoints and normalized to 18S ribosomal RNA levels. Treatment with hBD2 increased APOBEC3G mRNA in JKT-FT7 CCR6 GFP cells but not in the JKT-FT7 cells (Figure 4b). These results are consistent with our findings that APOBEC3G protein induction is restricted to CCR6+ cells [71]. Similar to our findings in primary cells, we observed a rapid but transient increase in APOBEC3G mRNA following treatment with hBD2 in JKT-FT7 CCR6 GFP cells and a 2-fold increase occurring at 8 h (Figure 4b).
3.5. NFAT and IRF-4 Binding after hBD2 Treatment
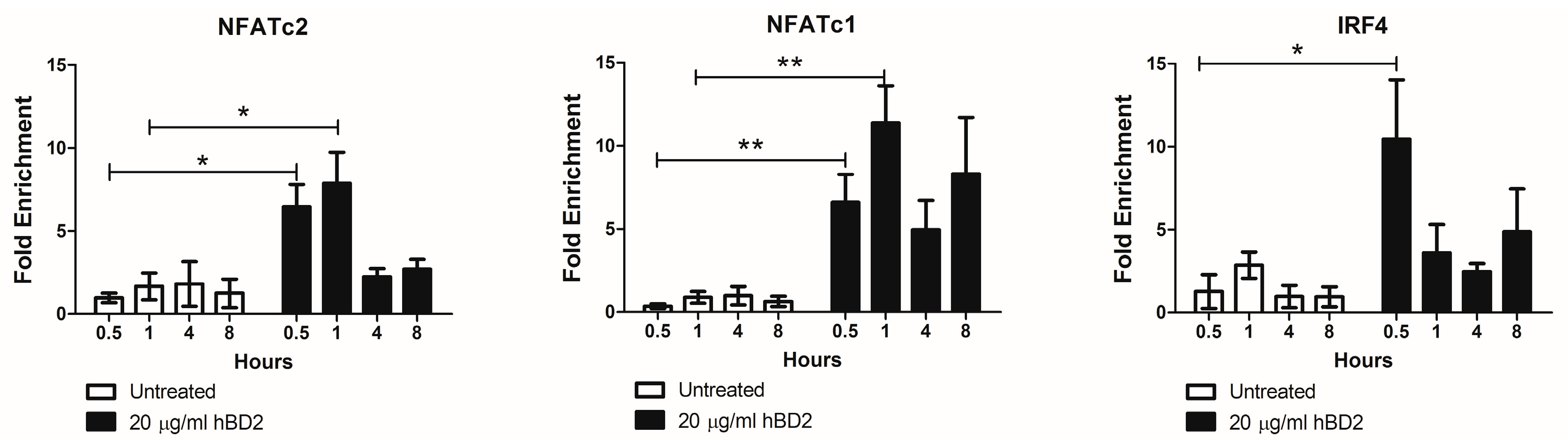
The transcription factors NFATc2, NFATc1, and IRF-4 regulate APOBEC3G in CD4+ T cells [88]. Therefore, we next determined by ChIP assay whether hBD2 (20 μg/mL) treatment induces NFATc2, NFATc1, and IRF-4 binding to the APOBEC3G promoter. We observed a 6.5-fold and 7.9-fold enrichment in NFATc2 and a 6.6 and 11.4-fold enrichment in NFATc1 bound to APOBEC3G promoter in hBD2 (20 μg/mL) treated JKT-FT7 CCR6 GFP cells at 30 and 60 min, respectively. hBD2 treatment also yielded a 9.5-fold enrichment of IRF-4 bound to APOBEC3G promoter at 30 min (Figure 5). Thus, the increased nuclear localization of NFAT and increased IRF-4 expression after hBD2 treatment induced transcription of APOBEC3G in JKT-FT7 CCR6 GFP cells.
4. Discussion
CCR6+CD4+ T cells are comprised of a heterogeneous population of cells including Th17 cells that contribute to mucosal immune defenses. CCR6 is a critical receptor in gut homeostasis that is involved in trafficking of immature DCs, B cells, and T cells to sites of inflammation and into inductive sites within the GALT. A lack of CCR6 or its ligands, MIP-3α or hBD2, is associated with immune dysfunction including reduced levels of intestinal B cells and regulatory T cells, diminished production of antigen specific IgA, and defects in T cell priming [4,5,6,7,8]. Structural defects are also associated with a lack of CCR6, MIP-3α, or hBD2 including smaller Peyer’s patches with fewer B cells and follicular domes, reduced numbers of M cells, and a block in the development of isolated lymphoid follicles [6,7,9]. Similar defects in mucosal lymphoid development are observed in the absence of MIP-3α or hBD2, suggesting non-redundant roles [3]. MIP-3α recruits immune cells to inductive sites via CCR6 and is expressed by follicle-associated epithelium and CD90+ Th17 cells [89,90]. Although MIP-3α inhibits HIV-1 replication in vitro, MIP-3α is strongly chemotactic for CCR6+ cells and therefore in vivo MIP-3α may contribute to the spread of infection by recruiting these highly susceptible cells to sites of infection [71,89]. In SIV-infected rhesus macaques, CCR6+CD4+ T cells are recruited from the periphery to the gut mucosa by MIP-3α. This disruption in CCR6+CD4+ T cell homeostasis may expand the number of highly susceptible viral targets in the gut mucosa [24]. Interestingly, glycerol monolaurate, an antimicrobial that inhibits production of proinflammatory cytokines and MIP-3α, protects rhesus macaques from mucosal SIV transmission [89]. In contrast, hBD2 may prevent the spread of infection, as it directly inhibits HIV and induces APOBEC3G, but is weakly chemotactic for CCR6+ cells compared with MIP-3α [70,71].
CCR6+CD4+ T cells and Th17 cells are redistributed from peripheral blood and preferentially depleted from the GALT, which correlates with disease progression [1,2,13,14,17,21,24,90]. The connections between CCR6, its ligands, and Th17 cells are multiple and highly relevant in the context of HIV infection. Th17 cells are an important component of mucosal immune defenses against extracellular and intracellular bacteria and fungi [32,34]. IL-17 limits microbes through the induction of pro-inflammatory cytokines, chemokines that recruit innate and adaptive immune cells, proteins involved in enterocyte homeostasis and antimicrobial peptides such as hBD2 [36,37,38,39,41,42,91,92]. Limiting microbial threats, maintenance, and re-establishment of the mucosal barrier are vital for intestinal homeostasis and prevention of microbial translocation. It has been shown that CCR6+CCR4+, CCR6+CXCR3+ and CCR6+CD90+ Th17 cell subsets are highly permissive to HIV-1 infection [2,90]. Th17 cells can also convert into follicular T helper cells (TFH), a cell type that is highly infectible by HIV and SIV, despite expressing low levels of CCR5 [93,94,95,96]. It is possible that Th17 CCR6+ cells may get initially infected, and then convert to TFH cells. Interestingly, CCR6+CCR4+ Th17 cells are specific for Candida albicans and CCR6+CXCR3+ Th17 cells are specific for Mycobacterium tuberculosis, two HIV-1 associated infections [11]. Thus, the preferential depletion of CCR6+ T cells may be a key event in the pathogenesis of HIV infection (Figure 6) [11,97].
We have previously reported that hBD2 inhibits HIV-1 replication post-entry requiring induction of APOBEC3G via CCR6 [71]. Here, we found that hBD2 selectively inhibits HIV replication in CCR6+ CD4+ cells, but not in CCR6−CD4+ cells. As expected, the levels of HIV replication in CCR6−CD4+ cells were significantly lower than in CCR6+CD4+ cells; however, that low level of replication could not be inhibited by treating cells with azidothymidine (AZT), a reverse transcriptase inhibitor. One intriguing possibility is that this low level of reverse-transcription independent replication may lead to HIV latency, making the CCR6−CD4+ cells a possible reservoir. In this study, we analyzed the status (LMM versus HMM) of APOBEC3G and identified the hBD2-induced signaling pathway and transcription factors that result in its increased expression and selective protection of CCR6+CD4+ cells. We found that hBD2 causes a dramatic shift from the HMM to the highly active LMM form in CCR6+ Jurkat cells, but not in primary CD4+ T cells. It is possible that the lack of shift from HMM to LMM is due to the lack of depolimerization in CCR6− cells, however due to cell number limitations, the molecular mass of APOBEC3G was not determined in primary cells separated into CCR6+ and CCR6− subsets. Alternatively, it is possible that differences in signaling pathways between Jurkat and primary cells may be responsible for this difference. Treatment with hBD2 resulted in ERK1/2 phosphorylation in PBMC, CD4+ T cells, and JKT-FT7 CCR6 GFP cells and was required for APOBEC3G expression. We observed rapid transcriptional activation of APOBEC3G, which was sensitive to treatment with the transcriptional inhibitor actinomycin D, in both primary CD4+ T cells and JKT-FT7 CCR6 GFP cells. In contrast, hBD2 did not induce APOBEC3G in the CCR6− JKT-FT7 cells. The rapid transcriptional induction of APOBEC3G mRNA confirms the results from our ChIP experiments. We found enrichment of APOBEC3G promoter sequence pulled down with NFATc2, NFATc1, and IRF-4 antibodies at early timepoints. NFAT proteins cooperate with additional transcription factors and Farrow et al. identified an NFAT/IRF-4 composite site in the APOBEC3G promoter [88]. NFAT and IRF-4 induce APOBEC3G expression, which is markedly enhanced upon cooperative binding [88]. Our results suggest that the early increase in APOBEC3G mRNA after hBD2 treatment is due to the presence of both NFAT and IRF-4.
