
Immunologic and Virologic Mechanisms for Partial Protection from Intravenous Challenge by an Integration-Defective SIV Vaccine †

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
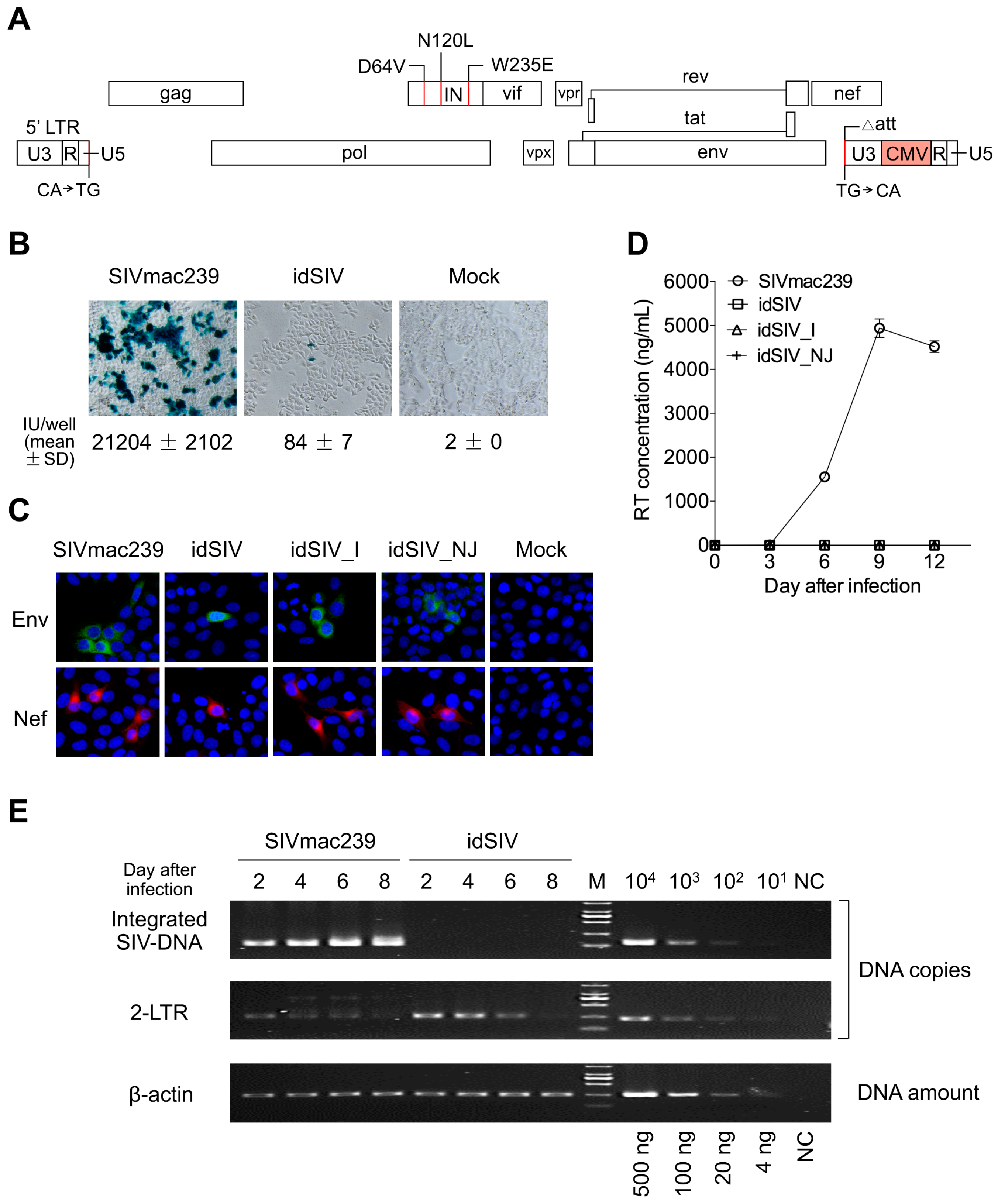
2.1. Generation and Characterization of a Novel Integration-Defective SIV Vaccine
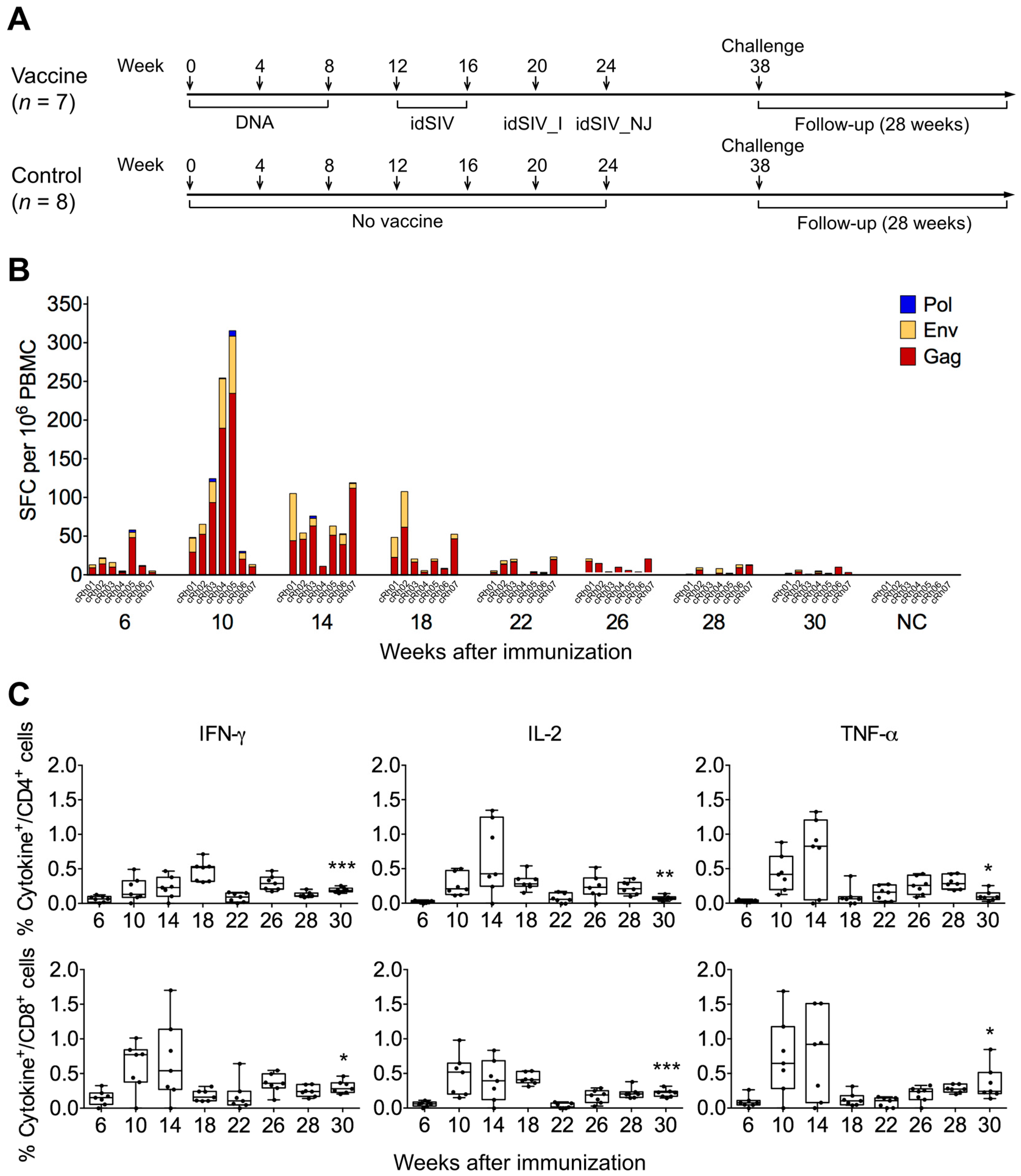
2.2. Elicitation of Cellular Responses
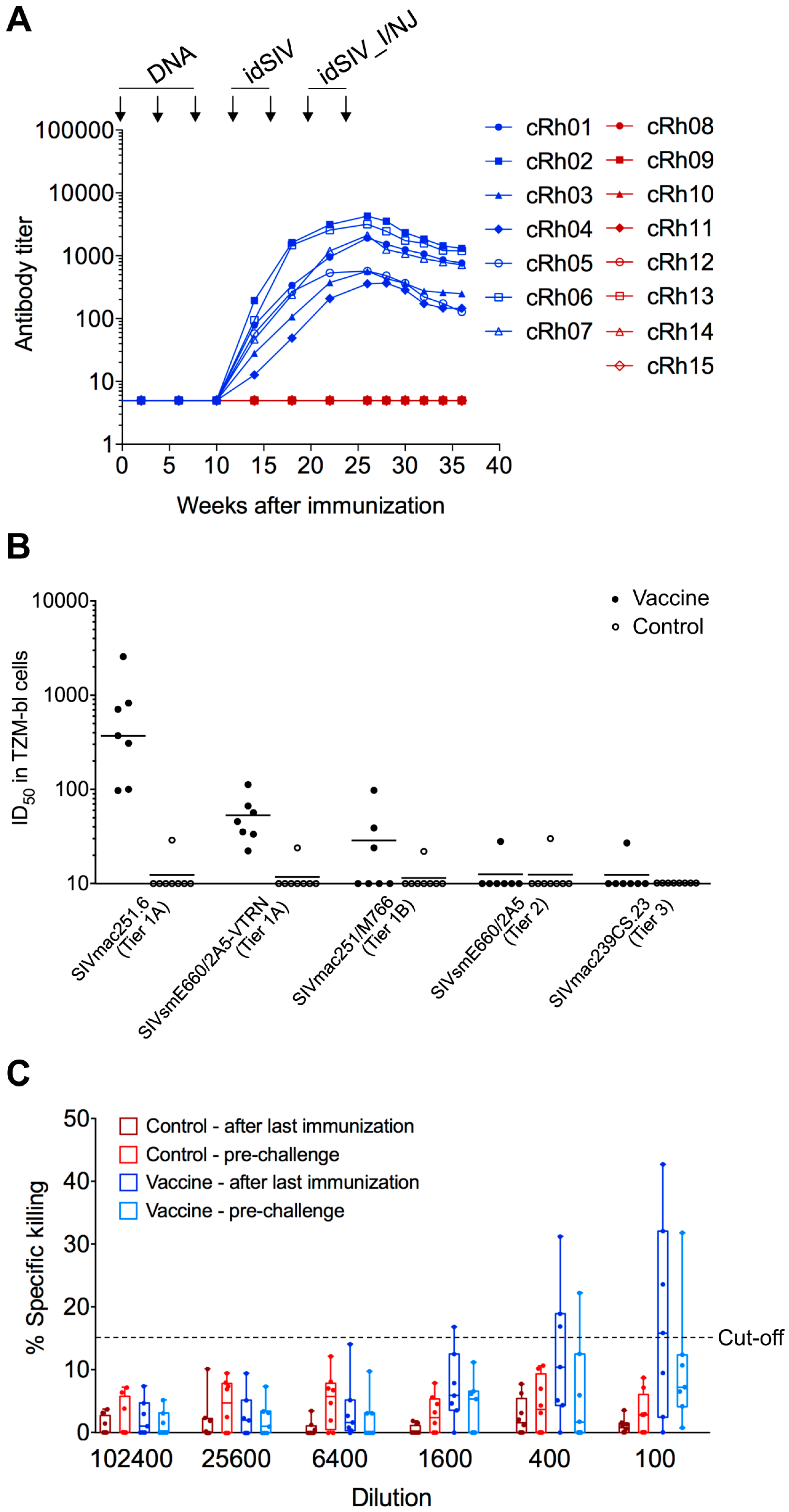
2.3. Humoral Responses in idSIV Vaccinated Macaques
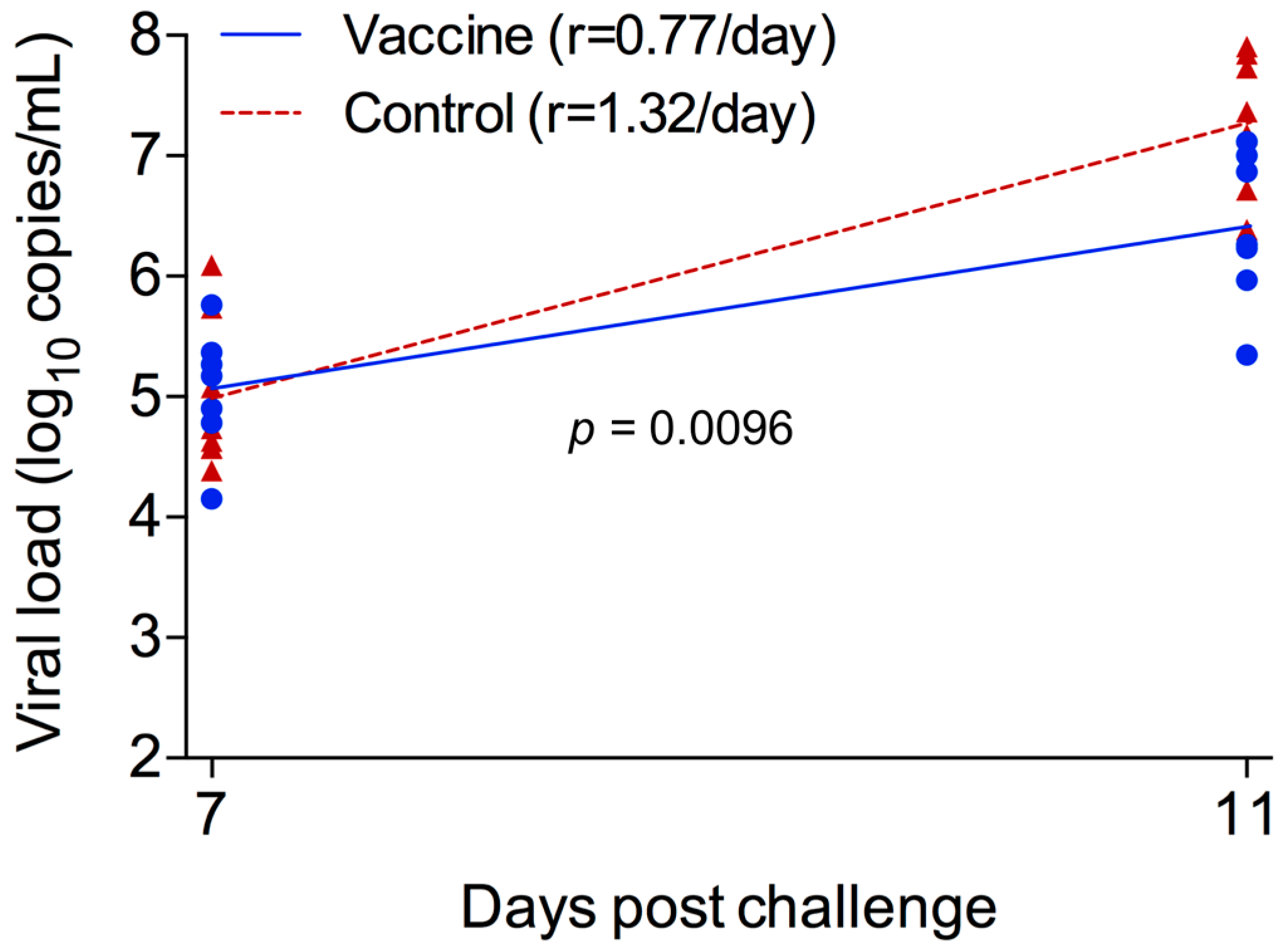
2.4. Reduced Peak Viremia in Vaccinated Monkeys after Challenge
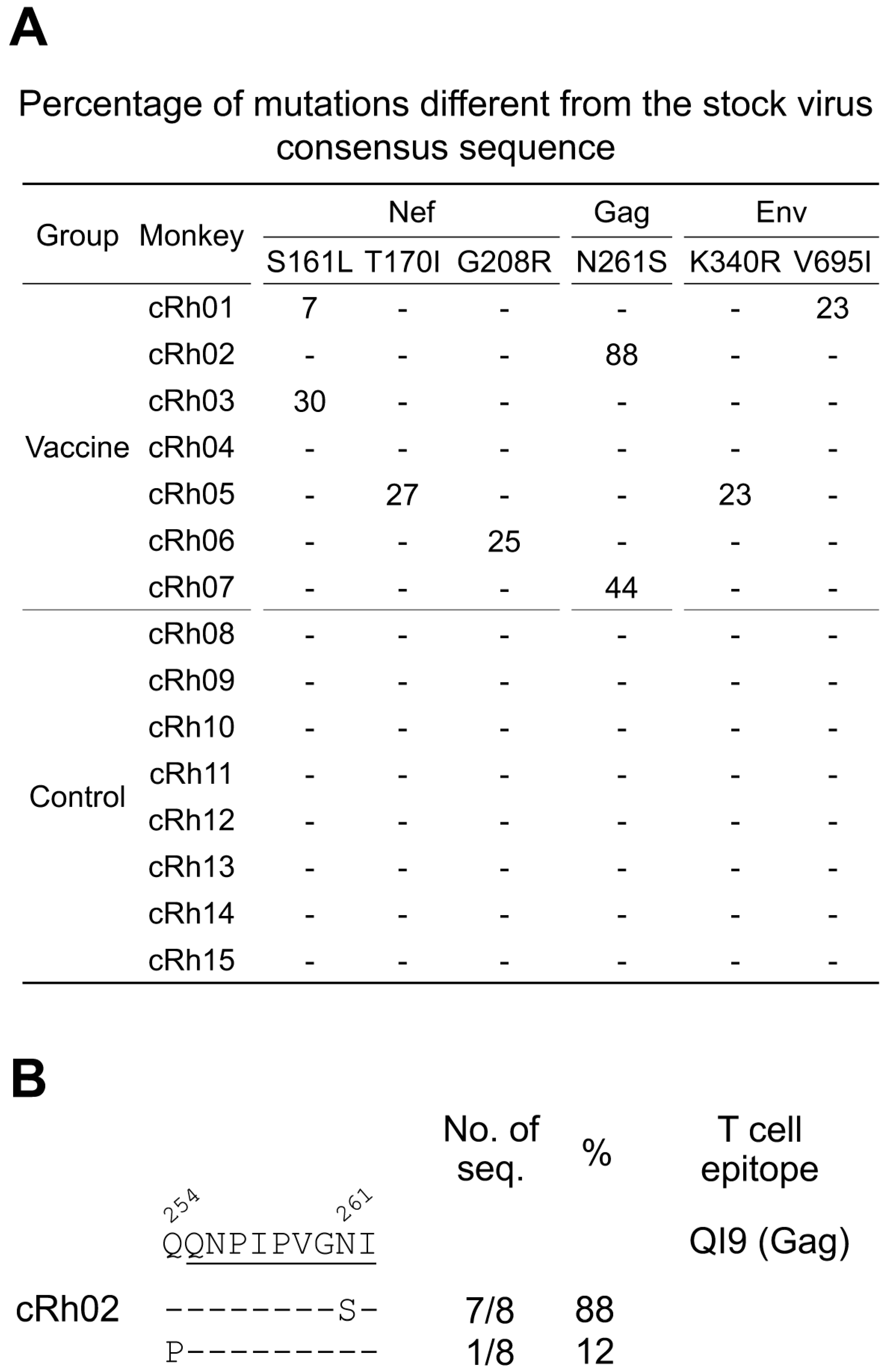
2.5. Identification of Selection Signatures in Breakthrough Viruses
3. Discussion
4. Materials and Methods
4.1. Generation of the Full-Length idSIV Genome
4.2. Generation of Purified idSIV Particles
4.3. Detection of Integrated and Unintegrated Proviral Genomes
4.4. Viral Infectivity Assay
4.5. Immunization and Challenge of Rhesus Macaques
4.6. Determination of T Cell Immune Response
4.7. SIV Specific Binding Antibody Assay
4.8. Neutralization Assay
4.9. ADCC Assays
4.10. Plasma Viral Load Assay
4.11. Viral Sequence Analysis
4.12. Statistical Analysis
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Burton, D.R.; Ahmed, R.; Barouch, D.H.; Butera, S.T.; Crotty, S.; Godzik, A.; Kaufmann, D.E.; McElrath, M.J.; Nussenzweig, M.C.; Pulendran, B.; et al. A blueprint for HIV vaccine discovery. Cell Host Microbe 2012, 12, 396–407. [Google Scholar] [CrossRef] [PubMed]
- Haynes, B.F.; Shaw, G.M.; Korber, B.; Kelsoe, G.; Sodroski, J.; Hahn, B.H.; Borrow, P.; McMichael, A.J. HIV-host interactions: Implications for vaccine design. Cell Host Microbe 2016, 19, 292–303. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, M.R.; Weiler, A.M.; Weisgrau, K.L.; Piaskowski, S.M.; Furlott, J.R.; Weinfurter, J.T.; Kaizu, M.; Soma, T.; Leon, E.J.; MacNair, C.; et al. Macaques vaccinated with live-attenuated SIV control replication of heterologous virus. J. Exp. Med. 2008, 205, 2537–2550. [Google Scholar] [CrossRef] [PubMed]
