Synthesis of a Bone-Targeted Bortezomib with In Vivo Anti-Myeloma Effects in Mice

 ,
,
Abstract
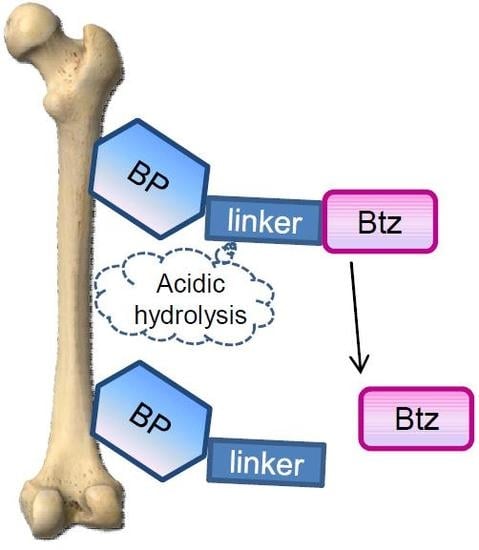
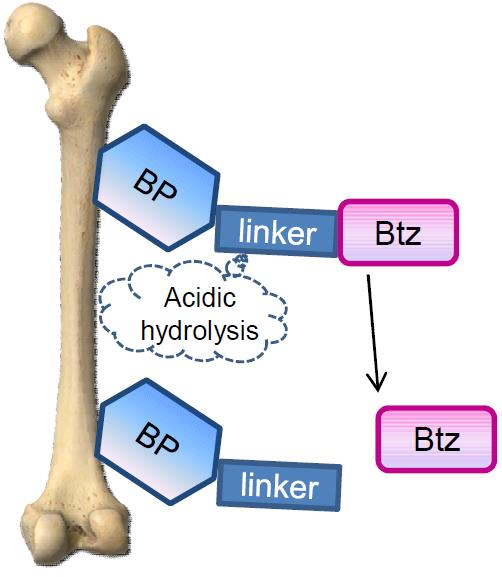
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
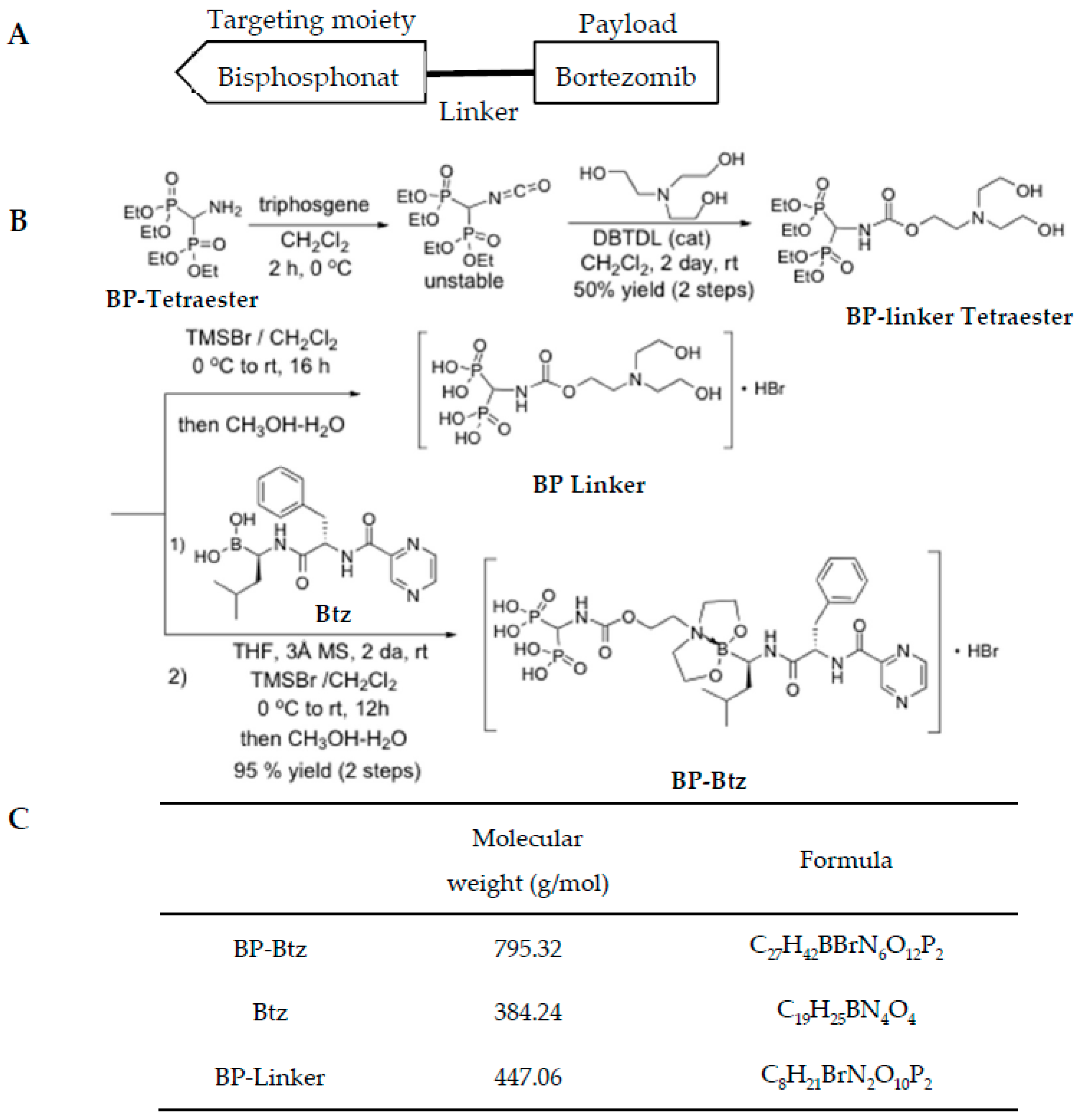
2.1. Preparation of BP-Btz
2.1.1. 2-(bis(2-hydroxyethyl)amino)ethyl (bis(diethoxyphosphoryl)methyl)carbamate (BP-Linker Tetraester)
2.1.2. (((2-(8-((R)-3-methyl-1-((S)-3-phenyl-2-(pyrazine-2-carboxamido)propanamido)butyl)tetrahydro-8H-4I4,8I4-[1,3,2]oxazaborolo[2,3-b]-[1,3,2]oxazaborol-4-yl)ethoxy)carbonyl)amino)methylene)bis(phosphonic acid (BP-Btz)
2.1.3. 2-(bis(2-hydroxyethyl)amino)ethyl (bis(dihydroxyphosphoryl)methyl)carbamate (1 BP-Linker)
2.2. Animal Experiments
2.2.1. Enzyme Linked Immunosorbent Assay (ELISA)
2.2.2. IVIS Imaging
2.2.3. MicroCT
2.2.4. Histology and Histomorphometric Analysis
2.2.5. Cell Cultures
2.2.6. Routine Blood Counting
2.3. Statistical Analysis
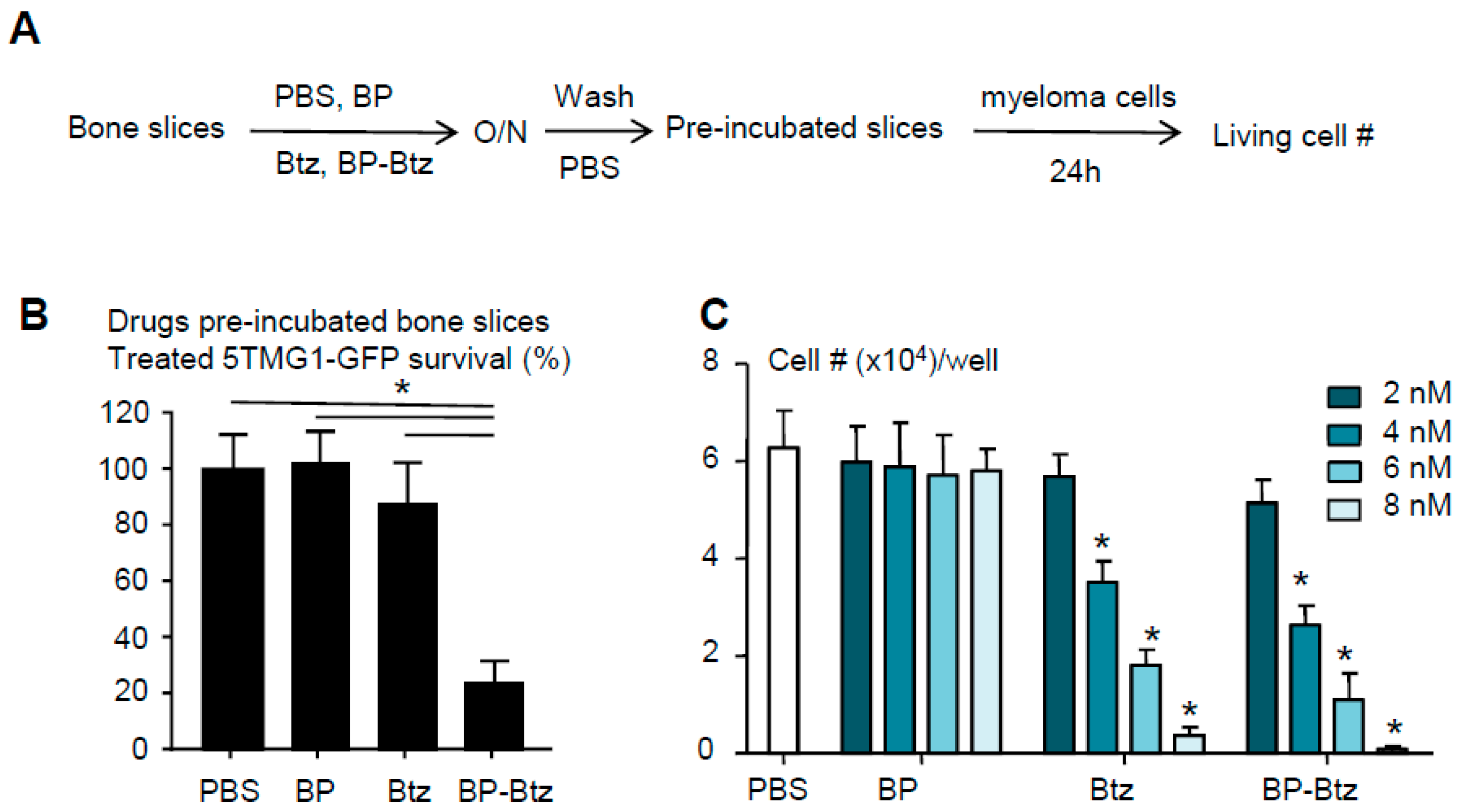
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- San Miguel, J.; Blade, J.; Boccadoro, M.; Cavenagh, J.; Glasmacher, A.; Jagannath, S.; Lonial, S.; Orlowski, R.Z.; Sonneveld, P.; Ludwig, H. A practical update on the use of bortezomib in the management of multiple myeloma. Oncologist 2006, 11, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Shaji, K.; Kumar, N.S.C. National Comprehensive Cancer Network (nccn) clinical practice guidelines in oncology. In Multiple Myeloma; V.4.2018; National Comprehensive Cancer Network: Fort Washington, PA, USA, 2018. [Google Scholar]
- Dingli, D.; Ailawadhi, S.; Bergsagel, P.L.; Buadi, F.K.; Dispenzieri, A.; Fonseca, R.; Gertz, M.A.; Gonsalves, W.I.; Hayman, S.R.; Kapoor, P.; et al. Therapy for relapsed multiple myeloma: Guidelines from the mayo stratification for myeloma and risk-adapted therapy. Mayo Clin. Proc. 2017, 92, 578–598. [Google Scholar] [CrossRef] [PubMed]