The preferential infection of CCR6+ Th17 cells may be attributable to the higher expression of the viral receptors CD4, CXCR4, and α4β7 compared with CCR6− Th17 cells resulting in enhanced HIV envelope binding [14]. Despite similar CCR5 expression levels, CCR6+ Th17 cells may be more susceptible to R5 tropic virus, partly due to low expression of CCR5 ligands and decreased expression of antiviral factors [14,16]. The preferential infection and depletion of CCR6+CD4+ T cell subsets may initiate or contribute to the failure of the gut mucosal immune system. HIV-1-infected long-term non-progressors and SIV-infected sooty mangabeys, which do not progress to AIDS and lack both microbial translocation and chronic immune activation, maintain levels of Th17 cells in both the blood and gut [21,28,30]. These findings suggest that protecting CCR6+CD4+ T cell subsets may be critical in preventing disease progression, and hBD2 produced from mucosal epithelial cells could contribute to preserving this subset (Figure 6). Consistent with this hypothesis, a recent study has highlighted that hBD2 is a key factor differentially expressed in primary endometrial epithelial cells (which produce protective factors), as compared to endometrial stromal fibroblasts (which enhance HIV infection in vitro) [98].
In agreement with previously reported findings, we found that CCR6+CD4+ T cells have higher levels of replication when infected with pNL4-3-deltaE-EGFP HIV pseudotyped with the AMLV envelope which does not use HIV receptors or CCR6 for entry [16,17]. In addition to the previously reported elevated levels of HIV receptors and decreased CCR5 ligands, these findings suggest that additional post-entry cellular factors contribute to the preferential infection of CCR6+ cells. The CCR6-mediated intracellular inhibition of HIV-1 by hBD2 described herein, provides insight into signaling pathways that may guide the development of therapeutics that selectively target and protect CCR6+ cells.
5. Conclusions
Our study shows that hBD2 protects CCR6+CD4+ T cells from HIV infection. The protection is associated with increased APOBEC3G transcription, due to ERK1/2 signaling. Since there is increasing evidence that CCR6+CD4+ cells are lost early in HIV infection, our results are important to devise novel strategy to combat HIV infection. One attractive possibility is to design drugs that bind to CCR6 inducing APOBEC3G transcription. hBD2 has low chemotactic activity, so that it is conceivable that a novel CCR6-binding drug could induce APOBEC3G expression, and have low chemotactic index. Such drug could target HIV even in the latent reservoir, which has been shown to include CCR6+ cells.
Acknowledgments
We thank Marvin Reitz for his critical reading of this manuscript. We are grateful to Richard Koup, Ferenc Livak, Nicholas Carbonetti, Yutaka Tagaya and Jennifer Bharucha for helpful discussions and suggestions. We thank Holly Morgan Barnard for her artwork. We also thank the AIDS Research and Reference Reagent Program, Division of AIDS. This work was supported by the National Institute of Neurological Disorders And Stroke award R01NS066842 to Alfredo Garzino-Demo. Aaron Christensen-Quick was a trainee under Institutional Training Grant 1T32AI095190-01A1 from the National Institute of Allergy and Infectious Diseases. Mark K. Lafferty was a trainee under Institutional Training Grant T32AI007540 from the National Institute of Allergy and Infectious Diseases. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Allergy and Infectious Diseases, the National Institute of Neurological Disorders And Stroke, or the National Institutes of Health. The authors declare no conflict of interest exists.
Author Contributions
M.K.L. and A.G.-D. conceived and designed the experiments; M.K.L., L.S., and A.C.-Q. performed the experiments; M.K.L. analyzed the data; W.L. contributed reagents/materials; M.K.L. and A.G.-D. wrote the paper.
Conflicts of Interest
The authors declare no conflict of interest. The funding sponsors had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, and in the decision to publish the results.
References
- Lecureuil, C.; Combadiere, B.; Mazoyer, E.; Bonduelle, O.; Samri, A.; Autran, B.; Debre, P.; Combadiere, C. Trapping and apoptosis of novel subsets of memory T lymphocytes expressing CCR6 in the spleen of HIV-infected patients. Blood 2007, 109, 3649–3657. [Google Scholar] [CrossRef] [PubMed]
- Gosselin, A.; Monteiro, P.; Chomont, N.; Diaz-Griffero, F.; Said, E.A.; Fonseca, S.; Wacleche, V.; El-Far, M.; Boulassel, M.R.; Routy, J.P.; et al. Peripheral blood CCR4+CCR6+ and CXCR3+CCR6+CD4+ T cells are highly permissive to HIV-1 infection. J. Immunol. 2010, 184, 1604–1616. [Google Scholar] [CrossRef] [PubMed]
- Bouskra, D.; Brezillon, C.; Berard, M.; Werts, C.; Varona, R.; Boneca, I.G.; Eberl, G. Lymphoid tissue genesis induced by commensals through NOD1 regulates intestinal homeostasis. Nature 2008, 456, 507–510. [Google Scholar] [CrossRef] [PubMed]
- Cook, D.N.; Prosser, D.M.; Forster, R.; Zhang, J.; Kuklin, N.A.; Abbondanzo, S.J.; Niu, X.D.; Chen, S.C.; Manfra, D.J.; Wiekowski, M.T.; et al. CCR6 mediates dendritic cell localization, lymphocyte homeostasis, and immune responses in mucosal tissue. Immunity 2000, 12, 495–503. [Google Scholar] [CrossRef]
- Le Borgne, M.; Etchart, N.; Goubier, A.; Lira, S.A.; Sirard, J.C.; van Rooijen, N.; Caux, C.; Ait-Yahia, S.; Vicari, A.; Kaiserlian, D.; et al. Dendritic cells rapidly recruited into epithelial tissues via CCR6/CCL20 are responsible for CD8+ T cell crosspriming in vivo. Immunity 2006, 24, 191–201. [Google Scholar] [CrossRef] [PubMed]
- Lugering, A.; Floer, M.; Westphal, S.; Maaser, C.; Spahn, T.W.; Schmidt, M.A.; Domschke, W.; Williams, I.R.; Kucharzik, T. Absence of CCR6 inhibits CD4+ regulatory T-cell development and M-cell formation inside peyer's patches. Am. J. Pathol. 2005, 166, 1647–1654. [Google Scholar] [CrossRef]
- McDonald, K.G.; McDonough, J.S.; Wang, C.; Kucharzik, T.; Williams, I.R.; Newberry, R.D. CC chemokine receptor 6 expression by B lymphocytes is essential for the development of isolated lymphoid follicles. Am. J. Pathol. 2007, 170, 1229–1240. [Google Scholar] [CrossRef] [PubMed]