- Daniel, M.D.; Kirchhoff, F.; Czajak, S.C.; Sehgal, P.K.; Desrosiers, R.C. Protective effects of a live attenuated SIV vaccine with a deletion in the nef gene. Science 1992, 258, 1938–1941. [Google Scholar] [CrossRef] [PubMed]
- Willer, D.O.; Guan, Y.; Luscher, M.A.; Li, B.; Pilon, R.; Fournier, J.; Parenteau, M.; Wainberg, M.A.; Sandstrom, P.; MacDonald, K.S. Multi-low-dose mucosal simian immunodeficiency virus SIVmac239 challenge of cynomolgus macaques immunized with “hyperattenuated” SIV constructs. J. Virol. 2010, 84, 2304–2317. [Google Scholar] [CrossRef] [PubMed]
- Baba, T.W.; Liska, V.; Khimani, A.H.; Ray, N.B.; Dailey, P.J.; Penninck, D.; Bronson, R.; Greene, M.F.; McClure, H.M.; Martin, L.N.; et al. Live attenuated, multiply deleted simian immunodeficiency virus causes AIDS in infant and adult macaques. Nat. Med. 1999, 5, 194–203. [Google Scholar] [CrossRef] [PubMed]
- Hansen, S.G.; Ford, J.C.; Lewis, M.S.; Ventura, A.B.; Hughes, C.M.; Coyne-Johnson, L.; Whizin, N.; Oswald, K.; Shoemaker, R.; Swanson, T.; et al. Profound early control of highly pathogenic SIV by an effector memory T-cell vaccine. Nature 2011, 473, 523–527. [Google Scholar] [CrossRef] [PubMed]
- Hansen, S.G.; Sacha, J.B.; Hughes, C.M.; Ford, J.C.; Burwitz, B.J.; Scholz, I.; Gilbride, R.M.; Lewis, M.S.; Gilliam, A.N.; Ventura, A.B.; et al. Cytomegalovirus vectors violate CD8+ T cell epitope recognition paradigms. Science 2013, 340, 1237874. [Google Scholar] [CrossRef] [PubMed]
- Sloan, R.D.; Wainberg, M.A. The role of unintegrated DNA in HIV infection. Retrovirology 2011, 8, 52. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Ourmanov, I.; Hirsch, V.M. Persistent transcription of a nonintegrating mutant of simian immunodeficiency virus in rhesus macrophages. Virology 2008, 372, 291–299. [Google Scholar] [CrossRef] [PubMed]