- Hideshima, T.; Richardson, P.G.; Anderson, K.C. Mechanism of action of proteasome inhibitors and deacetylase inhibitors and the biological basis of synergy in multiple myeloma. Mol. Cancer Ther. 2011, 10, 2034–2042. [Google Scholar] [CrossRef] [PubMed]
- Obeng, E.A.; Carlson, L.M.; Gutman, D.M.; Harrington, W.J., Jr.; Lee, K.P.; Boise, L.H. Proteasome inhibitors induce a terminal unfolded protein response in multiple myeloma cells. Blood 2006, 107, 4907–4916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zinszner, H.; Kuroda, M.; Wang, X.; Batchvarova, N.; Lightfoot, R.T.; Remotti, H.; Stevens, J.L.; Ron, D. Chop is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes. Dev. 1998, 12, 982–995. [Google Scholar] [CrossRef] [PubMed]
- Brewer, J.W.; Diehl, J.A. Perk mediates cell-cycle exit during the mammalian unfolded protein response. Proc. Natl. Acad. Sci. USA 2000, 97, 12625–12630. [Google Scholar] [CrossRef] [PubMed]
- Mohty, B.; El-Cheikh, J.; Yakoub-Agha, I.; Moreau, P.; Harousseau, J.L.; Mohty, M. Peripheral neuropathy and new treatments for multiple myeloma: Background and practical recommendations. Haematologica 2010, 95, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Cole, L.E.; Vargo-Gogola, T.; Roeder, R.K. Targeted delivery to bone and mineral deposits using bisphosphonate ligands. Adv. Drug Deliv. Rev. 2016, 99, 12–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agyin, J.K.; Santhamma, B.; Roy, S.S. Design, synthesis, and biological evaluation of bone-targeted proteasome inhibitors for multiple myeloma. Bioorg. Med. Chem. Lett. 2013, 23, 6455–6458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dallas, S.L.; Garrett, I.R.; Oyajobi, B.O.; Dallas, M.R.; Boyce, B.F.; Bauss, F.; Radl, J.; Mundy, G.R. Ibandronate reduces osteolytic lesions but not tumor burden in a murine model of myeloma bone disease. Blood 1999, 93, 1697–1706. [Google Scholar] [PubMed]