- Sallusto, F.; Lanzavecchia, A.; Mackay, C.R. Chemokines and chemokine receptors in T-cell priming and Th1/Th2-mediated responses. Immunol. Today 1998, 19, 568–574. [Google Scholar] [CrossRef]
- Williams, I.R. CCR6 and CCL20: Partners in intestinal immunity and lymphorganogenesis. Ann. N. Y. Acad. Sci. 2006, 1072, 52–61. [Google Scholar] [CrossRef] [PubMed]
- Liao, F.; Rabin, R.L.; Smith, C.S.; Sharma, G.; Nutman, T.B.; Farber, J.M. CC-chemokine receptor 6 is expressed on diverse memory subsets of T cells and determines responsiveness to macrophage inflammatory protein 3 alpha. J. Immunol. 1999, 162, 186–194. [Google Scholar] [PubMed]
- Acosta-Rodriguez, E.V.; Rivino, L.; Geginat, J.; Jarrossay, D.; Gattorno, M.; Lanzavecchia, A.; Sallusto, F.; Napolitani, G. Surface phenotype and antigenic specificity of human interleukin 17-producing T helper memory cells. Nat. Immunol. 2007, 8, 639–646. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.P.; Zhang, H.H.; Foley, J.F.; Hedrick, M.N.; Farber, J.M. Human T cells that are able to produce IL-17 express the chemokine receptor CCR6. J. Immunol. 2008, 180, 214–221. [Google Scholar] [CrossRef] [PubMed]
- El Hed, A.; Khaitan, A.; Kozhaya, L.; Manel, N.; Daskalakis, D.; Borkowsky, W.; Valentine, F.; Littman, D.R.; Unutmaz, D. Susceptibility of human Th17 cells to human immunodeficiency virus and their perturbation during infection. J. Infect. Dis. 2010, 201, 843–854. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, Y.; Tuen, M.; Shen, G.; Nawaz, F.; Arthos, J.; Wolff, M.J.; Poles, M.A.; Hioe, C.E. Preferential HIV infection of CCR6+ Th17 cells is associated with higher levels of virus receptor expression and lack of CCR5 ligands. J. Virol. 2013, 87, 10843–10854. [Google Scholar] [CrossRef] [PubMed]
- Guan, Y.; Abdelwahab, S.; Kamin-Lewis, R.; DeVico, A.L.; Lewis, G.K. Self-protection of individual CD4+ T cells against R5 HIV-1 infection by the synthesis of anti-viral CCR5 ligands. PLoS ONE 2008, 3, e3481. [Google Scholar] [CrossRef] [PubMed]
- Christensen-Quick, A.; Lafferty, M.; Sun, L.; Marchionni, L.; DeVico, A.; Garzino-Demo, A. Human Th17 cells lack HIV-inhibitory rnases and are highly permissive to productive HIV infection. J. Virol. 2016, 90, 7833–7847. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, P.; Gosselin, A.; Wacleche, V.S.; El-Far, M.; Said, E.A.; Kared, H.; Grandvaux, N.; Boulassel, M.R.; Routy, J.P.; Ancuta, P. Memory CCR6+CD4+ T cells are preferential targets for productive HIV type 1 infection regardless of their expression of integrin beta7. J. Immunol. 2011, 186, 4618–4630. [Google Scholar] [CrossRef] [PubMed]
- Khoury, G.; Anderson, J.L.; Fromentin, R.; Hartogenesis, W.; Smith, M.Z.; Bacchetti, P.; Hecht, F.M.; Chomont, N.; Cameron, P.U.; Deeks, S.G.; et al. Persistence of integrated HIV DNA in CXCR3+ CCR6+ memory CD4+ T cells in HIV-infected individuals on antiretroviral therapy. AIDS 2016, 30, 1511–1520. [Google Scholar] [CrossRef] [PubMed]
- Wacleche, V.S.; Goulet, J.P.; Gosselin, A.; Monteiro, P.; Soudeyns, H.; Fromentin, R.; Jenabian, M.A.; Vartanian, S.; Deeks, S.G.; Chomont, N.; et al. New insights into the heterogeneity of Th17 subsets contributing to HIV-1 persistence during antiretroviral therapy. Retrovirology 2016, 13, 59. [Google Scholar] [CrossRef] [PubMed]
- Gosselin, A.; Wiche Salinas, T.R.; Planas, D.; Wacleche, V.S.; Zhang, Y.; Fromentin, R.; Chomont, N.; Cohen, E.A.; Shacklett, B.; Mehraj, V.; et al. HIV persists in CCR6+CD4+ T cells from colon and blood during antiretroviral therapy. AIDS 2017, 31, 35–48. [Google Scholar] [CrossRef] [PubMed]
- Brenchley, J.M.; Paiardini, M.; Knox, K.S.; Asher, A.I.; Cervasi, B.; Asher, T.E.; Scheinberg, P.; Price, D.A.; Hage, C.A.; Kholi, L.M.; et al. Differential Th17 CD4 T-cell depletion in pathogenic and nonpathogenic lentiviral infections. Blood 2008, 112, 2826–2835. [Google Scholar] [CrossRef] [PubMed]
- Cecchinato, V.; Trindade, C.J.; Laurence, A.; Heraud, J.M.; Brenchley, J.M.; Ferrari, M.G.; Zaffiri, L.; Tryniszewska, E.; Tsai, W.P.; Vaccari, M.; et al. Altered balance between Th17 and Th1 cells at mucosal sites predicts aids progression in simian immunodeficiency virus-infected macaques. Mucosal Immunol. 2008, 1, 279–288. [Google Scholar] [CrossRef] [PubMed]
- Stieh, D.J.; Matias, E.; Xu, H.; Fought, A.J.; Blanchard, J.L.; Marx, P.A.; Veazey, R.S.; Hope, T.J. Th17 cells are preferentially infected very early after vaginal transmission of SIV in macaques. Cell Host Microbe 2016, 19, 529–540. [Google Scholar] [CrossRef] [PubMed]
- McGary, C.S.; Alvarez, X.; Harrington, S.; Cervasi, B.; Ryan, E.S.; Iriele, R.I.; Paganini, S.; Harper, J.L.; Easley, K.; Silvestri, G.; et al. The loss of CCR6+ and CD161+ CD4+ T-cell homeostasis contributes to disease progression in SIV-infected rhesus macaques. Mucosal Immunol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Brenchley, J.M.; Schacker, T.W.; Ruff, L.E.; Price, D.A.; Taylor, J.H.; Beilman, G.J.; Nguyen, P.L.; Khoruts, A.; Larson, M.; Haase, A.T.; et al. Cd4+ T cell depletion during all stages of HIV disease occurs predominantly in the gastrointestinal tract. J. Exp. Med. 2004, 200, 749–759. [Google Scholar] [CrossRef] [PubMed]
- Mehandru, S.; Poles, M.A.; Tenner-Racz, K.; Horowitz, A.; Hurley, A.; Hogan, C.; Boden, D.; Racz, P.; Markowitz, M. Primary HIV-1 infection is associated with preferential depletion of CD4+ T lymphocytes from effector sites in the gastrointestinal tract. J. Exp. Med. 2004, 200, 761–770. [Google Scholar] [CrossRef] [PubMed]
- Mehandru, S.; Poles, M.A.; Tenner-Racz, K.; Jean-Pierre, P.; Manuelli, V.; Lopez, P.; Shet, A.; Low, A.; Mohri, H.; Boden, D.; et al. Lack of mucosal immune reconstitution during prolonged treatment of acute and early HIV-1 infection. PLoS Med. 2006, 3, e484. [Google Scholar] [CrossRef]
- Brenchley, J.M.; Price, D.A.; Schacker, T.W.; Asher, T.E.; Silvestri, G.; Rao, S.; Kazzaz, Z.; Bornstein, E.; Lambotte, O.; Altmann, D.; et al. Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nat. Med. 2006, 12, 1365–1371. [Google Scholar] [CrossRef] [PubMed]
- Raffatellu, M.; Santos, R.L.; Verhoeven, D.E.; George, M.D.; Wilson, R.P.; Winter, S.E.; Godinez, I.; Sankaran, S.; Paixao, T.A.; Gordon, M.A.; et al. Simian immunodeficiency virus-induced mucosal interleukin-17 deficiency promotes salmonella dissemination from the gut. Nat. Med. 2008, 14, 421–428. [Google Scholar] [CrossRef] [PubMed]