- Kelly, J.; Beddall, M.H.; Yu, D.; Iyer, S.R.; Marsh, J.W.; Wu, Y. Human macrophages support persistent transcription from unintegrated HIV-1 DNA. Virology 2008, 372, 300–312. [Google Scholar] [CrossRef] [PubMed]
- Cara, A.; Klotman, M.E. Retroviral E-DNA: Persistence and gene expression in nondividing immune cells. J. Leukoc. Biol. 2006, 80, 1013–1017. [Google Scholar] [CrossRef] [PubMed]
- Yanez-Munoz, R.J.; Balaggan, K.S.; MacNeil, A.; Howe, S.J.; Schmidt, M.; Smith, A.J.; Buch, P.; MacLaren, R.E.; Anderson, P.N.; Barker, S.E.; et al. Effective gene therapy with nonintegrating lentiviral vectors. Nat. Med. 2006, 12, 348–353. [Google Scholar] [CrossRef] [PubMed]
- Michelini, Z.; Negri, D.R.; Baroncelli, S.; Spada, M.; Leone, P.; Bona, R.; Klotman, M.E.; Cara, A. Development and use of SIV-based integrase defective lentiviral vector for immunization. Vaccine 2009, 27, 4622–4629. [Google Scholar] [CrossRef] [PubMed]
- Philippe, S.; Sarkis, C.; Barkats, M.; Mammeri, H.; Ladroue, C.; Petit, C.; Mallet, J.; Serguera, C. Lentiviral vectors with a defective integrase allow efficient and sustained transgene expression in vitro and in vivo. Proc. Natl. Acad. Sci. USA 2006, 103, 17684–17689. [Google Scholar] [CrossRef] [PubMed]
- Negri, D.; Blasi, M.; LaBranche, C.; Parks, R.; Balachandran, H.; Lifton, M.; Shen, X.; Denny, T.; Ferrari, G.; Vescio, M.F.; et al. Immunization with an SIV-based IDLV expressing HIV-1 env 1086 clade C elicits durable humoral and cellular responses in rhesus macaques. Mol. Ther. 2016, 24, 2021–2032. [Google Scholar] [CrossRef] [PubMed]
- Cara, A.; Cereseto, A.; Lori, F.; Reitz, M.S., Jr. HIV-1 protein expression from synthetic circles of DNA mimicking the extrachromosomal forms of viral DNA. J. Biol. Chem. 1996, 271, 5393–5397. [Google Scholar] [CrossRef] [PubMed]
- Coutant, F.; Sanchez David, R.Y.; Felix, T.; Boulay, A.; Caleechurn, L.; Souque, P.; Thouvenot, C.; Bourgouin, C.; Beignon, A.S.; Charneau, P. A nonintegrative lentiviral vector-based vaccine provides long-term sterile protection against malaria. PLoS ONE 2012, 7, e48644. [Google Scholar] [CrossRef] [PubMed]