- Oyajobi, B.O.; Munoz, S.; Kakonen, R.; Williams, P.J.; Gupta, A.; Wideman, C.L.; Story, B.; Grubbs, B.; Armstrong, A.; Dougall, W.C.; et al. Detection of myeloma in skeleton of mice by whole-body optical fluorescence imaging. Mol. Cancer Ther. 2007, 6, 1701–1708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oyajobi, B.O.; Franchin, G.; Williams, P.J.; Pulkrabek, D.; Gupta, A.; Munoz, S.; Grubbs, B.; Zhao, M.; Chen, D.; Sherry, B.; et al. Dual effects of macrophage inflammatory protein-1alpha on osteolysis and tumor burden in the murine 5tgm1 model of myeloma bone disease. Blood 2003, 102, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Sun, W.; Li, J.; Wang, M.; Zhang, H.; Pei, L.; Boyce, B.F.; Wang, Z.; Xing, L. Clomipramine causes osteoporosis by promoting osteoclastogenesis via e3 ligase itch, which is prevented by zoledronic acid. Sci. Rep. 2017, 7, 41358. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Hilton, M.J.; Anolik, J.H.; Welle, S.L.; Zhao, C.; Yao, Z.; Li, X.; Wang, Z.; Boyce, B.F.; Xing, L. Notch inhibits osteoblast formation in inflammatory arthritis via noncanonical nf-kappab. J. Clin. Investig. 2014, 124, 3200–3214. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Gangal, G.; Uludag, H. ‘Magic bullets’ for bone diseases: Progress in rational design of bone-seeking medicinal agents. Chem. Soc. Rev. 2007, 36, 507–531. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.S.; Houghton, T.J.; Kang, T.; Dietrich, E.; Delorme, D.; Ferreira, S.S.; Caron, L.; Viens, F.; Arhin, F.F.; Sarmiento, I.; et al. Bisphosphonated fluoroquinolone esters as osteotropic prodrugs for the prevention of osteomyelitis. Bioorg. Med. Chem. 2008, 16, 9217–9229. [Google Scholar] [CrossRef] [PubMed]