- Ciccone, E.J.; Greenwald, J.H.; Lee, P.I.; Biancotto, A.; Read, S.W.; Yao, M.A.; Hodge, J.N.; Thompson, W.L.; Kovacs, S.B.; Chairez, C.L.; et al. CD4+ T cells, including Th17 and cycling subsets, are intact in the gut mucosa of HIV-1-infected long-term nonprogressors. J. Virol. 2011, 85, 5880–5888. [Google Scholar] [CrossRef] [PubMed]
- Chege, D.; Sheth, P.M.; Kain, T.; Kim, C.J.; Kovacs, C.; Loutfy, M.; Halpenny, R.; Kandel, G.; Chun, T.W.; Ostrowski, M.; et al. Sigmoid Th17 populations, the HIV latent reservoir, and microbial translocation in men on long-term antiretroviral therapy. AIDS 2011, 25, 741–749. [Google Scholar] [CrossRef] [PubMed]
- Curtis, M.M.; Way, S.S. Interleukin-17 in host defence against bacterial, mycobacterial and fungal pathogens. Immunology 2009, 126, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Conti, H.R.; Shen, F.; Nayyar, N.; Stocum, E.; Sun, J.N.; Lindemann, M.J.; Ho, A.W.; Hai, J.H.; Yu, J.J.; Jung, J.W.; et al. Th17 cells and IL-17 receptor signaling are essential for mucosal host defense against oral candidiasis. J. Exp. Med. 2009, 206, 299–311. [Google Scholar] [CrossRef] [PubMed]
- Klatt, N.R.; Brenchley, J.M. Th17 cell dynamics in HIV infection. Curr. Opin. HIV AIDS 2010, 5, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Santos, N.; Huppler, A.R.; Peterson, A.C.; Khader, S.A.; McKenna, K.C.; Gaffen, S.L. Th17 cells confer long-term adaptive immunity to oral mucosal Candida albicans infections. Mucosal Immunol. 2013, 6, 900–910. [Google Scholar] [CrossRef] [PubMed]
- Fossiez, F.; Djossou, O.; Chomarat, P.; Flores-Romo, L.; Ait-Yahia, S.; Maat, C.; Pin, J.J.; Garrone, P.; Garcia, E.; Saeland, S.; et al. T cell interleukin-17 induces stromal cells to produce proinflammatory and hematopoietic cytokines. J. Exp. Med. 1996, 183, 2593–2603. [Google Scholar] [CrossRef] [PubMed]
- LeGrand, A.; Fermor, B.; Fink, C.; Pisetsky, D.S.; Weinberg, J.B.; Vail, T.P.; Guilak, F. Interleukin-1, tumor necrosis factor alpha, and interleukin-17 synergistically up-regulate nitric oxide and prostaglandin E2 production in explants of human osteoarthritic knee menisci. Arthritis Rheum. 2001, 44, 2078–2083. [Google Scholar] [CrossRef]
- Moseley, T.A.; Haudenschild, D.R.; Rose, L.; Reddi, A.H. Interleukin-17 family and IL-17 receptors. Cytokine Growth Factor Rev. 2003, 14, 155–174. [Google Scholar] [CrossRef]
- Ye, P.; Rodriguez, F.H.; Kanaly, S.; Stocking, K.L.; Schurr, J.; Schwarzenberger, P.; Oliver, P.; Huang, W.; Zhang, P.; Zhang, J.; et al. Requirement of interleukin 17 receptor signaling for lung CXC chemokine and granulocyte colony-stimulating factor expression, neutrophil recruitment, and host defense. J. Exp. Med. 2001, 194, 519–527. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Thai, P.; Zhao, Y.H.; Ho, Y.S.; DeSouza, M.M.; Wu, R. Stimulation of airway mucin gene expression by interleukin (IL)-17 through IL-6 paracrine/autocrine loop. J. Biol. Chem. 2003, 278, 17036–17043. [Google Scholar] [CrossRef] [PubMed]
- Kao, C.Y.; Chen, Y.; Thai, P.; Wachi, S.; Huang, F.; Kim, C.; Harper, R.W.; Wu, R. IL-17 markedly up-regulates beta-defensin-2 expression in human airway epithelium via JAK and NF-kappaB signaling pathways. J. Immunol. 2004, 173, 3482–3491. [Google Scholar] [CrossRef] [PubMed]
- Kao, C.Y.; Huang, F.; Chen, Y.; Thai, P.; Wachi, S.; Kim, C.; Tam, L.; Wu, R. Up-regulation of CC chemokine ligand 20 expression in human airway epithelium by IL-17 through a JAK-independent but MEK/NF-KappaB-dependent signaling pathway. J. Immunol. 2005, 175, 6676–6685. [Google Scholar] [CrossRef] [PubMed]
- Kinugasa, T.; Sakaguchi, T.; Gu, X.; Reinecker, H.C. Claudins regulate the intestinal barrier in response to immune mediators. Gastroenterology 2000, 118, 1001–1011. [Google Scholar] [CrossRef]
- Liang, S.C.; Tan, X.Y.; Luxenberg, D.P.; Karim, R.; Dunussi-Joannopoulos, K.; Collins, M.; Fouser, L.A. Interleukin (IL)-22 and IL-17 are coexpressed by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J. Exp. Med. 2006, 203, 2271–2279. [Google Scholar] [CrossRef] [PubMed]
- Wolk, K.; Kunz, S.; Witte, E.; Friedrich, M.; Asadullah, K.; Sabat, R. IL-22 increases the innate immunity of tissues. Immunity 2004, 21, 241–254. [Google Scholar] [CrossRef] [PubMed]
- Lehrer, R.I.; Ganz, T.; Selsted, M.E. Defensins: Endogenous antibiotic peptides of animal cells. Cell 1991, 64, 229–230. [Google Scholar] [CrossRef]
- Zasloff, M. Antibiotic peptides as mediators of innate immunity. Curr. Opin. Immunol. 1992, 4, 3–7. [Google Scholar] [CrossRef]
- Ganz, T. Defensins: Antimicrobial peptides of innate immunity. Nat. Rev. Immunol. 2003, 3, 710–720. [Google Scholar] [CrossRef] [PubMed]
- Selsted, M.E.; Ouellette, A.J. Mammalian defensins in the antimicrobial immune response. Nat. Immunol. 2005, 6, 551–557. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Xu, Z.; Peng, L.; Fang, X.; Yin, X.; Xu, N.; Cen, P. Recent advances in the research and development of human defensins. Peptides 2006, 27, 931–940. [Google Scholar] [CrossRef] [PubMed]
- Pazgier, M.; Hoover, D.M.; Yang, D.; Lu, W.; Lubkowski, J. Human beta-defensins. Cell. Mol. Life Sci. 2006, 63, 1294–1313. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Chou, Y.Y.; Chang, T.L. Defensins in viral infections. J. Innate Immun. 2009, 1, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Wilson, S.S.; Wiens, M.E.; Smith, J.G. Antiviral mechanisms of human defensins. J. Mol. Biol. 2013, 425, 4965–4980. [Google Scholar] [CrossRef] [PubMed]
- Bevins, C.L. Innate immune functions of alpha-defensins in the small intestine. Dig. Dis. 2013, 31, 299–304. [Google Scholar] [CrossRef] [PubMed]
- Daher, K.A.; Selsted, M.E.; Lehrer, R.I. Direct inactivation of viruses by human granulocyte defensins. J. Virol. 1986, 60, 1068–1074. [Google Scholar] [PubMed]
- Chang, T.L.; Klotman, M.E. Defensins: Natural anti-HIV peptides. AIDS Rev. 2004, 6, 161–168. [Google Scholar] [PubMed]