- Couzin, J.; Kaiser, J. Gene therapy. As gelsinger case ends, gene therapy suffers another blow. Science 2005, 307, 1028. [Google Scholar] [CrossRef] [PubMed]
- Hacein-Bey-Abina, S.; Von Kalle, C.; Schmidt, M.; McCormack, M.P.; Wulffraat, N.; Leboulch, P.; Lim, A.; Osborne, C.S.; Pawliuk, R.; Morillon, E.; et al. LMO2-associated clonal t cell proliferation in two patients after gene therapy for SCID-X1. Science 2003, 302, 415–419. [Google Scholar] [CrossRef] [PubMed]
- Engelman, A. In vivo analysis of retroviral integrase structure and function. Adv. Virus Res. 1999, 52, 411–426. [Google Scholar] [PubMed]
- Lesbats, P.; Engelman, A.N.; Cherepanov, P. Retroviral DNA integration. Chem. Rev. 2016, 63, 836–843. [Google Scholar] [CrossRef] [PubMed]
- O’ Connor, D.H.; Allen, T.M.; Vogel, T.U.; Jing, P.; DeSouza, I.P.; Dodds, E.; Dunphy, E.J.; Melsaether, C.; Mothe, B.; Yamamoto, H.; et al. Acute phase cytotoxic T lymphocyte escape is a hallmark of simian immunodeficiency virus infection. Nat. Med. 2002, 8, 493–499. [Google Scholar] [CrossRef] [PubMed]
- Cale, E.M.; Hraber, P.; Giorgi, E.E.; Fischer, W.; Bhattacharya, T.; Leitner, T.; Yeh, W.W.; Gleasner, C.; Green, L.D.; Han, C.S.; et al. Epitope-specific CD8+ T lymphocytes cross-recognize mutant simian immunodeficiency virus (SIV) sequences but fail to contain very early evolution and eventual fixation of epitope escape mutations during SIV infection. J. Virol. 2011, 85, 3746–3757. [Google Scholar] [CrossRef] [PubMed]
- Allen, T.M.; O’ Connor, D.H.; Jing, P.; Dzuris, J.L.; Mothe, B.R.; Vogel, T.U.; Dunphy, E.; Liebl, M.E.; Emerson, C.; Wilson, N.; et al. Tat-specific cytotoxic T lymphocytes select for SIV escape variants during resolution of primary viraemia. Nature 2000, 407, 386–390. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.A.; Kilgore, K.M.; Kasturi, S.P.; Pulendran, B.; Hunter, E.; Amara, R.R.; Derdeyn, C.A. Signatures in simian immunodeficiency virus SIVsmE660 envelope gp120 are associated with mucosal transmission but not vaccination breakthrough in rhesus macaques. J. Virol. 2016, 90, 1880–1887. [Google Scholar] [CrossRef] [PubMed]