- Terpos, E.; Heath, D.J.; Rahemtulla, A.; Zervas, K.; Chantry, A.; Anagnostopoulos, A.; Pouli, A.; Katodritou, E.; Verrou, E.; Vervessou, E.C.; et al. Bortezomib reduces serum dickkopf-1 and receptor activator of nuclear factor-kappab ligand concentrations and normalises indices of bone remodelling in patients with relapsed multiple myeloma. Br. J. Haematol. 2006, 135, 688–692. [Google Scholar] [CrossRef] [PubMed]
- Roodman, G.D. Bone building with bortezomib. J. Clin. Investig. 2008, 118, 462–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weathington, N.M.; Mallampalli, R.K. Emerging therapies targeting the ubiquitin proteasome system in cancer. J. Clin. Investig. 2014, 124, 6–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richardson, P.G.; Sonneveld, P.; Schuster, M.W.; Irwin, D.; Stadtmauer, E.A.; Facon, T.; Harousseau, J.L.; Ben-Yehuda, D.; Lonial, S.; Goldschmidt, H.; et al. Bortezomib or high-dose dexamethasone for relapsed multiple myeloma. N. Engl. J. Med. 2005, 352, 2487–2498. [Google Scholar] [CrossRef] [PubMed]
- Bonvini, P.; Zorzi, E.; Basso, G.; Rosolen, A. Bortezomib-mediated 26s proteasome inhibition causes cell-cycle arrest and induces apoptosis in cd-30+ anaplastic large cell lymphoma. Leukemia 2007, 21, 838–842. [Google Scholar] [CrossRef] [PubMed]
- Nawrocki, S.T.; Carew, J.S.; Pino, M.S.; Highshaw, R.A.; Dunner, K., Jr.; Huang, P.; Abbruzzese, J.L.; McConkey, D.J. Bortezomib sensitizes pancreatic cancer cells to endoplasmic reticulum stress-mediated apoptosis. Cancer Res. 2005, 65, 11658–11666. [Google Scholar] [CrossRef] [PubMed]
- Shakespeare, W.C.; Wang, Y.; Bohacek, R.; Keenan, T.; Sundaramoorthi, R.; Metcalf, C., 3rd; Dilauro, A.; Roeloffzen, S.; Liu, S.; Saltmarsh, J.; et al. Sar of carbon-linked, 2-substituted purines: Synthesis and characterization of ap23451 as a novel bone-targeted inhibitor of src tyrosine kinase with in vivo anti-resorptive activity. Chem. Biol. Drug Des. 2008, 71, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Boyce, B.; Shakespeare, W.C.; Xing, L.; Wang, Y.; Sundaramoorthi, R.; Keenan, T.; Metcalf, C.; Bohacek, R.; van Schravendijk, M.R.; Dalgarno, D.; et al. Development of a novel bone-targeted src tyrosine kinase inhibitor ap23451 having potental in vitro and in vivo anti-resorptive properties. J. Bone Miner. Res. 2002, S159. [Google Scholar]
- Argyriou, A.A.; Cavaletti, G.; Bruna, J.; Kyritsis, A.P.; Kalofonos, H.P. Bortezomib-induced peripheral neurotoxicity: An update. Arch. Toxicol. 2014, 88, 1669–1679. [Google Scholar] [CrossRef] [PubMed]
- Cavaletti, G.; Gilardini, A.; Canta, A.; Rigamonti, L.; Rodriguez-Menendez, V.; Ceresa, C.; Marmiroli, P.; Bossi, M.; Oggioni, N.; D’Incalci, M.; et al. Bortezomib-induced peripheral neurotoxicity: A neurophysiological and pathological study in the rat. Exp. Neurol. 2007, 204, 317–325. [Google Scholar] [CrossRef] [PubMed]





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, H.; Xiao, L.; Tao, J.; Srinivasan, V.; Boyce, B.F.; Ebetino, F.H.; Oyajobi, B.O.; Boeckman, R.K., Jr.; Xing, L. Synthesis of a Bone-Targeted Bortezomib with In Vivo Anti-Myeloma Effects in Mice. Pharmaceutics 2018, 10, 154. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics10030154
Wang H, Xiao L, Tao J, Srinivasan V, Boyce BF, Ebetino FH, Oyajobi BO, Boeckman RK Jr., Xing L. Synthesis of a Bone-Targeted Bortezomib with In Vivo Anti-Myeloma Effects in Mice. Pharmaceutics. 2018; 10(3):154. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics10030154
Chicago/Turabian StyleWang, Hua, Lifeng Xiao, Jianguo Tao, Venkat Srinivasan, Brendan F. Boyce, Frank H. Ebetino, Babatunde O. Oyajobi, Robert K. Boeckman, Jr., and Lianping Xing. 2018. "Synthesis of a Bone-Targeted Bortezomib with In Vivo Anti-Myeloma Effects in Mice" Pharmaceutics 10, no. 3: 154. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics10030154