- Garzino-Demo, A. Chemokines and defensins as HIV suppressive factors: An evolving story. Curr. Pharm. Des. 2007, 13, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Chertov, O.; Bykovskaia, S.N.; Chen, Q.; Buffo, M.J.; Shogan, J.; Anderson, M.; Schroder, J.M.; Wang, J.M.; Howard, O.M.; et al. Beta-defensins: Linking innate and adaptive immunity through dendritic and T cell CCR6. Science 1999, 286, 525–528. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Hoover, D.M.; Yang, D.; Boulegue, C.; Santamaria, F.; Oppenheim, J.J.; Lubkowski, J.; Lu, W. Engineering disulfide bridges to dissect antimicrobial and chemotactic activities of human beta-defensin 3. Proc. Natl. Acad. Sci. USA 2003, 100, 8880–8885. [Google Scholar] [CrossRef] [PubMed]
- Niyonsaba, F.; Ogawa, H.; Nagaoka, I. Human beta-defensin-2 functions as a chemotactic agent for tumour necrosis factor-alpha-treated human neutrophils. Immunology 2004, 111, 273–281. [Google Scholar] [CrossRef] [PubMed]
- Soruri, A.; Grigat, J.; Forssmann, U.; Riggert, J.; Zwirner, J. Beta-defensins chemoattract macrophages and mast cells but not lymphocytes and dendritic cells: CCR6 is not involved. Eur. J. Immunol. 2007, 37, 2474–2486. [Google Scholar] [CrossRef] [PubMed]
- Barabas, N.; Rohrl, J.; Holler, E.; Hehlgans, T. Beta-defensins activate macrophages and synergize in pro-inflammatory cytokine expression induced by TLR ligands. Immunobiology 2013, 218, 1005–1011. [Google Scholar] [CrossRef] [PubMed]
- Rohrl, J.; Yang, D.; Oppenheim, J.J.; Hehlgans, T. Human beta-defensin 2 and 3 and their mouse orthologs induce chemotaxis through interaction with CCR2. J. Immunol. 2010, 184, 6688–6694. [Google Scholar] [CrossRef] [PubMed]
- Boniotto, M.; Jordan, W.J.; Eskdale, J.; Tossi, A.; Antcheva, N.; Crovella, S.; Connell, N.D.; Gallagher, G. Human beta-defensin 2 induces a vigorous cytokine response in peripheral blood mononuclear cells. Antimicrob. Agents Chemother. 2006, 50, 1433–1441. [Google Scholar] [CrossRef] [PubMed]
- Niyonsaba, F.; Ushio, H.; Nakano, N.; Ng, W.; Sayama, K.; Hashimoto, K.; Nagaoka, I.; Okumura, K.; Ogawa, H. Antimicrobial peptides human beta-defensins stimulate epidermal keratinocyte migration, proliferation and production of proinflammatory cytokines and chemokines. J. Investig. Dermatol. 2007, 127, 594–604. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Niyonsaba, F.; Ushio, H.; Hara, M.; Yokoi, H.; Matsumoto, K.; Saito, H.; Nagaoka, I.; Ikeda, S.; Okumura, K.; et al. Antimicrobial peptides human beta-defensin (HBD)-3 and HBD-4 activate mast cells and increase skin vascular permeability. Eur. J. Immunol. 2007, 37, 434–444. [Google Scholar] [CrossRef] [PubMed]
- Otte, J.M.; Werner, I.; Brand, S.; Chromik, A.M.; Schmitz, F.; Kleine, M.; Schmidt, W.E. Human beta defensin 2 promotes intestinal wound healing in vitro. J. Cell. Biochem. 2008, 104, 2286–2297. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, A.; Jin, G.; Sieg, S.; McCormick, T.S. The yin and yang of human beta-defensins in health and disease. Front. Immunol. 2012, 3, 294. [Google Scholar] [CrossRef] [PubMed]
- Quinones-Mateu, M.E.; Lederman, M.M.; Feng, Z.; Chakraborty, B.; Weber, J.; Rangel, H.R.; Marotta, M.L.; Mirza, M.; Jiang, B.; Kiser, P.; et al. Human epithelial beta-defensins 2 and 3 inhibit HIV-1 replication. AIDS 2003, 17, F39–F48. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Finnegan, C.M.; Kish-Catalone, T.; Blumenthal, R.; Garzino-Demo, P.; La Terra Maggiore, G.M.; Berrone, S.; Kleinman, C.; Wu, Z.; Abdelwahab, S.; et al. Human beta-defensins suppress human immunodeficiency virus infection: Potential role in mucosal protection. J. Virol. 2005, 79, 14318–14329. [Google Scholar] [CrossRef] [PubMed]
- Lafferty, M.K.; Sun, L.; DeMasi, L.; Lu, W.; Garzino-Demo, A. CCR6 ligands inhibit HIV by inducing APOBEC3G. Blood 2010, 115, 1564–1571. [Google Scholar] [CrossRef] [PubMed]
- Sheehy, A.M.; Gaddis, N.C.; Choi, J.D.; Malim, M.H. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature 2002, 418, 646–650. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Yang, B.; Pomerantz, R.J.; Zhang, C.; Arunachalam, S.C.; Gao, L. The cytidine deaminase CEM15 induces hypermutation in newly synthesized HIV-1 DNA. Nature 2003, 424, 94–98. [Google Scholar] [CrossRef] [PubMed]
- Bishop, K.N.; Holmes, R.K.; Malim, M.H. Antiviral potency of APOBEC proteins does not correlate with cytidine deamination. J. Virol. 2006, 80, 8450–8458. [Google Scholar] [CrossRef] [PubMed]
- Bishop, K.N.; Verma, M.; Kim, E.Y.; Wolinsky, S.M.; Malim, M.H. APOBEC3G inhibits elongation of HIV-1 reverse transcripts. PLoS Pathog. 2008, 4, e1000231. [Google Scholar] [CrossRef] [PubMed]
- De Pasquale, M.; Kourteva, Y.; Allos, T.; D’Aquila, R.T. Lower HIV provirus levels are associated with more APOBEC3G protein in blood resting memory CD4+ T lymphocytes of controllers in vivo. PLoS ONE 2013, 8, e76002. [Google Scholar] [CrossRef]
- Kourteva, Y.; De Pasquale, M.; Allos, T.; McMunn, C.; D’Aquila, R.T. APOBEC3G expression and hypermutation are inversely associated with human immunodeficiency virus type 1 (HIV-1) burden in vivo. Virology 2012, 430, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Vetter, M.L.; D’Aquila, R.T. Cytoplasmic APOBEC3G restricts incoming Vif-positive human immunodeficiency virus type 1 and increases two-long terminal repeat circle formation in activated T-helper-subtype cells. J. Virol. 2009, 83, 8646–8654. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Chiu, Y.L.; Witkowska, H.E.; Hall, S.C.; Santiago, M.; Soros, V.B.; Esnault, C.; Heidmann, T.; Greene, W.C. High-molecular-mass APOBEC3G complexes restrict alu retrotransposition. Proc. Natl. Acad. Sci. USA 2006, 103, 15588–15593. [Google Scholar] [CrossRef] [PubMed]
- Opi, S.; Takeuchi, H.; Kao, S.; Khan, M.A.; Miyagi, E.; Goila-Gaur, R.; Iwatani, Y.; Levin, J.G.; Strebel, K. Monomeric APOBEC3G is catalytically active and has antiviral activity. J. Virol. 2006, 80, 4673–4682. [Google Scholar] [CrossRef] [PubMed]
- Goila-Gaur, R.; Khan, M.A.; Miyagi, E.; Kao, S.; Opi, S.; Takeuchi, H.; Strebel, K. HIV-1 Vif promotes the formation of high molecular mass APOBEC3G complexes. Virology 2008, 372, 136–146. [Google Scholar] [CrossRef] [PubMed]
- Shultz, L.D.; Saito, Y.; Najima, Y.; Tanaka, S.; Ochi, T.; Tomizawa, M.; Doi, T.; Sone, A.; Suzuki, N.; Fujiwara, H.; et al. Generation of functional human T-cell subsets with hla-restricted immune responses in HLA class I expressing NOD/SCID/IL2r gamma(null) humanized mice. Proc. Natl. Acad. Sci. USA 2010, 107, 13022–13027. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Huang, J.; Zhang, C.; Huang, S.; Nunnari, G.; Wang, F.X.; Tong, X.; Gao, L.; Nikisher, K.; Zhang, H. Alpha interferon potently enhances the anti-human immunodeficiency virus type 1 activity of APOBEC3G in resting primary CD4 T cells. J. Virol. 2006, 80, 7645–7657. [Google Scholar] [CrossRef] [PubMed]