- Rolland, M.; Edlefsen, P.T.; Larsen, B.B.; Tovanabutra, S.; Sanders-Buell, E.; Hertz, T.; deCamp, A.C.; Carrico, C.; Menis, S.; Magaret, C.A.; et al. Increased HIV-1 vaccine efficacy against viruses with genetic signatures in Env V2. Nature 2012, 490, 417–420. [Google Scholar] [CrossRef] [PubMed]
- Patel, V.; Jalah, R.; Kulkarni, V.; Valentin, A.; Rosati, M.; Alicea, C.; von Gegerfelt, A.; Huang, W.; Guan, Y.; Keele, B.F.; et al. DNA and virus particle vaccination protects against acquisition and confers control of viremia upon heterologous simian immunodeficiency virus challenge. Proc. Natl. Acad. Sci. USA 2013, 110, 2975–2980. [Google Scholar] [CrossRef] [PubMed]
- Vaccari, M.; Keele, B.F.; Bosinger, S.E.; Doster, M.N.; Ma, Z.M.; Pollara, J.; Hryniewicz, A.; Ferrari, G.; Guan, Y.; Forthal, D.N.; et al. Protection afforded by an HIV vaccine candidate in macaques depends on the dose of SIVmac251 at challenge exposure. J. Virol. 2013, 87, 3538–3548. [Google Scholar] [CrossRef] [PubMed]
- Burton, S.L.; Kilgore, K.M.; Smith, S.A.; Reddy, S.; Hunter, E.; Robinson, H.L.; Silvestri, G.; Amara, R.R.; Derdeyn, C.A. Breakthrough of SIV strain smE660 challenge in SIV strain mac239-vaccinated rhesus macaques despite potent autologous neutralizing antibody responses. Proc. Natl. Acad. Sci. USA 2015, 112, 10780–10785. [Google Scholar] [CrossRef] [PubMed]
- Roederer, M.; Keele, B.F.; Schmidt, S.D.; Mason, R.D.; Welles, H.C.; Fischer, W.; Labranche, C.; Foulds, K.E.; Louder, M.K.; Yang, Z.Y.; et al. Immunological and virological mechanisms of vaccine-mediated protection against SIV and HIV. Nature 2014, 505, 502–508. [Google Scholar] [CrossRef] [PubMed]
- Evans, D.T.; O’ Connor, D.H.; Jing, P.; Dzuris, J.L.; Sidney, J.; da Silva, J.; Allen, T.M.; Horton, H.; Venham, J.E.; Rudersdorf, R.A.; et al. Virus-specific cytotoxic T-lymphocyte responses select for amino-acid variation in simian immunodeficiency virus Env and Nef. Nat. Med. 1999, 5, 1270–1276. [Google Scholar] [CrossRef] [PubMed]
- Brockman, M.A.; Schneidewind, A.; Lahaie, M.; Schmidt, A.; Miura, T.; Desouza, I.; Ryvkin, F.; Derdeyn, C.A.; Allen, S.; Hunter, E.; et al. Escape and compensation from early HLA-B57-mediated cytotoxic T-lymphocyte pressure on human immunodeficiency virus type 1 Gag alter capsid interactions with cyclophilin A. J. Virol. 2007, 81, 12608–12618. [Google Scholar] [CrossRef] [PubMed]
- Crawford, H.; Prado, J.G.; Leslie, A.; Hue, S.; Honeyborne, I.; Reddy, S.; van der Stok, M.; Mncube, Z.; Brander, C.; Rousseau, C.; et al. Compensatory mutation partially restores fitness and delays reversion of escape mutation within the immunodominant HLA-B*5703-restricted Gag epitope in chronic human immunodeficiency virus type 1 infection. J. Virol. 2007, 81, 8346–8351. [Google Scholar] [CrossRef] [PubMed]
- Brockman, M.A.; Brumme, Z.L.; Brumme, C.J.; Miura, T.; Sela, J.; Rosato, P.C.; Kadie, C.M.; Carlson, J.M.; Markle, T.J.; Streeck, H.; et al. Early selection in Gag by protective HLA alleles contributes to reduced HIV-1 replication capacity that may be largely compensated for in chronic infection. J. Virol. 2010, 84, 11937–11949. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Hora, B.; Bhattacharya, T.; Goonetilleke, N.; Liu, M.K.; Wiehe, K.; Li, H.; Iyer, S.S.; McMichael, A.J.; Perelson, A.S.; et al. Reversion and T cell escape mutations compensate the fitness loss of a CD8+ T cell escape mutant in their cognate transmitted/founder virus. PLoS ONE 2014, 9, e102734. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Zuo, T.; Hora, B.; Song, H.; Kong, W.; Yu, X.; Goonetilleke, N.; Bhattacharya, T.; Perelson, A.S.; Haynes, B.F.; et al. Preexisting compensatory amino acids compromise fitness costs of a HIV-1 T cell escape mutation. Retrovirology 2014, 11, 101. [Google Scholar] [CrossRef] [PubMed]