- Rose, K.M.; Marin, M.; Kozak, S.L.; Kabat, D. Transcriptional regulation of APOBEC3G, a cytidine deaminase that hypermutates human immunodeficiency virus. J. Biol. Chem. 2004, 279, 41744–41749. [Google Scholar] [CrossRef] [PubMed]
- Stopak, K.S.; Chiu, Y.L.; Kropp, J.; Grant, R.M.; Greene, W.C. Distinct patterns of cytokine regulation of APOBEC3G expression and activity in primary lymphocytes, macrophages, and dendritic cells. J. Biol. Chem. 2007, 282, 3539–3546. [Google Scholar] [CrossRef] [PubMed]
- Lin, A.; Minden, A.; Martinetto, H.; Claret, F.X.; Lange-Carter, C.; Mercurio, F.; Johnson, G.L.; Karin, M. Identification of a dual specificity kinase that activates the jun kinases and p38-MPK2. Science 1995, 268, 286–290. [Google Scholar] [CrossRef] [PubMed]
- Farrow, M.A.; Kim, E.Y.; Wolinsky, S.M.; Sheehy, A.M. NFAT and IRF proteins regulate transcription of the anti-HIV protein, APOBEC3G. J. Biol. Chem. 2011, 286, 2567–2577. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Estes, J.D.; Schlievert, P.M.; Duan, L.; Brosnahan, A.J.; Southern, P.J.; Reilly, C.S.; Peterson, M.L.; Schultz-Darken, N.; Brunner, K.G.; et al. Glycerol monolaurate prevents mucosal SIV transmission. Nature 2009, 458, 1034–1038. [Google Scholar] [CrossRef] [PubMed]
- Guillot-Delost, M.; Le Gouvello, S.; Mesel-Lemoine, M.; Cherai, M.; Baillou, C.; Simon, A.; Levy, Y.; Weiss, L.; Louafi, S.; Chaput, N.; et al. Human CD90 identifies Th17/Tc17 T cell subsets that are depleted in HIV-infected patients. J. Immunol. 2011, 188, 981–991. [Google Scholar] [CrossRef] [PubMed]
- Shen, F.; Ruddy, M.J.; Plamondon, P.; Gaffen, S.L. Cytokines link osteoblasts and inflammation: Microarray analysis of interleukin-17- and TNF-alpha-induced genes in bone cells. J. Leukoc. Biol. 2005, 77, 388–399. [Google Scholar] [CrossRef] [PubMed]
- Ruddy, M.J.; Shen, F.; Smith, J.B.; Sharma, A.; Gaffen, S.L. Interleukin-17 regulates expression of the CXC chemokine LIX/CXCL5 in osteoblasts: Implications for inflammation and neutrophil recruitment. J. Leukoc. Biol. 2004, 76, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Hirota, K.; Turner, J.E.; Villa, M.; Duarte, J.H.; Demengeot, J.; Steinmetz, O.M.; Stockinger, B. Plasticity of Th17 cells in peyer’s patches is responsible for the induction of T cell-dependent IgA responses. Nat. Immunol. 2013, 14, 372–379. [Google Scholar] [CrossRef] [PubMed]
- Lindqvist, M.; van Lunzen, J.; Soghoian, D.Z.; Kuhl, B.D.; Ranasinghe, S.; Kranias, G.; Flanders, M.D.; Cutler, S.; Yudanin, N.; Muller, M.I.; et al. Expansion of HIV-specific T follicular helper cells in chronic HIV infection. J. Clin. Investig. 2012, 122, 3271–3280. [Google Scholar] [CrossRef] [PubMed]
- Petrovas, C.; Yamamoto, T.; Gerner, M.Y.; Boswell, K.L.; Wloka, K.; Smith, E.C.; Ambrozak, D.R.; Sandler, N.G.; Timmer, K.J.; Sun, X.; et al. CD4 T follicular helper cell dynamics during SIV infection. J. Clin. Investig. 2012, 122, 3281–3294. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Weatherall, C.; Bailey, M.; Alcantara, S.; De Rose, R.; Estaquier, J.; Wilson, K.; Suzuki, K.; Corbeil, J.; Cooper, D.A.; et al. Simian immunodeficiency virus infects follicular helper CD4 T cells in lymphoid tissues during pathogenic infection of pigtail macaques. J. Virol. 2013, 87, 3760–3773. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Fan, X.; Auclair, S.; Ferguson, M.; Sun, J.; Soong, L.; Hou, W.; Redfield, R.R.; Birx, D.L.; Ratto-Kim, S.; et al. Sequential dysfunction and progressive depletion of candida albicans-specific CD4 T cell response in HIV-1 infection. PLoS Pathog. 2016, 12, e1005663. [Google Scholar] [CrossRef] [PubMed]
- Neidleman, J.A.; Chen, J.C.; Kohgadai, N.; Muller, J.A.; Laustsen, A.; Thavachelvam, K.; Jang, K.S.; Sturzel, C.M.; Jones, J.J.; Ochsenbauer, C.; et al. Mucosal stromal fibroblasts markedly enhance HIV infection of CD4+ T cells. PLoS Pathog. 2017, 13, e1006163. [Google Scholar] [CrossRef] [PubMed]
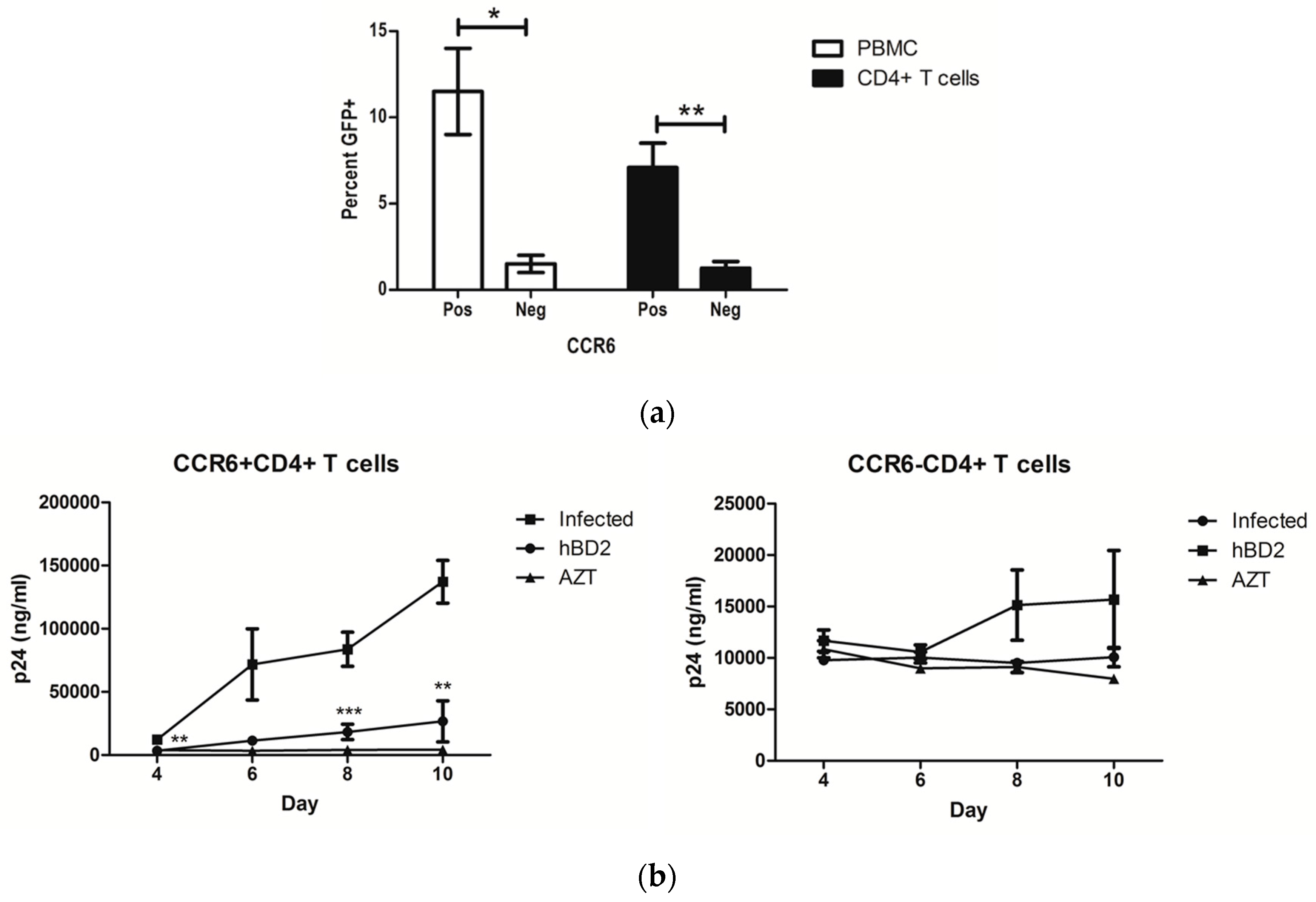
Figure 1.
CCR6+CD4+ T cells are more permissive to HIV than CCR6−CD4+ T cells. (a) Peripheral blood mononuclear cells (PBMC) and CD4+ T cells were infected with amphotropic murine leukemia virus (AMLV) pseudotyped pNL4-3ΔE-EGFP virus for 3 days. Percent GFP+CD4+CD3+ cells are shown for CCR6+ and CCR6− cells of four independent experiments (±standard error of the mean (SEM)). (b) CCR6+ and CCR6−CD4+ T cells were treated for 4 h with 20 μg/mL human beta-defensin 2 (hBD2), washed, and infected with 100 TCID50 of HIV-1BaL, corresponding to 1–2 ng of input p24. Some cells were treated after infection with 2.67 µg/mL of azidothymidine (AZT), for the duration of the experiment. After 6 days, HIV p24 in tissue culture supernatant was quantified by ELISA. Shown here are mean p24 ng/mL and percent inhibition (±SEM) of three independent experiments performed in triplicate. Statistical analysis was performed by paired 2-tail Student t test. * p < 0.05, ** p < 0.01, *** p < 0.001.
Figure 1.
CCR6+CD4+ T cells are more permissive to HIV than CCR6−CD4+ T cells. (a) Peripheral blood mononuclear cells (PBMC) and CD4+ T cells were infected with amphotropic murine leukemia virus (AMLV) pseudotyped pNL4-3ΔE-EGFP virus for 3 days. Percent GFP+CD4+CD3+ cells are shown for CCR6+ and CCR6− cells of four independent experiments (±standard error of the mean (SEM)). (b) CCR6+ and CCR6−CD4+ T cells were treated for 4 h with 20 μg/mL human beta-defensin 2 (hBD2), washed, and infected with 100 TCID50 of HIV-1BaL, corresponding to 1–2 ng of input p24. Some cells were treated after infection with 2.67 µg/mL of azidothymidine (AZT), for the duration of the experiment. After 6 days, HIV p24 in tissue culture supernatant was quantified by ELISA. Shown here are mean p24 ng/mL and percent inhibition (±SEM) of three independent experiments performed in triplicate. Statistical analysis was performed by paired 2-tail Student t test. * p < 0.05, ** p < 0.01, *** p < 0.001.

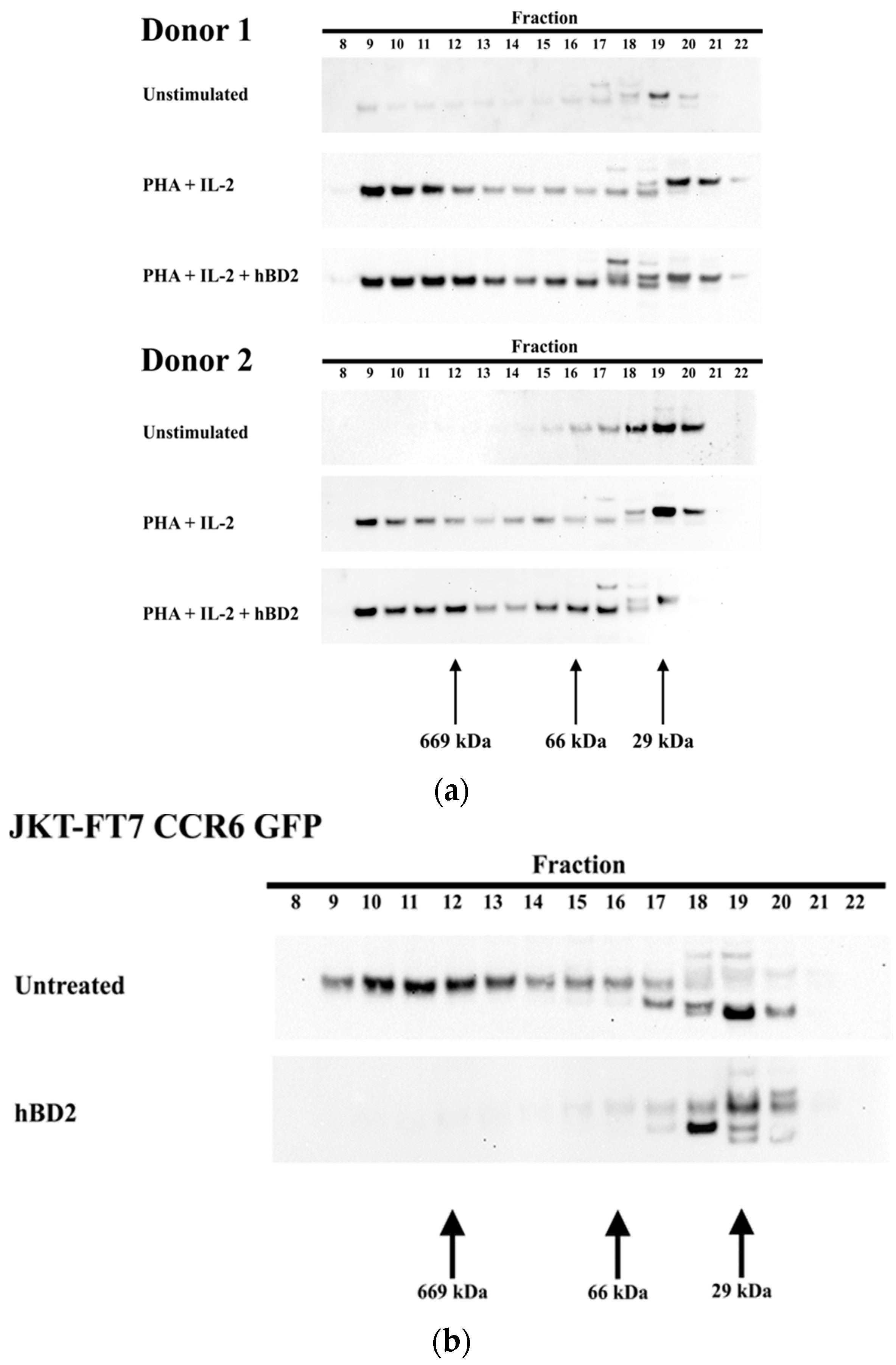
Figure 2.