- Kloverpris, H.N.; Leslie, A.; Goulder, P. Role of HLA adaptation in HIV evolution. Front. Immunol. 2015, 6, 665. [Google Scholar] [CrossRef] [PubMed]
- Cara, A.; Maggiorella, M.T.; Bona, R.; Sernicola, L.; Baroncelli, S.; Negri, D.R.; Leone, P.; Fagrouch, Z.; Heeney, J.; Titti, F.; et al. Circular viral DNA detection and junction sequence analysis from PBMC of SHIV-infected cynomolgus monkeys with undetectable virus plasma RNA. Virology 2004, 324, 531–539. [Google Scholar] [CrossRef] [PubMed]
- Fenizia, C.; Keele, B.F.; Nichols, D.; Cornara, S.; Binello, N.; Vaccari, M.; Pegu, P.; Robert-Guroff, M.; Ma, Z.M.; Miller, C.J.; et al. TRIM5α does not affect simian immunodeficiency virus SIV(mac251) replication in vaccinated or unvaccinated indian rhesus macaques following intrarectal challenge exposure. J. Virol. 2011, 85, 12399–12409. [Google Scholar] [CrossRef] [PubMed]
- Vaccari, M.; Gordon, S.N.; Fourati, S.; Schifanella, L.; Liyanage, N.P.; Cameron, M.; Keele, B.F.; Shen, X.; Tomaras, G.D.; Billings, E.; et al. Adjuvant-dependent innate and adaptive immune signatures of risk of SIVmac251 acquisition. Nat. Med. 2016, 22, 762–770. [Google Scholar] [CrossRef] [PubMed]
- Montefiori, D.C. Measuring HIV neutralization in a luciferase reporter gene assay. Methods Mol. Biol. 2009, 485, 395–405. [Google Scholar] [PubMed]
- Liao, H.X.; Bonsignori, M.; Alam, S.M.; McLellan, J.S.; Tomaras, G.D.; Moody, M.A.; Kozink, D.M.; Hwang, K.K.; Chen, X.; Tsao, C.Y.; et al. Vaccine induction of antibodies against a structurally heterogeneous site of immune pressure within HIV-1 envelope protein variable regions 1 and 2. Immunity 2013, 38, 176–186. [Google Scholar] [CrossRef] [PubMed]
- Pollara, J.; Bonsignori, M.; Moody, M.A.; Liu, P.; Alam, S.M.; Hwang, K.K.; Gurley, T.C.; Kozink, D.M.; Armand, L.C.; Marshall, D.J.; et al. HIV-1 vaccine-induced C1 and V2 Env-specific antibodies synergize for increased antiviral activities. J. Virol. 2014, 88, 7715–7726. [Google Scholar] [CrossRef] [PubMed]
- Palmer, S.; Kearney, M.; Maldarelli, F.; Halvas, E.K.; Bixby, C.J.; Bazmi, H.; Rock, D.; Falloon, J.; Davey, R.T., Jr.; Dewar, R.L.; et al. Multiple, linked human immunodeficiency virus type 1 drug resistance mutations in treatment-experienced patients are missed by standard genotype analysis. J. Clin. Microbiol. 2005, 43, 406–413. [Google Scholar] [CrossRef] [PubMed]
- Keele, B.F.; Giorgi, E.E.; Salazar-Gonzalez, J.F.; Decker, J.M.; Pham, K.T.; Salazar, M.G.; Sun, C.; Grayson, T.; Wang, S.; Li, H.; et al. Identification and characterization of transmitted and early founder virus envelopes in primary HIV-1 infection. Proc. Natl. Acad. Sci. USA 2008, 105, 7552–7557. [Google Scholar] [CrossRef] [PubMed]






© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, C.; Jiang, C.; Gao, N.; Zhang, K.; Liu, D.; Wang, W.; Cong, Z.; Qin, C.; Ganusov, V.V.; Ferrari, G.; et al. Immunologic and Virologic Mechanisms for Partial Protection from Intravenous Challenge by an Integration-Defective SIV Vaccine. Viruses 2017, 9, 135. https://0-doi-org.brum.beds.ac.uk/10.3390/v9060135
Wang C, Jiang C, Gao N, Zhang K, Liu D, Wang W, Cong Z, Qin C, Ganusov VV, Ferrari G, et al. Immunologic and Virologic Mechanisms for Partial Protection from Intravenous Challenge by an Integration-Defective SIV Vaccine. Viruses. 2017; 9(6):135. https://0-doi-org.brum.beds.ac.uk/10.3390/v9060135
Chicago/Turabian StyleWang, Chu, Chunlai Jiang, Nan Gao, Kaikai Zhang, Donglai Liu, Wei Wang, Zhe Cong, Chuan Qin, Vitaly V. Ganusov, Guido Ferrari, and et al. 2017. "Immunologic and Virologic Mechanisms for Partial Protection from Intravenous Challenge by an Integration-Defective SIV Vaccine" Viruses 9, no. 6: 135. https://0-doi-org.brum.beds.ac.uk/10.3390/v9060135