Induction of low-molecular-mass (LMM) and high-molecular-mass (HMM) APOBEC3G. (a) Unstimulated CD4+ T cells or CD4+ T cells activated with phytohemagglutinin (PHA) (2.5 µg/mL) and IL-2 (10 ng/mL) for 48 h and treated with hBD2 (20 µg/mL) for 8 h. (b) JKT-FT7 CCR6 GFP cells treated with hBD2 (20 µg/mL) for 8 h. Cell lysates were loaded on a gel size-exclusion column for fast protein liquid chromatography (FPLC) analysis. Twenty-four 1-mL fractions were collected. Eluted fractions were subjected to sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), followed by an immunoblotting assay with anti-apolipoprotein B mRNA editing enzyme (APOBEC3G) antibody.
Figure 2.
Induction of low-molecular-mass (LMM) and high-molecular-mass (HMM) APOBEC3G. (a) Unstimulated CD4+ T cells or CD4+ T cells activated with phytohemagglutinin (PHA) (2.5 µg/mL) and IL-2 (10 ng/mL) for 48 h and treated with hBD2 (20 µg/mL) for 8 h. (b) JKT-FT7 CCR6 GFP cells treated with hBD2 (20 µg/mL) for 8 h. Cell lysates were loaded on a gel size-exclusion column for fast protein liquid chromatography (FPLC) analysis. Twenty-four 1-mL fractions were collected. Eluted fractions were subjected to sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), followed by an immunoblotting assay with anti-apolipoprotein B mRNA editing enzyme (APOBEC3G) antibody.

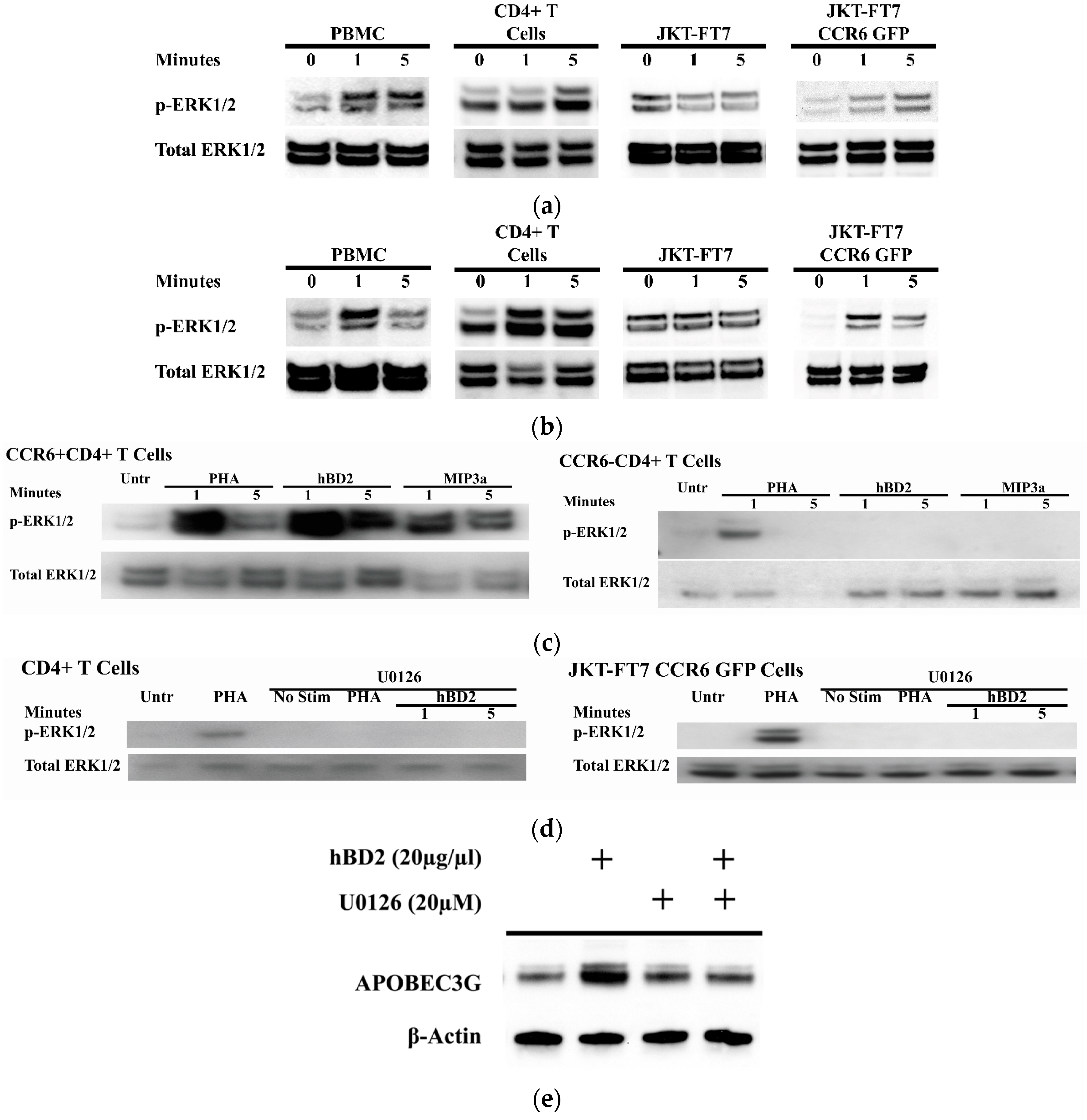
Figure 3.
CCR6 ligands activate the extracellular signal-regulated kinases 1/2 (ERK1/2) mitogen-activated protein kinases (MAPK) pathway. Activated PBMC, CD4+ T cells, JKT-FT7, or JKT-FT7 CCR6 GFP cells were treated with (a) hBD2 (20 μg/mL) or (b) MIP-3α (5 μg/mL) for 1 and 5 min. Phosphorylated ERK1/2 (p-ERK1/2) and total ERK1/2 were detected at the indicated time points by Western blotting. (c) Activated CCR6+CD4+ T cells or CCR6−CD4+ T cells were treated with PHA (2.5 μg/mL), hBD2 (20 μg/mL) or macrophage Inflammatory Protein-3 (MIP-3α) (5 μg/mL) for 1 and 5 min. Phosphorylated ERK1/2 (p-ERK1/2) and total ERK1/2 were detected at the indicated time points by Western blotting. (d) Activated CD4+ T cells and JKT-FT7 CCR6 GFP were pre-treated with U0126 (20 µM) for 1 h and then treated with PHA (2.5 μg/mL) or hBD2 (20 µg/mL) and probed for phosphorylated ERK1/2 and total ERK1/2. (e) Lysates from CD4+ T cells pre-treated with U0126 (20 µM) and subsequently treated with hBD2 (20 µg/mL) were probed for APOBEC3G and β-actin by Western blotting. Images shown are representative of 2–4 independent experiments from different donors or cell passages.
Figure 3.
CCR6 ligands activate the extracellular signal-regulated kinases 1/2 (ERK1/2) mitogen-activated protein kinases (MAPK) pathway. Activated PBMC, CD4+ T cells, JKT-FT7, or JKT-FT7 CCR6 GFP cells were treated with (a) hBD2 (20 μg/mL) or (b) MIP-3α (5 μg/mL) for 1 and 5 min. Phosphorylated ERK1/2 (p-ERK1/2) and total ERK1/2 were detected at the indicated time points by Western blotting. (c) Activated CCR6+CD4+ T cells or CCR6−CD4+ T cells were treated with PHA (2.5 μg/mL), hBD2 (20 μg/mL) or macrophage Inflammatory Protein-3 (MIP-3α) (5 μg/mL) for 1 and 5 min. Phosphorylated ERK1/2 (p-ERK1/2) and total ERK1/2 were detected at the indicated time points by Western blotting. (d) Activated CD4+ T cells and JKT-FT7 CCR6 GFP were pre-treated with U0126 (20 µM) for 1 h and then treated with PHA (2.5 μg/mL) or hBD2 (20 µg/mL) and probed for phosphorylated ERK1/2 and total ERK1/2. (e) Lysates from CD4+ T cells pre-treated with U0126 (20 µM) and subsequently treated with hBD2 (20 µg/mL) were probed for APOBEC3G and β-actin by Western blotting. Images shown are representative of 2–4 independent experiments from different donors or cell passages.

Figure 4.
hBD2 induces transcription of APOBEC3G mRNA. (a) Primary CD4+ T cells were treated with hBD2 (20 μg/mL), pre-treated with actinomycin D (10 μg/mL), or pre-treated with actinomycin D (10 μg/mL) followed by treatment with hBD2 (20 μg/mL). APOBEC3G mRNA was assessed by quantitative real-time PCR comparing treated samples with untreated samples at matched timepoints. The data was normalized to 18S ribosomal RNA and reflects the results from two different donors. Error bars representing SEM were included. (b) JKT-FT7 and JKT-FT7 CCR6 GFP cells were treated with hBD2 (20 μg/mL). A3G mRNA was assessed by quantitative real-time PCR comparing treated samples with untreated samples at matched timepoints. The data reflects the results of three independent experiments. Error bars representing SEM were included. Statistical analysis was performed by paired 2-tail Student t test. * p < 0.05, *** p < 0.001.
Figure 4.
hBD2 induces transcription of APOBEC3G mRNA. (a) Primary CD4+ T cells were treated with hBD2 (20 μg/mL), pre-treated with actinomycin D (10 μg/mL), or pre-treated with actinomycin D (10 μg/mL) followed by treatment with hBD2 (20 μg/mL). APOBEC3G mRNA was assessed by quantitative real-time PCR comparing treated samples with untreated samples at matched timepoints. The data was normalized to 18S ribosomal RNA and reflects the results from two different donors. Error bars representing SEM were included. (b) JKT-FT7 and JKT-FT7 CCR6 GFP cells were treated with hBD2 (20 μg/mL). A3G mRNA was assessed by quantitative real-time PCR comparing treated samples with untreated samples at matched timepoints. The data reflects the results of three independent experiments. Error bars representing SEM were included. Statistical analysis was performed by paired 2-tail Student t test. * p < 0.05, *** p < 0.001.

Figure 5.
hBD2 enhances binding of NFAT and IRF-4 to the APOBEC3G promoter. JKT-FT7 CCR6 GFP cells were treated with hBD2 (20 μg/mL) for the indicated timepoints. Chromatin immunoprecipitation (ChIP) assays were performed using antibodies against NFATc2, NFATc1, or IRF-4. Immunoprecipitated DNA was detected by quantitative real-time PCR performed in triplicate. Shown is fold enrichment determined by comparison with IgG control of three independent experiments. Error bars representing SEM were included. Statistical analysis was performed by paired 2-tail Student t test. * p < 0.05, ** p < 0.01.
Figure 5.
hBD2 enhances binding of NFAT and IRF-4 to the APOBEC3G promoter. JKT-FT7 CCR6 GFP cells were treated with hBD2 (20 μg/mL) for the indicated timepoints. Chromatin immunoprecipitation (ChIP) assays were performed using antibodies against NFATc2, NFATc1, or IRF-4. Immunoprecipitated DNA was detected by quantitative real-time PCR performed in triplicate. Shown is fold enrichment determined by comparison with IgG control of three independent experiments. Error bars representing SEM were included. Statistical analysis was performed by paired 2-tail Student t test. * p < 0.05, ** p < 0.01.

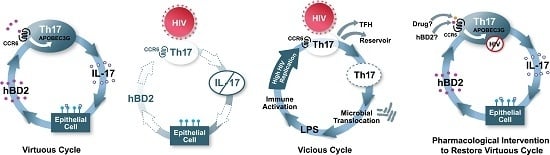
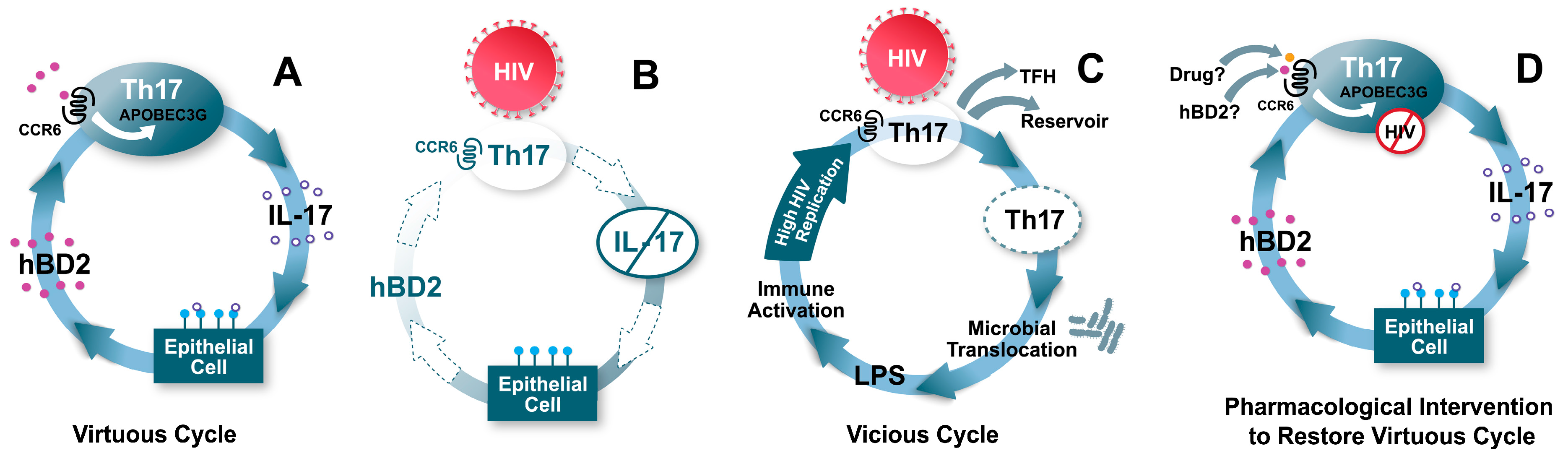
Figure 6.
Model: hBD2 contributes to a “virtuous cycle”, maintaining mucosal integrity and protecting CCR6+ T cells. (A) CCR6+ T cells include Th17 cells, which produce IL-17. IL-17 has several effects on mucosal epithelia, including regulation of tight junctions, and induction of the antimicrobial peptide hBD2. hBD2 binding to CCR6 induces intracellular signaling events leading to increased transcription of APOBEC3G, which inhibits HIV intracellularly. (B) When HIV reaches CCR6+ Th17 cells due to breaches in the mucosa, their death causes loss of production of IL-17, and hBD2 production is also decreased as a consequence. Thus, CCR6+ T cells become more permissive to HIV infection. (C) The loss of mucosal integrity due to lower levels of Th17 and hBD2 result in microbial translocation. High levels of lipopolysaccharide (LPS) and other microbial factors cause immune activation, which further enhance HIV replication, eventuating in a self-perpetuating “vicious cycle”. (D) Induction or administration of adequate levels of hBD2, or a pharmaceutical drug targeting CCR6 could result in high levels of expression of APOBEC3G and protection of CCR6+ Th17 cells.
Figure 6.
Model: hBD2 contributes to a “virtuous cycle”, maintaining mucosal integrity and protecting CCR6+ T cells. (A) CCR6+ T cells include Th17 cells, which produce IL-17. IL-17 has several effects on mucosal epithelia, including regulation of tight junctions, and induction of the antimicrobial peptide hBD2. hBD2 binding to CCR6 induces intracellular signaling events leading to increased transcription of APOBEC3G, which inhibits HIV intracellularly. (B) When HIV reaches CCR6+ Th17 cells due to breaches in the mucosa, their death causes loss of production of IL-17, and hBD2 production is also decreased as a consequence. Thus, CCR6+ T cells become more permissive to HIV infection. (C) The loss of mucosal integrity due to lower levels of Th17 and hBD2 result in microbial translocation. High levels of lipopolysaccharide (LPS) and other microbial factors cause immune activation, which further enhance HIV replication, eventuating in a self-perpetuating “vicious cycle”. (D) Induction or administration of adequate levels of hBD2, or a pharmaceutical drug targeting CCR6 could result in high levels of expression of APOBEC3G and protection of CCR6+ Th17 cells.

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Lafferty, M.K.; Sun, L.; Christensen-Quick, A.; Lu, W.; Garzino-Demo, A. Human Beta Defensin 2 Selectively Inhibits HIV-1 in Highly Permissive CCR6+CD4+ T Cells. Viruses 2017, 9, 111. https://0-doi-org.brum.beds.ac.uk/10.3390/v9050111
AMA Style
Lafferty MK, Sun L, Christensen-Quick A, Lu W, Garzino-Demo A. Human Beta Defensin 2 Selectively Inhibits HIV-1 in Highly Permissive CCR6+CD4+ T Cells. Viruses. 2017; 9(5):111. https://0-doi-org.brum.beds.ac.uk/10.3390/v9050111
Chicago/Turabian StyleLafferty, Mark K., Lingling Sun, Aaron Christensen-Quick, Wuyuan Lu, and Alfredo Garzino-Demo. 2017. "Human Beta Defensin 2 Selectively Inhibits HIV-1 in Highly Permissive CCR6+CD4+ T Cells" Viruses 9, no. 5: 111. https://0-doi-org.brum.beds.ac.uk/10.3390/v9050111
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.